
From Klebsiella pneumoniae Colonization to Dissemination: An Overview of Studies Implementing Murine Models

 and
and
Abstract
1. Introduction
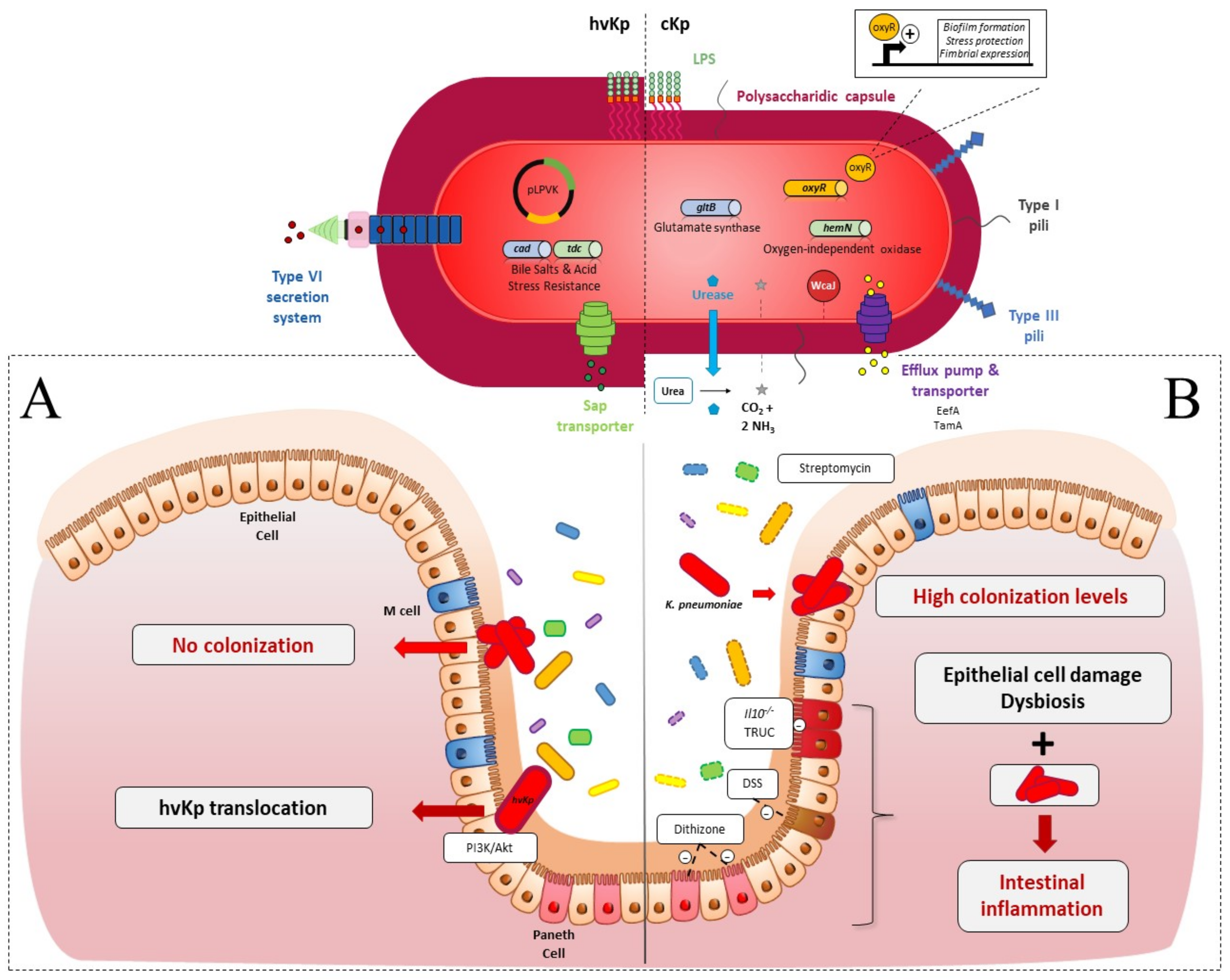
2. Intestinal Colonization
2.1. Colonization Resistance of the Intestinal Microbiota to K. pneumoniae
2.2. K. pneumoniae Colonization Factors
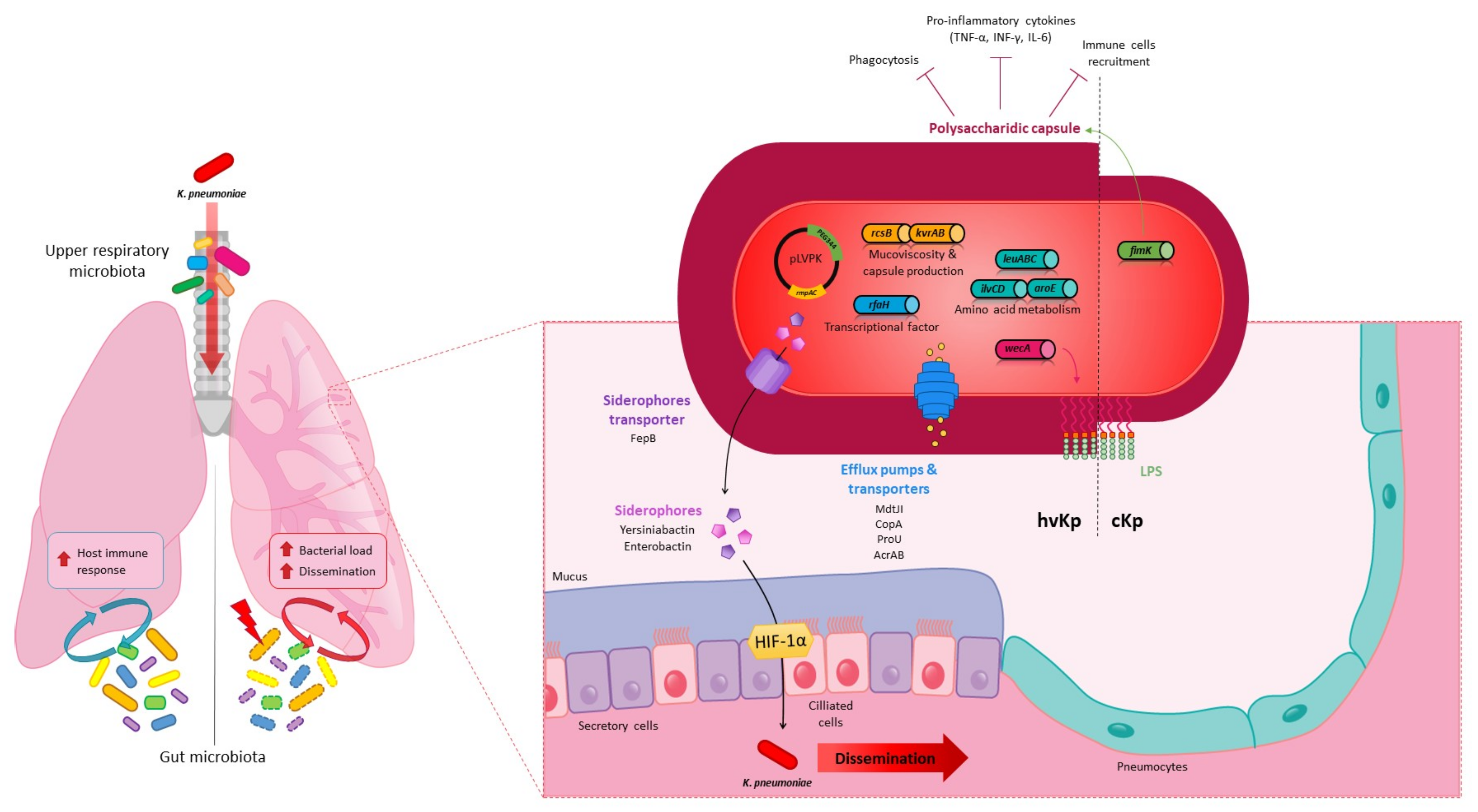
3. Systemic Infections
3.1. Spreading from the Intestinal Tract
3.2. Spreading from Extraintestinal Sites
4. Pulmonary Infections
4.1. In Vivo Models
4.2. K. pneumoniae Pulmonary Virulence Factors
4.3. K. pneumoniae in the Lungs: Host Immune and Microbiota Responses
5. Urinary Tract Infection
6. Role of Animal Models in the Development of New Therapies against K. pneumoniae Infections
6.1. Animal Models to Assess the Efficacy of Natural or Chemical Agents
6.2. Animal Models with Controlled Intestinal Microbiota
6.3. Immunized Animal Model Mimicking the Vaccination Processes
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bagley, S.T. Habitat Association of Klebsiella Species. Infect. Control 1985, 6, 52–58. [Google Scholar] [CrossRef]
- Podschun, R.; Pietsch, S.; Höller, C.; Ullmann, U. Incidence of Klebsiella Species in Surface Waters and Their Expression of Virulence Factors. Appl. Environ. Microbiol. 2001, 67, 3325–3327. [Google Scholar] [CrossRef] [PubMed]
- Donskey, C.J. The Role of the Intestinal Tract as a Reservoir and Source for Transmission of Nosocomial Pathogens. Clin. Infect. Dis. 2004, 39, 219–226. [Google Scholar] [CrossRef]
- Paczosa, M.K.; Mecsas, J. Klebsiella Pneumoniae: Going on the Offense with a Strong Defense. Microbiol. Mol. Biol. Rev. 2016, 80, 629–661. [Google Scholar] [CrossRef]
- Podschun, R.; Ullmann, U. Klebsiella Spp. as Nosocomial Pathogens: Epidemiology, Taxonomy, Typing Methods, and Pathogenicity Factors. Clin. Microbiol. Rev. 1998, 11, 589–603. [Google Scholar] [CrossRef]
- Gonzalez-Ferrer, S.; Peñaloza, H.F.; Budnick, J.A.; Bain, W.G.; Nordstrom, H.R.; Lee, J.S.; Van Tyne, D. Finding Order in the Chaos: Outstanding Questions in Klebsiella Pneumoniae Pathogenesis. Infect. Immun. 2021, 89, e00693-20. [Google Scholar] [CrossRef]
- Holt, K.E.; Wertheim, H.; Zadoks, R.N.; Baker, S.; Whitehouse, C.A.; Dance, D.; Jenney, A.; Connor, T.R.; Hsu, L.Y.; Severin, J.; et al. Genomic Analysis of Diversity, Population Structure, Virulence, and Antimicrobial Resistance in Klebsiella Pneumoniae, an Urgent Threat to Public Health. Proc. Natl. Acad. Sci. USA 2015, 112, E3574. [Google Scholar] [CrossRef] [PubMed]
- Grall, N.; Andremont, A.; Armand-Lefèvre, L. Résistance aux carbapénèmes: Vers une nouvelle impasse? J. Anti-Infect. 2011, 13, 87–102. [Google Scholar] [CrossRef]
- Wyres, K.L.; Holt, K.E. Klebsiella Pneumoniae as a Key Trafficker of Drug Resistance Genes from Environmental to Clinically Important Bacteria. Curr. Opin. Microbiol. 2018, 45, 131–139. [Google Scholar] [CrossRef]
