
Comparative Genomics of Clinical Isolates of the Emerging Tick-Borne Pathogen Neoehrlichia mikurensis

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
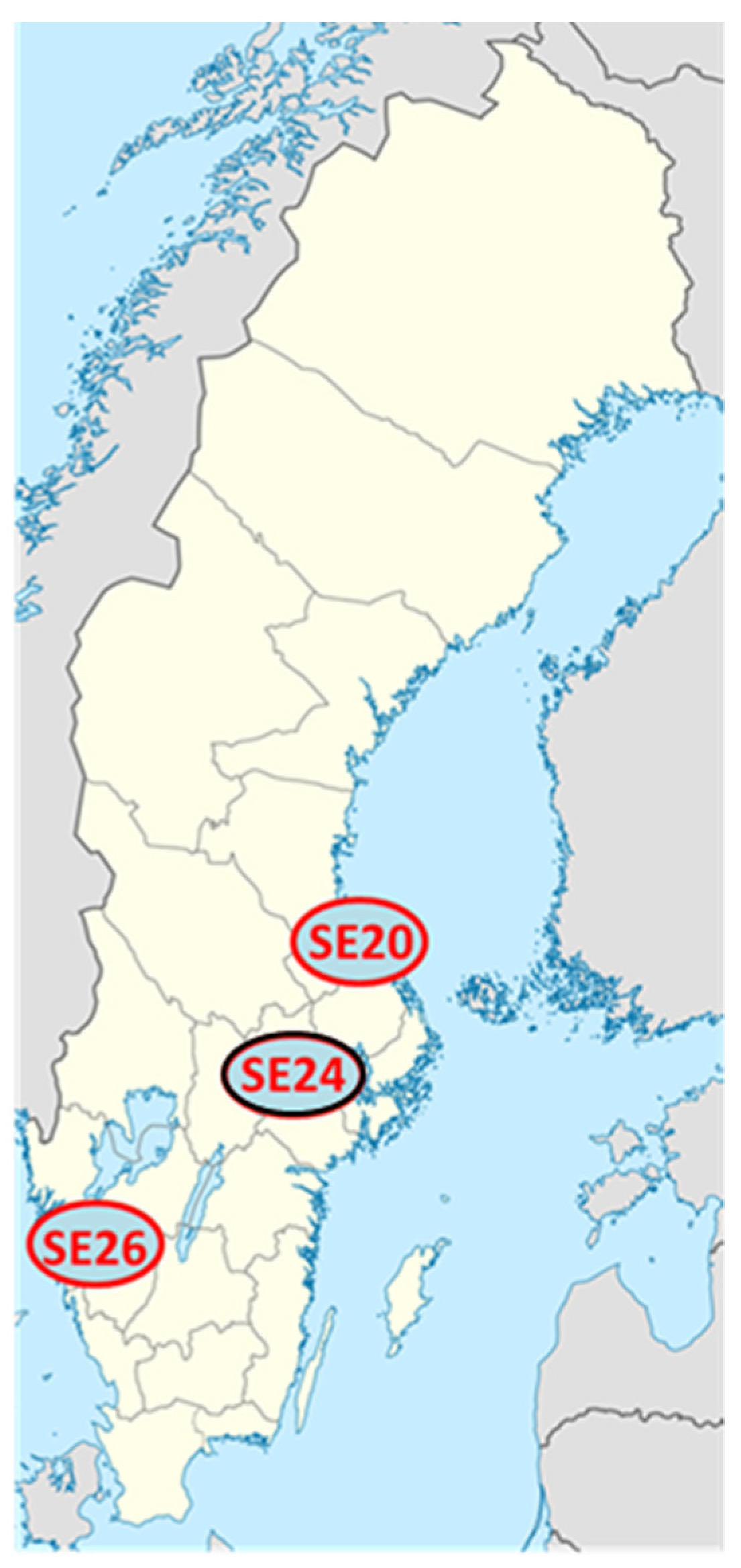
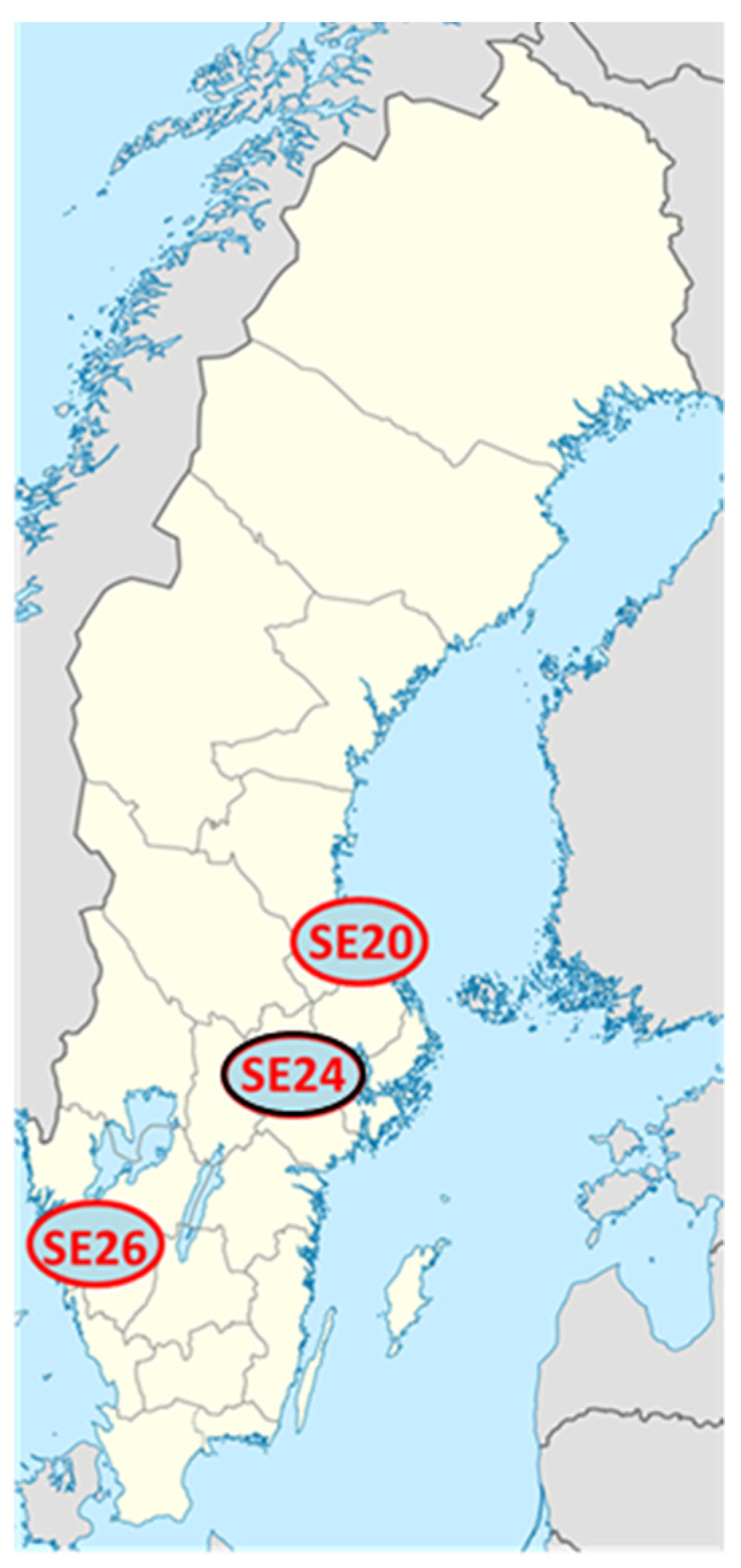
2.1. Clinical Isolates of Ca. N. mikurensis
2.2. Tick Cell Line Cultivated Isolate of Ca. N. mikurensis
2.3. Bacterial DNA Extraction
2.4. 10X Chromium Library and Sequencing
2.5. Genomic Analyses and Comparisons
2.6. Phylogenetic Analyses
3. Results and Discussion
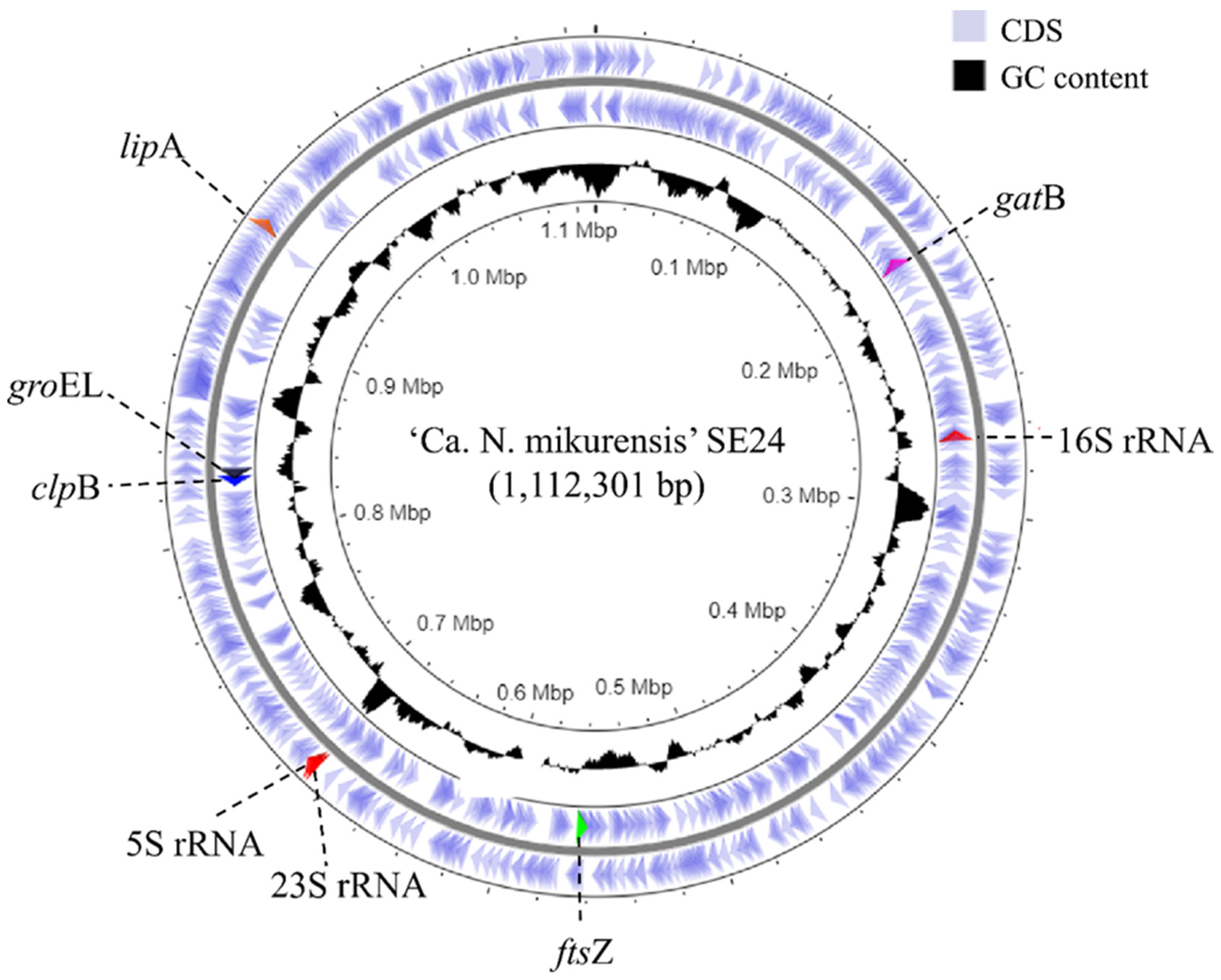
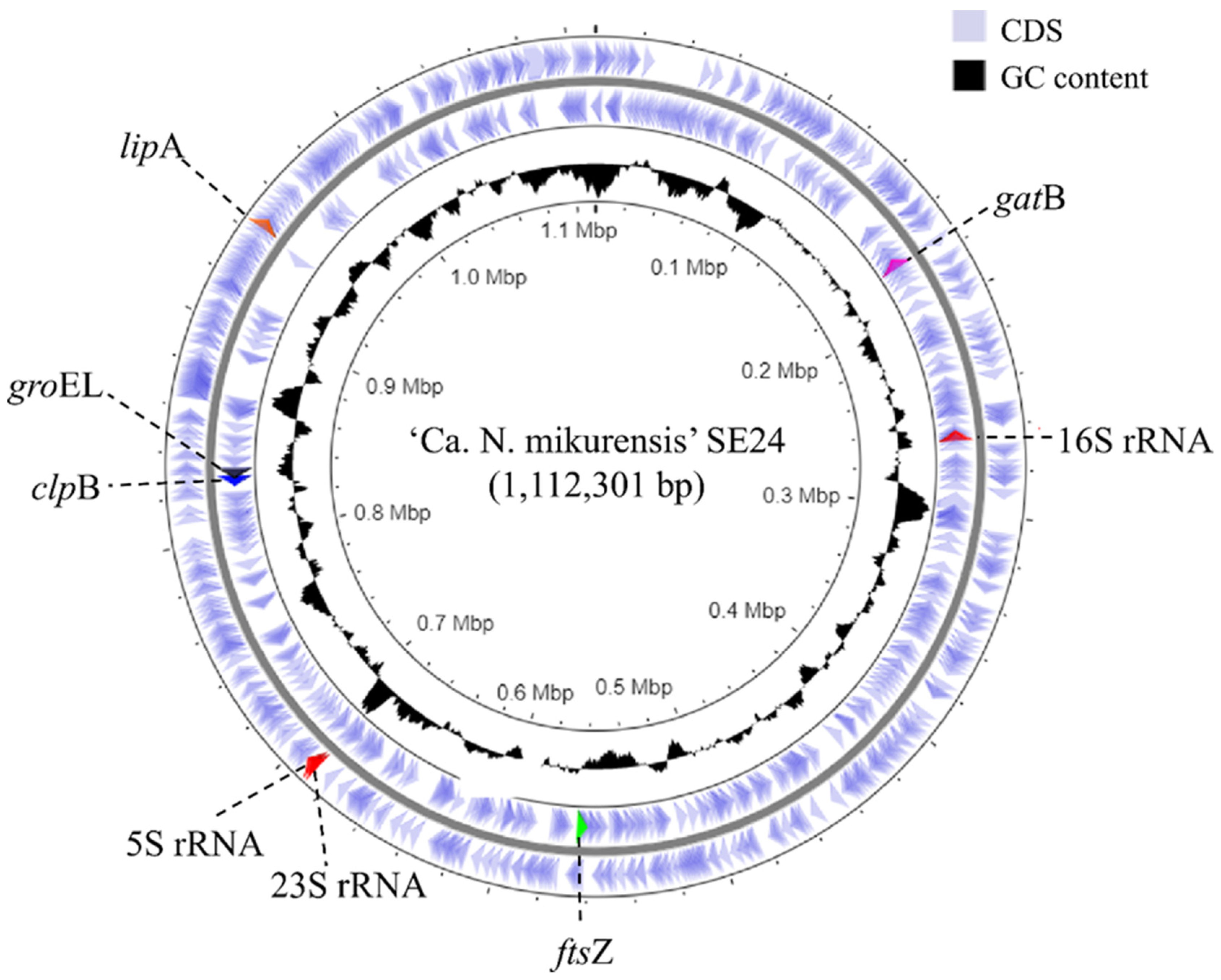
3.1. Genome Assembly and De Novo Annotation
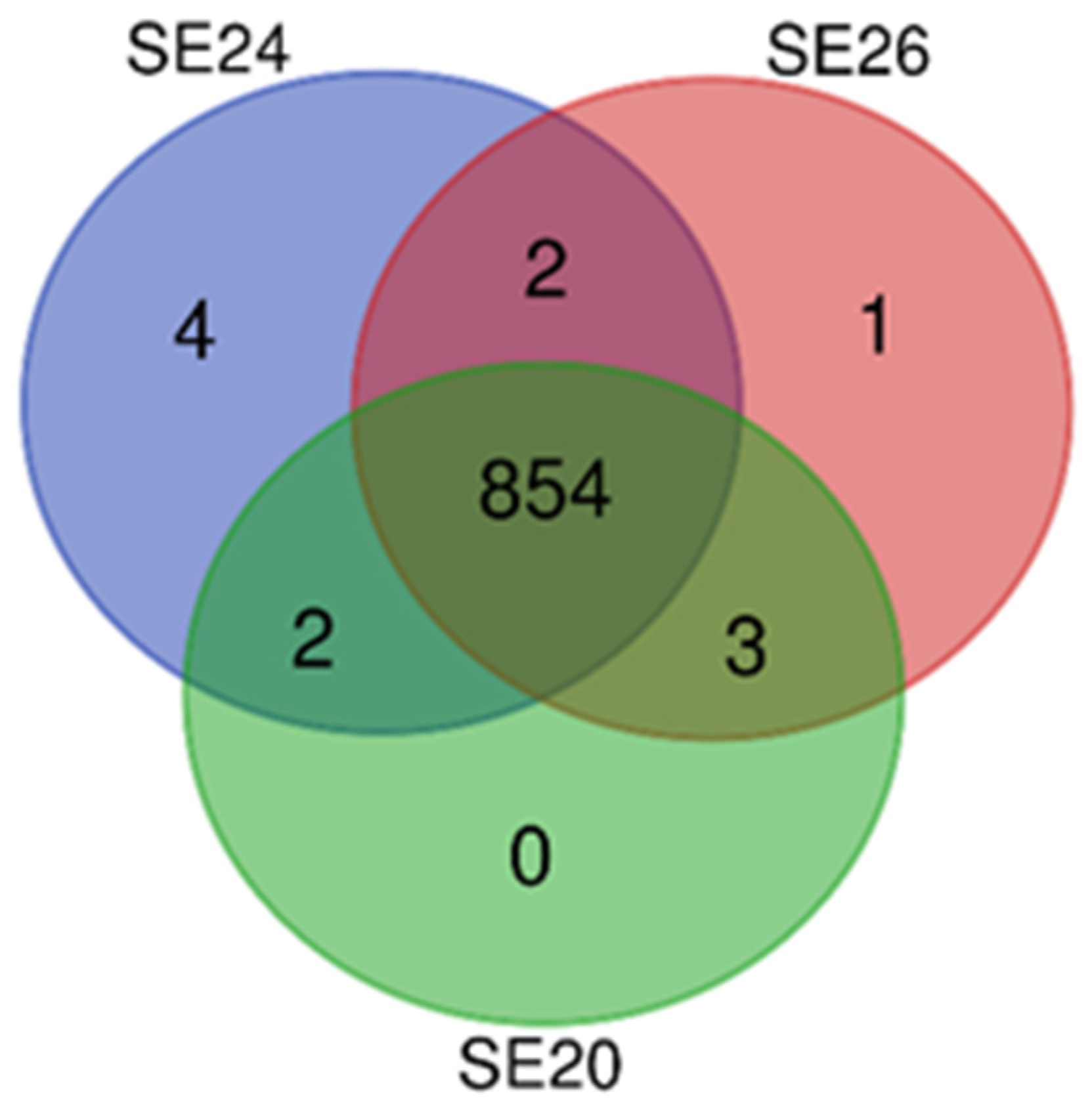
3.2. Intra-Species Genomic Comparisons
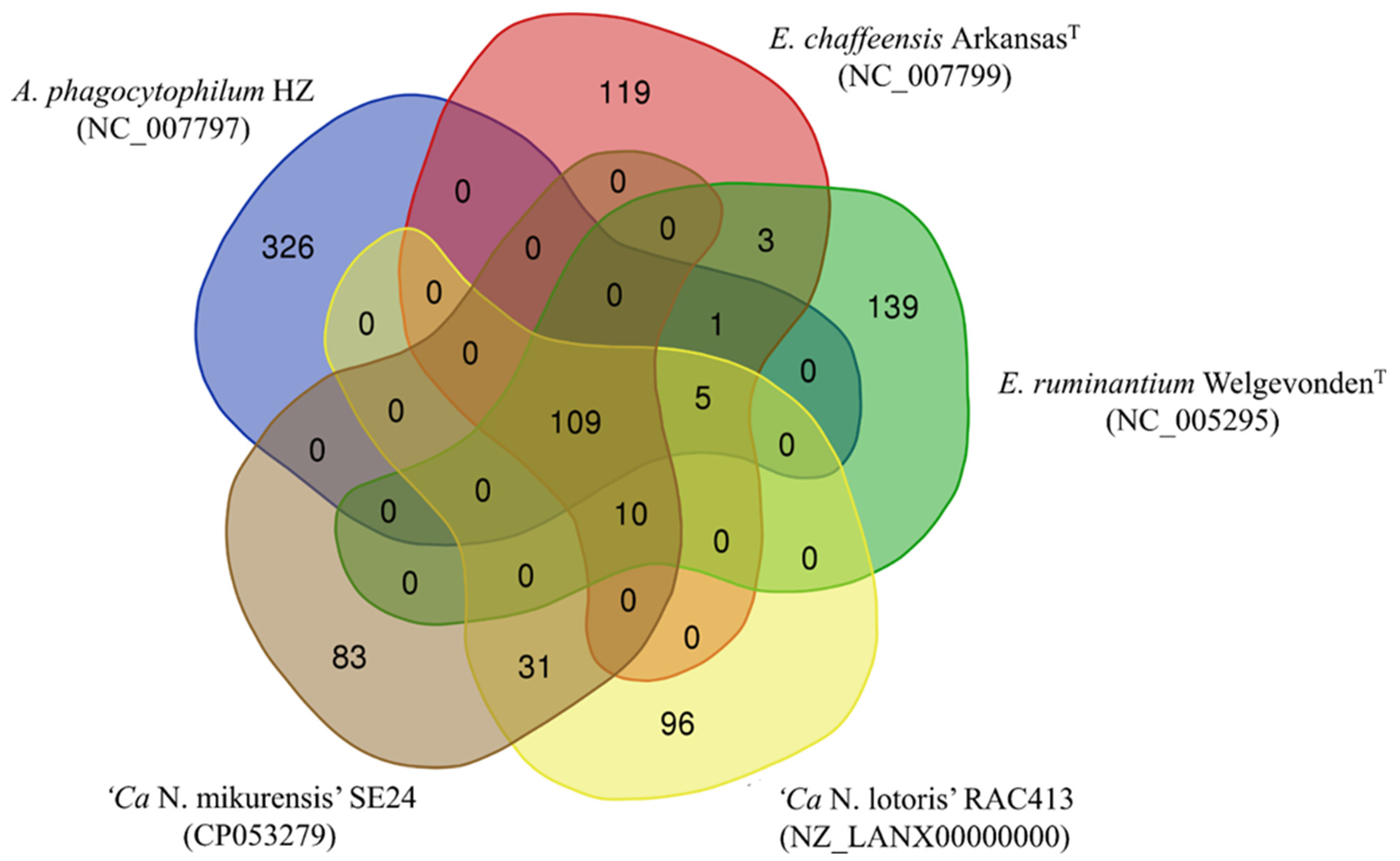
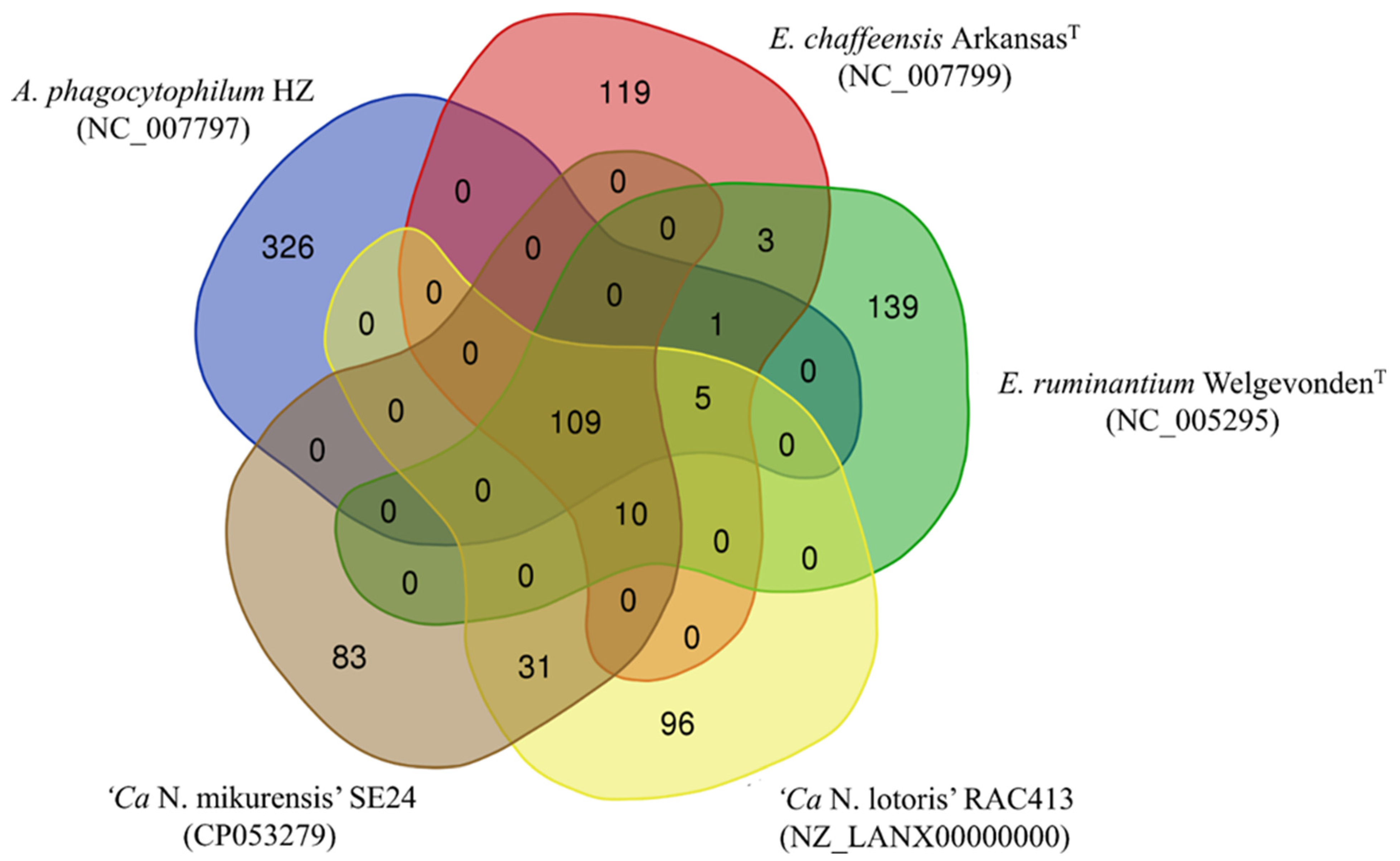
3.3. Comparison of the Ca. N. mikurensis Genome with Other Genomes within the Anaplasmataceae Family
3.4. Protein Comparisons between Anaplasmataceae Species
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kawahara, M.; Rikihisa, Y.; Isogai, E.; Takahashi, M.; Misumi, H.; Suto, C.; Shibata, S.; Zhang, C.; Tsuji, M. Ultrastructure and phylogenetic analysis of ‘Candidatus Neoehrlichia mikurensis’ in the family Anaplasmataceae, isolated from wild rats and found in Ixodes ovatus ticks. Int. J. Syst. Evol. Microbiol. 2004, 54, 1837–1843. [Google Scholar] [CrossRef]
- Schouls, L.M.; Van De Pol, I.; Rijpkema, S.G.; Schot, C.S. Detection and identification of Ehrlichia, Borrelia burgdorferi sensu lato, and Bartonella species in Dutch Ixodes ricinus ticks. J. Clin. Microbiol. 1999, 37, 2215–2222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rar, V.A.; Livanova, N.N.; Panov, V.V.; Doroschenko, E.K.; Pukhovskaya, N.M.; Vysochina, N.P.; Ivanov, L.I. Genetic diversity of Anaplasma and Ehrlichia in the Asian part of Russia. Ticks Tick Borne Dis. 2010, 1, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Andersson, M.; Raberg, L. Wild rodents and novel human pathogen candidatus Neoehrlichia mikurensis, Southern Sweden. Emerg. Infect. Dis. 2011, 17, 1716–1718. [Google Scholar] [CrossRef] [PubMed]