- Wyres, K.L.; Lam, M.M.C.; Holt, K.E. Population Genomics of Klebsiella Pneumoniae. Nat. Rev. Microbiol. 2020, 18, 344–359. [Google Scholar] [CrossRef]
- Gorrie, C.L.; Mirčeta, M.; Wick, R.R.; Edwards, D.J.; Thomson, N.R.; Strugnell, R.A.; Pratt, N.F.; Garlick, J.S.; Watson, K.M.; Pilcher, D.V.; et al. Gastrointestinal Carriage Is a Major Reservoir of Klebsiella Pneumoniae Infection in Intensive Care Patients. Clin. Infect. Dis. 2017, 65, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Peña, C.; Pujol, M.; Ricart, A.; Ardanuy, C.; Ayats, J.; Liñares, J.; Garrigosa, F.; Ariza, J.; Gudiol, F. Risk Factors for Faecal Carriage of Klebsiella Pneumoniae Producing Extended Spectrum β-Lactamase (ESBL-KP) in the Intensive Care Unit. J. Hosp. Infect. 1997, 35, 9–16. [Google Scholar] [CrossRef]
- Jung, H.-J.; Littmann, E.R.; Seok, R.; Leiner, I.M.; Taur, Y.; Peled, J.; van den Brink, M.; Ling, L.; Chen, L.; Kreiswirth, B.N.; et al. Genome-Wide Screening for Enteric Colonization Factors in Carbapenem-Resistant ST258 Klebsiella Pneumoniae. mBio 2019, 10, e02663-18. [Google Scholar] [CrossRef]
- Martin, R.M.; Cao, J.; Brisse, S.; Passet, V.; Wu, W.; Zhao, L.; Malani, P.N.; Rao, K.; Bachman, M.A. Molecular Epidemiology of Colonizing and Infecting Isolates of Klebsiella Pneumoniae. mSphere 2016, 1, e00261-16. [Google Scholar] [CrossRef]
- Taur, Y.; Xavier, J.B.; Lipuma, L.; Ubeda, C.; Goldberg, J.; Gobourne, A.; Lee, Y.J.; Dubin, K.A.; Socci, N.D.; Viale, A.; et al. Intestinal Domination and the Risk of Bacteremia in Patients Undergoing Allogeneic Hematopoietic Stem Cell Transplantation. Clin. Infect. Dis. 2012, 55, 905–914. [Google Scholar] [CrossRef]
- Kabha, K.; Nissimov, L.; Athamna, A.; Keisari, Y.; Parolis, H.; Parolis, L.A.; Grue, R.M.; Schlepper-Schafer, J.; Ezekowitz, A.R.; Ohman, D.E. Relationships among Capsular Structure, Phagocytosis, and Mouse Virulence in Klebsiella Pneumoniae. Infect. Immun. 1995, 63, 847–852. [Google Scholar] [CrossRef]
- Russo, T.A.; Marr, C.M. Hypervirulent Klebsiella Pneumoniae. Clin. Microbiol. Rev. 2019, 32, 42. [Google Scholar] [CrossRef]
- Pomakova, D.K.; Hsiao, C.-B.; Beanan, J.M.; Olson, R.; MacDonald, U.; Keynan, Y.; Russo, T.A. Clinical and Phenotypic Differences between Classic and Hypervirulent Klebsiella Pneumoniae: An Emerging and under-Recognized Pathogenic Variant. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 981–989. [Google Scholar] [CrossRef]
- Shon, A.S.; Bajwa, R.P.S.; Russo, T.A. Hypervirulent (Hypermucoviscous) Klebsiella Pneumoniae. Virulence 2013, 4, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Huang, Z.Y.; Yu, T.; Tao, X.Y.; Hu, Y.M.; Wang, H.C.; Zou, M.X. Isolation and Characterization of a Sequence Type 25 Carbapenem-Resistant Hypervirulent Klebsiella Pneumoniae from the Mid-South Region of China. BMC Microbiol. 2019, 19, 219. [Google Scholar] [CrossRef]
- Wu, K.-M.; Li, L.-H.; Yan, J.-J.; Tsao, N.; Liao, T.-L.; Tsai, H.-C.; Fung, C.-P.; Chen, H.-J.; Liu, Y.-M.; Wang, J.-T.; et al. Genome Sequencing and Comparative Analysis of Klebsiella Pneumoniae NTUH-K2044, a Strain Causing Liver Abscess and Meningitis. J. Bacteriol. JB 2009, 191, 4492–4501. [Google Scholar] [CrossRef]
- Kislichkina, A.A.; Lev, A.I.; Komisarova, E.V.; Fursova, N.K.; Myakinina, V.P.; Mukhina, T.N.; Bogun, A.A.; Volozhantsev, N.V. Genome Sequencing and Comparative Analysis of Three Hypermucoviscous Klebsiella Pneumoniae Strains Isolated in Russia. Pathog. Dis. 2017, 75. [Google Scholar] [CrossRef]
- Li, W.; Sun, G.; Yu, Y.; Li, N.; Chen, M.; Jin, R.; Jiao, Y.; Wu, H. Increasing Occurrence of Antimicrobial- Resistant Hypervirulent (Hypermucoviscous) Klebsiella Pneumoniae Isolates in China. BMC Microbiol. 2014, 58, 225–232. [Google Scholar] [CrossRef]
- Martin, R.M.; Bachman, M.A. Colonization, Infection, and the Accessory Genome of Klebsiella Pneumoniae. Front. Cell. Infect. Microbiol. 2018, 8, 4. [Google Scholar] [CrossRef]
- Bengoechea, J.A.; Sa Pessoa, J. Klebsiella Pneumoniae Infection Biology: Living to Counteract Host Defences. FEMS Microbiol. Rev. 2019, 43, 123–144. [Google Scholar] [CrossRef]
- Caballero, S.; Carter, R.; Ke, X.; Sušac, B.; Leiner, I.M.; Kim, G.J.; Miller, L.; Ling, L.; Manova, K.; Pamer, E.G. Distinct but Spatially Overlapping Intestinal Niches for Vancomycin-Resistant Enterococcus Faecium and Carbapenem-Resistant Klebsiella Pneumoniae. PLoS Pathog. 2015, 11, e1005132. [Google Scholar] [CrossRef]
- Lau, H.Y.; Huffnagle, G.B.; Moore, T.A. Host and Microbiota Factors That Control Klebsiella Pneumoniae Mucosal Colonization in Mice. Microbes Infect. 2008, 10, 1283–1290. [Google Scholar] [CrossRef] [PubMed]
- Favre-Bonté, S.; Licht, T.R.; Forestier, C.; Krogfelt, K.A. Klebsiella Pneumoniae Capsule Expression Is Necessary for Colonization of Large Intestines of Streptomycin-Treated Mice. Infect. Immun. 1999, 67, 6152–6156. [Google Scholar] [CrossRef]
- Perez, F.; Pultz, M.J.; Endimiani, A.; Bonomo, R.A.; Donskey, C.J. Effect of Antibiotic Treatment on Establishment and Elimination of Intestinal Colonization by KPC-Producing Klebsiella Pneumoniae in Mice. Antimicrob. Agents Chemother. 2011, 55, 2585–2589. [Google Scholar] [CrossRef]
- Maroncle, N.; Rich, C.; Forestier, C. The Role of Klebsiella Pneumoniae Urease in Intestinal Colonization and Resistance to Gastrointestinal Stress. Res. Microbiol. 2006, 157, 184–193. [Google Scholar] [CrossRef]
- Struve, C.; Bojer, M.; Krogfelt, K.A. Characterization of Klebsiella Pneumoniae Type 1 Fimbriae by Detection of Phase Variation during Colonization and Infection and Impact on Virulence. Infect. Immun. 2008, 76, 4055–4065. [Google Scholar] [CrossRef]
- Khater, F.; Balestrino, D.; Charbonnel, N.; Dufayard, J.F.; Brisse, S.; Forestier, C. In Silico Analysis of Usher Encoding Genes in Klebsiella Pneumoniae and Characterization of Their Role in Adhesion and Colonization. PLoS ONE 2015, 10, e0116215. [Google Scholar] [CrossRef]