- Welinder-Olsson, C.; Kjellin, E.; Vaht, K.; Jacobsson, S.; Wenneras, C. First case of human “Candidatus Neoehrlichia mikurensis” infection in a febrile patient with chronic lymphocytic leukemia. J. Clin. Microbiol. 2010, 48, 1956–1959. [Google Scholar] [CrossRef] [Green Version]
- Von Loewenich, F.D.; Geissdorfer, W.; Disque, C.; Matten, J.; Schett, G.; Sakka, S.G.; Bogdan, C. Detection of “Candidatus Neoehrlichia mikurensis” in two patients with severe febrile illnesses: Evidence for a European sequence variant. J. Clin. Microbiol. 2010, 48, 2630–2635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pekova, S.; Vydra, J.; Kabickova, H.; Frankova, S.; Haugvicova, R.; Mazal, O.; Cmejla, R.; Hardekopf, D.W.; Jancuskova, T.; Kozak, T. Candidatus Neoehrlichia mikurensis infection identified in 2 hematooncologic patients: Benefit of molecular techniques for rare pathogen detection. Diagn. Microbiol. Infect. Dis. 2011, 69, 266–270. [Google Scholar] [CrossRef]
- Fehr, J.S.; Bloemberg, G.V.; Ritter, C.; Hombach, M.; Luscher, T.F.; Weber, R.; Keller, P.M. Septicemia caused by tick-borne bacterial pathogen Candidatus Neoehrlichia mikurensis. Emerg. Infect. Dis. 2010, 16, 1127–1129. [Google Scholar] [CrossRef] [Green Version]
- Grankvist, A.; Andersson, P.-O.; Mattsson, M.; Sender, M.; Vaht, K.; Hoper, L.; Sakiniene, E.; Trysberg, E.; Stenson, M.; Fehr, J.; et al. Infections with the Tick-Borne Bacterium “Candidatus Neoehrlichia mikurensis” Mimic Noninfectious Conditions in Patients with B Cell Malignancies or Autoimmune Diseases. Clin. Infect. Dis. 2014, 58, 1716–1722. [Google Scholar] [CrossRef]
- Wass, L.; Grankvist, A.; Bell-Sakyi, L.; Bergström, M.; Ulfhammer, E.; Lingblom, C.; Wennerås, C. Cultivation of the causative agent of human neoehrlichiosis from clinical isolates identifies vascular endothelium as a target of infection. Emerg. Microbes Infect. 2019, 8, 413–425. [Google Scholar] [CrossRef] [Green Version]