- Coudeyras, S.; Nakusi, L.; Charbonnel, N.; Forestier, C. A Tripartite Efflux Pump Involved in Gastrointestinal Colonization by Klebsiella Pneumoniae Confers a Tolerance Response to Inorganic Acid. Infect. Immun. 2008, 76, 4633–4641. [Google Scholar] [CrossRef]
- Hsieh, P.; Lin, H.; Lin, T.; Wang, J. CadC Regulates Cad and Tdc Operons in Response to Gastrointestinal Stresses and Enhances Intestinal Colonization of Klebsiella Pneumoniae. J. Infect. Dis. 2010, 202, 52–64. [Google Scholar] [CrossRef]
- Sequeira, R.P.; McDonald, J.A.K.; Marchesi, J.R.; Clarke, T.B. Commensal Bacteroidetes Protect against Klebsiella Pneumoniae Colonization and Transmission through IL-36 Signalling. Nat. Microbiol. 2020, 5, 304–313. [Google Scholar] [CrossRef]
- Lagrafeuille, R.; Miquel, S.; Balestrino, D.; Vareille-Delarbre, M.; Chain, F.; Langella, P.; Forestier, C. Opposing Effect of Lactobacillus on In Vitro Klebsiella Pneumoniae in Biofilm and in an In Vivo Intestinal Colonisation Model. Benef. Microbes 2018, 9, 87–100. [Google Scholar] [CrossRef]
- Schjorring, S.; Struve, C.; Krogfelt, K.A. Transfer of Antimicrobial Resistance Plasmids from Klebsiella Pneumoniae to Escherichia Coli in the Mouse Intestine. J. Antimicrob. Chemother. 2008, 62, 1086–1093. [Google Scholar] [CrossRef]
- Hoyen, C.K.; Pultz, N.J.; Paterson, D.L.; Aron, D.C.; Donskey, C.J. Effect of Parenteral Antibiotic Administration on Establishment of Intestinal Colonization in Mice by Klebsiella Pneumoniae Strains Producing Extended-Spectrum β-Lactamases. Antimicrob. Agents Chemother. 2003, 47, 3610–3612. [Google Scholar] [CrossRef]
- Deshpande, A.; Hurless, K.; Cadnum, J.L.; Chesnel, L.; Gao, L.; Chan, L.; Kundrapu, S.; Polinkovsky, A.; Donskey, C.J. Effect of Fidaxomicin versus Vancomycin on Susceptibility to Intestinal Colonization with Vancomycin-Resistant Enterococci and Klebsiella Pneumoniae in Mice. Antimicrob. Agents Chemother. 2016, 60, 3988–3993. [Google Scholar] [CrossRef] [PubMed]
- Pope, J.L.; Yang, Y.; Newsome, R.C.; Sun, W.; Sun, X.; Ukhanova, M.; Neu, J.; Issa, J.-P.; Mai, V.; Jobin, C. Microbial Colonization Coordinates the Pathogenesis of a Klebsiella Pneumoniae Infant Isolate. Sci. Rep. 2019, 9, 3380. [Google Scholar] [CrossRef]
- Atarashi, K.; Suda, W.; Luo, C.; Kawaguchi, T.; Motoo, I.; Narushima, S.; Kiguchi, Y.; Yasuma, K.; Watanabe, E.; Tanoue, T.; et al. Ectopic Colonization of Oral Bacteria in the Intestine Drives TH1 Cell Induction and Inflammation. Science 2017, 358, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Garrett, W.S.; Gallini, C.A.; Yatsunenko, T.; Michaud, M.; DuBois, A.; Delaney, M.L.; Punit, S.; Karlsson, M.; Bry, L.; Glickman, J.N.; et al. Enterobacteriaceae Act in Concert with the Gut Microbiota to Induce Spontaneous and Maternally Transmitted Colitis. Cell Host Microbe 2010, 8, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Maroncle, N.; Balestrino, D.; Rich, C.; Forestier, C. Identification of Klebsiella Pneumoniae Genes Involved in Intestinal Colonization and Adhesion Using Signature-Tagged Mutagenesis. Infect. Immun. 2002, 70, 4729–4734. [Google Scholar] [CrossRef] [PubMed]