- Grankvist, A.; Sandelin, L.L.; Andersson, J.; Fryland, L.; Wilhelmsson, P.; Lindgren, P.E.; Forsberg, P.; Wenneras, C. Infections with Candidatus Neoehrlichia mikurensis and Cytokine Responses in 2 Persons Bitten by Ticks, Sweden. Emerg. Infect. Dis. 2015, 21, 1462–1465. [Google Scholar] [CrossRef]
- Welc-Faleciak, R.; Sinski, E.; Kowalec, M.; Zajkowska, J.; Pancewicz, S.A. Asymptomatic “Candidatus Neoehrlichia mikurensis” infections in immunocompetent humans. J. Clin. Microbiol. 2014, 52, 3072–3074. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Jiang, J.-F.; Liu, W.; Zheng, Y.-C.; Huo, Q.-B.; Tang, K.; Zuo, S.-Y.; Liu, K.; Jiang, B.-G.; Yang, H.; et al. Human Infection withCandidatusNeoehrlichia mikurensis, China. Emerg. Infect. Dis. 2012, 18, 1636–1639. [Google Scholar] [CrossRef] [PubMed]
- Yabsley, M.J.; Murphy, S.M.; Luttrell, M.P.; Wilcox, B.R.; Howerth, E.W.; Munderloh, U.G. Characterization of ‘Candidatus Neoehrlichia lotoris’ (family Anaplasmataceae) from raccoons (Procyon lotor). Int. J. Syst. Evol. Microbiol. 2008, 58, 2794–2798. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Jiang, J.; Tang, F.; Sun, Y.; Li, Z.; Zhang, W.; Gong, Z.; Liu, K.; Yang, H.; Liu, W.; et al. Wide Distribution and Genetic Diversity of “Candidatus Neoehrlichia mikurensis” in Rodents from China. Appl. Environ. Microbiol. 2013, 79, 1024–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rar, V.A.; Epikhina, T.I.; Livanova, N.N.; Panov, V.V.; Doroshenko, E.K.; Pukhovskaia, N.M.; Vysochina, N.P.; Ivanov, L.I. Study of the heterogeneity of 16s rRNA gene and groESL operone in the dna samples of Anaplasma phagocytophilum, Ehrlichia muris, and “Candidatus Neoehrlichia mikurensis” determined in the Ixodes persulcatus ticks in the area of Urals, Siberia, and far east of Russia. Mol. Gen. Mikrobiol. Virusol. 2011, 2, 17–23. [Google Scholar]
- Grankvist, A.; Moore, E.R.; Svensson Stadler, L.; Pekova, S.; Bogdan, C.; Geissdorfer, W.; Grip-Linden, J.; Brandstrom, K.; Marsal, J.; Andreasson, K.; et al. Multilocus Sequence Analysis of Clinical “Candidatus Neoehrlichia mikurensis” Strains from Europe. J. Clin. Microbiol. 2015, 53, 3126–3132. [Google Scholar] [CrossRef] [Green Version]