- van Aartsen, J.J.; Stahlhut, S.G.; Harrison, E.M.; Crosatti, M.; Ou, H.-Y.; Krogfelt, K.A.; Struve, C.; Rajakumar, K. Characterization of a Novel Chaperone/Usher Fimbrial Operon Present on KpGI-5, a Methionine TRNA Gene-Associated Genomic Island in Klebsiella Pneumoniae. BMC Microbiol. 2012, 12, 59. [Google Scholar] [CrossRef]
- Hennequin, C.; Forestier, C. OxyR, a LysR-Type Regulator Involved in Klebsiella Pneumoniae Mucosal and Abiotic Colonization. IAI 2009, 77, 5449–5457. [Google Scholar] [CrossRef]
- Struve, C.; Forestier, C.; Krogfelt, K.A. Application of a Novel Multi-Screening Signature-Tagged Mutagenesis Assay for Identification of Klebsiella Pneumoniae Genes Essential in Colonization and Infection. Microbiology 2003, 149, 167–176. [Google Scholar] [CrossRef]
- Lueschow, S.R.; Stumphy, J.; Gong, H.; Kern, S.L.; Elgin, T.G.; Underwood, M.A.; Kalanetra, K.M.; Mills, D.A.; Wong, M.H.; Meyerholz, D.K.; et al. Loss of Murine Paneth Cell Function Alters the Immature Intestinal Microbiome and Mimics Changes Seen in Neonatal Necrotizing Enterocolitis. PLoS ONE 2018, 13, e0204967. [Google Scholar] [CrossRef]
- Zhang, C.; Sherman, M.P.; Prince, L.S.; Bader, D.; Weitkamp, J.-H.; Slaughter, J.C.; McElroy, S.J. Paneth Cell Ablation in the Presence of Klebsiella Pneumoniae Induces Necrotizing Enterocolitis (NEC)-like Injury in the Small Intestine of Immature Mice. Dis. Models Mech. 2012, 5, 522–532. [Google Scholar] [CrossRef]
- Tu, Y.-C.; Lu, M.-C.; Chiang, M.-K.; Huang, S.-P.; Peng, H.-L.; Chang, H.-Y.; Jan, M.-S.; Lai, Y.-C. Genetic Requirements for Klebsiella Pneumoniae-Induced Liver Abscess in an Oral Infection Model. Infect. Immun. 2009, 77, 2657–2671. [Google Scholar] [CrossRef]
- Tang, H.-L.; Chiang, M.-K.; Liou, W.-J.; Chen, Y.-T.; Peng, H.-L.; Chiou, C.-S.; Liu, K.-S.; Lu, M.-C.; Tung, K.-C.; Lai, Y.-C. Correlation between Klebsiella Pneumoniae Carrying PLVPK-Derived Loci and Abscess Formation. Eur. J. Clin. Microbiol. Infect. Dis. 2010, 29, 689–698. [Google Scholar] [CrossRef]
- Hsu, C.-R.; Chang, I.-W.; Hsieh, P.-F.; Lin, T.-L.; Liu, P.-Y.; Huang, C.-H.; Li, K.-T.; Wang, J.-T. A Novel Role for the Klebsiella Pneumoniae Sap (Sensitivity to Antimicrobial Peptides) Transporter in Intestinal Cell Interactions, Innate Immune Responses, Liver Abscess, and Virulence. J. Infect. Dis. 2019, 219, 1294–1306. [Google Scholar] [CrossRef]
- Hsieh, P.-F.; Lu, Y.-R.; Lin, T.-L.; Lai, L.-Y.; Wang, J.-T. Klebsiella Pneumoniae Type VI Secretion System Contributes to Bacterial Competition, Cell Invasion, Type-1 Fimbriae Expression, and In Vivo Colonization. J. Infect. Dis. 2019, 219, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.H.; Chen, Y.; Chu, W.H.W.; Sham, L.-T.; Gan, Y.-H. Cell Envelope Defects of Different Capsule-Null Mutants in K1 Hypervirulent Klebsiella Pneumoniae Can Affect Bacterial Pathogenesis. Mol. Microbiol. 2020, 113, 889–905. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.-R.; Pan, Y.-J.; Liu, J.-Y.; Chen, C.-T.; Lin, T.-L.; Wang, J.-T. Klebsiella Pneumoniae Translocates across the Intestinal Epithelium via Rho GTPase- and Phosphatidylinositol 3-Kinase/Akt-Dependent Cell Invasion. Infect. Immun. 2015, 83, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.-R.; Lin, T.-L.; Chen, Y.-C.; Chou, H.-C.; Wang, J.-T. The Role of Klebsiella Pneumoniae RmpA in Capsular Polysaccharide Synthesis and Virulence Revisited. Microbiology 2011, 157, 3446–3457. [Google Scholar] [CrossRef]
- Wu, J.H.; Tsai, C.G. Infectivity of Hepatic Strain Klebsiella Pneumoniae in Diabetic Mice. Exp. Biol. Med. 2005, 230, 757–761. [Google Scholar] [CrossRef]
- Lin, S.-H.; Chung, P.-H.; Wu, Y.-Y.; Fung, C.-P.; Hsu, C.-M.; Chen, L.-W. Inhibition of Nitric Oxide Production Reverses Diabetes-Induced Kupffer Cell Activation and Klebsiella Pneumonia Liver Translocation. PLoS ONE 2017, 12, e0177269. [Google Scholar] [CrossRef]
- Moore, T.A.; Perry, M.L.; Getsoian, A.G.; Newstead, M.W.; Standiford, T.J. Divergent Role of Gamma Interferon in a Murine Model of Pulmonary versus Systemic Klebsiella Pneumoniae Infection. Infect. Immun. 2002, 70, 6310–6318. [Google Scholar] [CrossRef]
- Wu, J.H.; Wu, A.M.; Tsai, C.G.; Chang, X.-Y.; Tsai, S.-F.; Wu, T.-S. Contribution of Fucose-Containing Capsules in Klebsiella Pneumoniae to Bacterial Virulence in Mice. Exp. Biol. Med. 2008, 233, 64–70. [Google Scholar] [CrossRef]
- Pan, P.-C.; Chen, H.-W.; Wu, P.-K.; Wu, Y.-Y.; Lin, C.-H.; Wu, J.H. Mutation in Fucose Synthesis Gene of Klebsiella Pneumoniae Affects Capsule Composition and Virulence in Mice. Exp. Biol. Med. 2011, 236, 219–226. [Google Scholar] [CrossRef]
- Diago-Navarro, E.; Calatayud-Baselga, I.; Sun, D.; Khairallah, C.; Mann, I.; Ulacia-Hernando, A.; Sheridan, B.; Shi, M.; Fries, B.C. Antibody-Based Immunotherapy to Treat and Prevent Infection with Hypervirulent Klebsiella Pneumoniae. Clin. Vaccine Immunol. 2017, 24, e00456-16. [Google Scholar] [CrossRef]
- Fung, C.-P.; Chang, F.-Y.; Lin, J.-C.; Ho, D.M.-T.; Chen, C.-T.; Chen, J.-H.; Yeh, K.-M.; Chen, T.-L.; Lin, Y.-T.; Siu, L.K. Immune Response and Pathophysiological Features of Klebsiella Pneumoniae Liver Abscesses in an Animal Model. Lab. Investig. 2011, 91, 1029–1039. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hsieh, P.-F.; Lin, T.-L.; Yang, F.-L.; Wu, M.-C.; Pan, Y.-J.; Wu, S.-H.; Wang, J.-T. Lipopolysaccharide O1 Antigen Contributes to the Virulence in Klebsiella Pneumoniae Causing Pyogenic Liver Abscess. PLoS ONE 2012, 7, e33155. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.-F.; Liu, J.-Y.; Pan, Y.-J.; Wu, M.-C.; Lin, T.-L.; Huang, Y.-T.; Wang, J.-T. Klebsiella Pneumoniae Peptidoglycan-Associated Lipoprotein and Murein Lipoprotein Contribute to Serum Resistance, Antiphagocytosis, and Proinflammatory Cytokine Stimulation. J. Infect. Dis. 2013, 208, 1580–1589. [Google Scholar] [CrossRef]