- Allsopp, B.A. Heartwater--Ehrlichia ruminantium infection. Rev. Sci. Tech. 2015, 34, 557–568. [Google Scholar] [CrossRef] [Green Version]
- Weisenfeld, N.I.; Kumar, V.; Shah, P.; Church, D.M.; Jaffe, D.B. Direct determination of diploid genome sequences. Genome Res. 2017, 27, 757–767. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [Green Version]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Haft, D.H.; DiCuccio, M.; Badretdin, A.; Brover, V.; Chetvernin, V.; O’Neill, K.; Li, W.; Chitsaz, F.; Derbyshire, M.K.; Gonzales, N.R.; et al. RefSeq: An update on prokaryotic genome annotation and curation. Nucleic Acids Res. 2018, 46, D851–D860. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; von Mering, C.; Bork, P. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.V.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [Green Version]
- Dunning Hotopp, J.C.; Lin, M.; Madupu, R.; Crabtree, J.; Angiuoli, S.V.; Eisen, J.A.; Seshadri, R.; Ren, Q.; Wu, M.; Utterback, T.R.; et al. Comparative genomics of emerging human ehrlichiosis agents. PLoS Genet. 2006, 2, e21. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Contreras-Moreira, B.; Vinuesa, P. GET_HOMOLOGUES, a Versatile Software Package for Scalable and Robust Microbial Pangenome Analysis. Appl. Environ. Microbiol. 2013, 79, 7696–7701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristensen, D.M.; Kannan, L.; Coleman, M.K.; Wolf, Y.; Sorokin, A.; Koonin, E.V.; Mushegian, A. A low-polynomial algorithm for assembling clusters of orthologous groups from intergenomic symmetric best matches. Bioinformatics 2010, 26, 1481–1487. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [Green Version]
- Pearson, W.R. An introduction to sequence similarity (“homology”) searching. Curr. Protoc. Bioinform. Chapter 2013, 3, 1–3. [Google Scholar] [CrossRef]