- De Souza, G.H.d.A. In Vitro and In Vivo Antibacterial Activity Assays of Carvacrol: A Candidate for Development of Innovative Treatments against KPC-Producing Klebsiella Pneumoniae. PLoS ONE 2021, 16, e0246003. [Google Scholar] [CrossRef]
- Zheng, Y.; Yue, C.; Zhang, H.; Chen, H.; Liu, Y.; Li, J. Deoxycholic Acid and Lithocholic Acid Alleviate Liver Injury and Inflammation in Mice with Klebsiella Pneumoniae-Induced Liver Abscess and Bacteremia. J. Inflamm. Res. 2021, 14, 777–789. [Google Scholar] [CrossRef]
- Young, T.M.; Bray, A.S.; Nagpal, R.K.; Caudell, D.L.; Yadav, H.; Zafar, M.A. Animal Model to Study Klebsiella Pneumoniae Gastrointestinal Colonization and Host-to-Host Transmission. Infect. Immun. 2020, 88, e00071-20. [Google Scholar] [CrossRef]
- Fang, C.-T.; Chuang, Y.-P.; Shun, C.-T.; Chang, S.-C.; Wang, J.-T. A Novel Virulence Gene in Klebsiella Pneumoniae Strains Causing Primary Liver Abscess and Septic Metastatic Complications. J. Exp. Med. 2004, 199, 697–705. [Google Scholar] [CrossRef]
- Hsieh, P.-F.; Hsu, C.-R.; Chen, C.-T.; Lin, T.-L.; Wang, J.-T. The Klebsiella Pneumoniae YfgL (BamB) Lipoprotein Contributes to Outer Membrane Protein Biogenesis, Type-1 Fimbriae Expression, Anti-Phagocytosis, and In Vivo Virulence. Virulence 2016, 7, 587–601. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-H.; Choi, H.-I.; Hong, S.-W.; Kim, K.; Gho, Y.S.; Jeon, S.G. Vaccination with Klebsiella Pneumoniae-Derived Extracellular Vesicles Protects against Bacteria-Induced Lethality via Both Humoral and Cellular Immunity. Exp. Mol. Med. 2015, 47, e183. [Google Scholar] [CrossRef] [PubMed]
- Yeh, K.-M.; Lin, J.-C.; Yin, F.-Y.; Fung, C.-P.; Hung, H.-C.; Siu, L.-K.; Chang, F.-Y. Revisiting the Importance of Virulence Determinant MagA and Its Surrounding Genes in Klebsiella Pneumoniae Causing Pyogenic Liver Abscesses: Exact Role in Serotype K1 Capsule Formation. J. Infect. Dis. 2010, 201, 1259–1267. [Google Scholar] [CrossRef]
- Russo, T.A.; Olson, R.; MacDonald, U.; Metzger, D.; Maltese, L.M.; Drake, E.J.; Gulick, A.M. Aerobactin Mediates Virulence and Accounts for Increased Siderophore Production under Iron-Limiting Conditions by Hypervirulent (Hypermucoviscous) Klebsiella Pneumoniae. Infect. Immun. 2014, 82, 2356–2367. [Google Scholar] [CrossRef]
- Cryz, S.J.; Mortimer, P.; Cross, A.S.; Fürer, E.; Germanier, R. Safety and Immunogenicity of a Polyvalent Klebsiella Capsular Polysaccharide Vaccine in Humans. Vaccine 1986, 4, 15–20. [Google Scholar] [CrossRef]