- Song, W.; Sun, H.-X.; Zhang, C.; Cheng, L.; Peng, Y.; Deng, Z.; Wang, D.; Wang, Y.; Hu, M.; Liu, W.; et al. Prophage Hunter: An integrative hunting tool for active prophages. Nucleic Acids Res. 2019, 47, W74–W80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overbeek, R.; Fonstein, M.; D’Souza, M.; Pusch, G.D.; Maltsev, N. The use of gene clusters to infer functional coupling. Proc. Natl. Acad. Sci. USA 1999, 96, 2896–2901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; López, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [CrossRef] [PubMed]
- Anisimova, M.; Gascuel, O. Approximate Likelihood-Ratio Test for Branches: A Fast, Accurate, and Powerful Alternative. Syst. Biol. 2006, 55, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Zheng, G.X.Y.; Lau, B.T.; Schnall-Levin, M.; Jarosz, M.; Bell, J.M.; Hindson, C.M.; Kyriazopoulou-Panagiotopoulou, S.; Masquelier, D.A.; Merrill, L.; Terry, J.M.; et al. Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat. Biotechnol. 2016, 34, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Collins, N.E.; Liebenberg, J.; de Villiers, E.P.; Brayton, K.A.; Louw, E.; Pretorius, A.; Faber, F.E.; van Heerden, H.; Josemans, A.; van Kleef, M.; et al. The genome of the heartwater agent Ehrlichia ruminantium contains multiple tandem repeats of actively variable copy number. Proc. Natl. Acad. Sci. USA 2005, 102, 838–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beare, P.A.; Sandoz, K.M.; Omsland, A.; Rockey, D.D.; Heinzen, R.A. Advances in genetic manipulation of obligate intracellular bacterial pathogens. Front. Microbiol. 2011, 2, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.; Zhang, C.; Gibson, K.; Rikihisa, Y. Analysis of complete genome sequence of Neorickettsia risticii: Causative agent of Potomac horse fever. Nucleic Acids Res. 2009, 37, 6076–6091. [Google Scholar] [CrossRef]
- Achard, M.E.; Hamilton, A.J.; Dankowski, T.; Heras, B.; Schembri, M.S.; Edwards, J.L.; Jennings, M.P.; McEwan, A.G. A periplasmic thioredoxin-like protein plays a role in defense against oxidative stress in Neisseria gonorrhoeae. Infect. Immun. 2009, 77, 4934–4939. [Google Scholar] [CrossRef] [Green Version]
- Cunha, B.A.; Chandrankunnel, J.G.; Hage, J.E. Ehrlichia chaffeensis human monocytic ehrlichiosis with pancytopenia. Scand. J. Infect. Dis. 2012, 44, 473–474. [Google Scholar] [CrossRef]
- Acinas, S.G.; Marcelino, L.A.; Klepac-Ceraj, V.; Polz, M.F. Divergence and redundancy of 16S rRNA sequences in genomes with multiple rrn operons. J. Bacteriol. 2004, 186, 2629–2635. [Google Scholar] [CrossRef] [Green Version]
- Krawiec, S.; Riley, M. Organization of the bacterial chromosome. Microbiol. Rev. 1990, 54, 502–539. [Google Scholar] [CrossRef]
- Klappenbach, J.A.; Dunbar, J.M.; Schmidt, T.M. rRNA Operon Copy Number Reflects Ecological Strategies of Bacteria. Appl. Environ. Microbiol. 2000, 66, 1328–1333. [Google Scholar] [CrossRef] [Green Version]
- Chakravortty, D.; Hensel, M. Inducible nitric oxide synthase and control of intracellular bacterial pathogens. Microbes Infect. 2003, 5, 621–627. [Google Scholar] [CrossRef]
- Patterson, L.L.; Byerly, C.D.; McBride, J.W. Anaplasmataceae: Dichotomous Autophagic Interplay for Infection. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Sample | Number of Reads | Fraction of Ca. N. mikurensis DNA in Sequenced Plasma Sample (%) | Bacterial Load in Extracted Plasma (c/mL) a |
|---|---|---|---|
| SE24-1 | 775,626,508 | 5.08 | 5.8 × 108 |
| SE24-2 | 729,557,012 | 0.10 | 5.8 × 108 |
| SE24-3 | 673,424,174 | 0.10 | 5.8 × 108 |
| SE20 | 748,875,060 | 1.25 | 1.2 × 108 |
| SE26 | 764,836,740 | 0.57 | 4.6 × 105 |
| Property | Organism | ||||
|---|---|---|---|---|---|
| Ca. Neoehrlichia mikurensis SE24 | Ca. Neoehrlichia lotoris | Ehrlichia ruminantium | Ehrlichia chaffeensis | Anaplasma phagocytophilum | |
| Accession number | CP053279 | NZ_LANX00000000 | NC_005295 | NC_007799 | NC_007797 |
| Size (bp) | 1,112,301 | 1,268,660 | 1,516,355 | 1,176,248 | 1,471,282 |
| GC content (%) | 26.9 | 27.7 | 27.5 | 30.1 | 41.6 |
| Genes, total (n) | 900 | 953 | 987 | 965 | 1152 |
| CDS, total (n) | 860 | 912 | 944 | 922 | 1108 |
| CDS with protein (n) | 845 | 908 | 919 | 886 | 1105 |
| Average CDS length (bp) | 960 | 1016 | 1007 | 995 | 929 |
| Assigned functions (n) | 776 | NR | 758 | 604 | 747 |
| Unknown functions (n) | 90 | NR | NR | 85 | 77 |
| Pseudogenes (n) | 15 | 4 | 25 | 36 | 103 |
| RNA genes (n) | 40 | 41 | 43 | 43 | 44 |
| rRNAs (n) | 3 | 3 | 3 | 3 | 3 |
| tRNAs (n) | 34 | 35 | 36 | 37 | 37 |
| ncRNAs (n) | 3 | 3 | 4 | 3 | 4 |
| Reference | This study | Daugherty, S.C et al. Direct submission | Collins et al. [44] | Dunning Hotopp et al. [26] | Dunning Hotopp et al. [26] |
| Functional Category | Ca. N. mikurensis Strain SE24 | Ca. N. mikurensis Strain SE20 | Ca. N. mikurensis Strain SE26 |
|---|---|---|---|
| Number of Genes (%) | |||
| Translation, ribosomal structure and biogenesis | 112 (13) | 115 (13) | 115 (13) |
| Energy production and conversion | 62 (7) | 62 (7) | 62 (7) |
| Posttranslational modification, protein turnover, chaperones | 58 (7) | 58 (7) | 58 (7) |
| Coenzyme transport and metabolism | 52 (6) | 54 (6) | 54 (6) |
| Replication, recombination and repair | 47 (5) | 47 (5) | 47 (5) |
| Cell wall/membrane/envelope biogenesis | 39 (5) | 39 (5) | 39 (5) |
| Nucleotide transport and metabolism | 37 (4) | 37 (4) | 37 (4) |
| Inorganic ion transport and metabolism | 34 (4) | 34 (4) | 34 (4) |
| Intracellular trafficking, secretion, and vesicular transport | 32 (4) | 32 (4) | 32 (4) |
| Amino acid transport and metabolism | 32 (4) | 32 (4) | 32 (4) |
| Lipid transport and metabolism | 26 (3) | 27 (3) | 27 (3) |
| Transcription | 23 (3) | 23 (3) | 23 (3) |
| Carbohydrate transport and metabolism | 19 (2) | 20 (2) | 20 (2) |
| Cell cycle control, cell division, chromosome partitioning | 11 (1) | 11 (1) | 11 (1) |
| Signal transduction mechanisms | 10 (1) | 10 (1) | 10 (1) |
| Secondary metabolites biosynthesis, transport and catabolism | 9 (1) | 9 (1) | 9 (1) |
| Defense mechanisms | 3 (0.3) | 3 (0.3) | 3 (0.3) |
| General function prediction only | 0 | 0 | 0 |
| Mobilome: prophages, transposons | 0 | 0 | 0 |
| Cell motility | 0 | 0 | 0 |
| Cytoskeleton | 0 | 0 | 0 |
| Extracellular structures | 0 | 0 | 0 |
| RNA processing and modification | 0 | 0 | 0 |
| Chromatin structure and dynamics | 0 | 0 | 0 |
| Nuclear structure | 0 | 0 | 0 |
| Function unknown | 90 (11) | 90 (11) | 90 (11) |
| No category assigned | 170 (20) | 166 (20) | 167 (20) |
| Protein | Function | Gene | Locus Tag |
|---|---|---|---|
| Argininosuccinate lyase | Amino-acid biosynthesis | argH1 | HL033_01080 |
| Argininosuccinate synthase | Amino-acid biosynthesis | argG | HL033_04485 |
| ParA family protein | Partitioning of plasmids | parA | HL033_02250 |
| Type I secretion system permease/ATPase | Protein secretion | prtD | HL033_03355 |
| 50S ribosomal protein L32 | Translation | rpmF | HL033_03630 |
| 50S ribosomal protein L34 | Translation | rpmH | HL033_03600 |
| 50S ribosomal protein L36 | Translation | rpmJ | HL033_04370 |
| DNA repair protein RadA | DNA repair | radA | HL033_02995 |
| DUF2671 domain-containing protein | Protein with domain of unknown function | unknown | HL033_00465 |
| Glutathione S-transferase family protein | Cellular detoxificaion | gstA | HL033_00805 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grankvist, A.; Jaén-Luchoro, D.; Wass, L.; Sikora, P.; Wennerås, C. Comparative Genomics of Clinical Isolates of the Emerging Tick-Borne Pathogen Neoehrlichia mikurensis. Microorganisms 2021, 9, 1488. https://doi.org/10.3390/microorganisms9071488
Grankvist A, Jaén-Luchoro D, Wass L, Sikora P, Wennerås C. Comparative Genomics of Clinical Isolates of the Emerging Tick-Borne Pathogen Neoehrlichia mikurensis. Microorganisms. 2021; 9(7):1488. https://doi.org/10.3390/microorganisms9071488
Chicago/Turabian StyleGrankvist, Anna, Daniel Jaén-Luchoro, Linda Wass, Per Sikora, and Christine Wennerås. 2021. "Comparative Genomics of Clinical Isolates of the Emerging Tick-Borne Pathogen Neoehrlichia mikurensis" Microorganisms 9, no. 7: 1488. https://doi.org/10.3390/microorganisms9071488
APA StyleGrankvist, A., Jaén-Luchoro, D., Wass, L., Sikora, P., & Wennerås, C. (2021). Comparative Genomics of Clinical Isolates of the Emerging Tick-Borne Pathogen Neoehrlichia mikurensis. Microorganisms, 9(7), 1488. https://doi.org/10.3390/microorganisms9071488