- Krapp, F.; Morris, A.R.; Ozer, E.A.; Hauser, A.R. Virulence Characteristics of Carbapenem-Resistant Klebsiella Pneumoniae Strains from Patients with Necrotizing Skin and Soft Tissue Infections. Sci. Rep. 2017, 7, 13533. [Google Scholar] [CrossRef] [PubMed]
- Bulger, J.; MacDonald, U.; Olson, R.; Beanan, J.; Russo, T.A. Metabolite Transporter PEG344 Is Required for Full Virulence of Hypervirulent Klebsiella Pneumoniae Strain HvKP1 after Pulmonary but Not Subcutaneous Challenge. Infect. Immun. 2017, 85, e00093-17. [Google Scholar] [CrossRef]
- Russo, T.A.; Olson, R.; Fang, C.-T.; Stoesser, N.; Miller, M.; MacDonald, U.; Hutson, A.; Barker, J.H.; La Hoz, R.M.; Johnson, J.R.; et al. Identification of Biomarkers for Differentiation of Hypervirulent Klebsiella Pneumoniae from Classical K. Pneumoniae. J. Clin. Microbiol. 2018, 56, e00776-18. [Google Scholar] [CrossRef] [PubMed]
- Russo, T.A.; Olson, R.; MacDonald, U.; Beanan, J.; Davidson, B.A. Aerobactin, but Not Yersiniabactin, Salmochelin, or Enterobactin, Enables the Growth/Survival of Hypervirulent (Hypermucoviscous) Klebsiella Pneumoniae Ex Vivo and In Vivo. Infect. Immun. 2015, 83, 3325–3333. [Google Scholar] [CrossRef]
- Lawlor, M.S.; Hsu, J.; Rick, P.D.; Miller, V.L. Identification of Klebsiella Pneumoniae Virulence Determinants Using an Intranasal Infection Model: Klebsiella Pneumoniae Intranasal STM. Mol. Microbiol. 2005, 58, 1054–1073. [Google Scholar] [CrossRef]
- Lawlor, M.S.; Handley, S.A.; Miller, V.L. Comparison of the Host Responses to Wild-Type and CpsB Mutant Klebsiella Pneumoniae Infections. Infect. Immun. 2006, 74, 5402–5407. [Google Scholar] [CrossRef] [PubMed]
- Palacios, M.; Broberg, C.A.; Walker, K.A.; Miller, V.L. A Serendipitous Mutation Reveals the Severe Virulence Defect of a Klebsiella Pneumoniae FepB Mutant. mSphere 2017, 2, e00341-17. [Google Scholar] [CrossRef] [PubMed]
- Lou, W.; Venkataraman, S.; Zhong, G.; Ding, B.; Tan, J.P.K.; Xu, L.; Fan, W.; Yang, Y.Y. Antimicrobial Polymers as Therapeutics for Treatment of Multidrug-Resistant Klebsiella Pneumoniae Lung Infection. Acta Biomater. 2018, 78, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Palacios, M.; Miner, T.A.; Frederick, D.R.; Sepulveda, V.E.; Quinn, J.D.; Walker, K.A.; Miller, V.L. Identification of Two Regulators of Virulence That Are Conserved in Klebsiella Pneumoniae Classical and Hypervirulent Strains. mBio 2018, 9, e01443-18. [Google Scholar] [CrossRef] [PubMed]
- Guilhen, C.; Miquel, S.; Charbonnel, N.; Joseph, L.; Carrier, G.; Forestier, C.; Balestrino, D. Colonization and Immune Modulation Properties of Klebsiella Pneumoniae Biofilm-Dispersed Cells. NPJ Biofilms Microbiomes 2019, 5, 25. [Google Scholar] [CrossRef]
- Vareille-Delarbre, M.; Miquel, S.; Garcin, S.; Bertran, T.; Balestrino, D.; Evrard, B.; Forestier, C. Immunomodulatory Effects of Lactobacillus Plantarum on Inflammatory Response Induced by Klebsiella Pneumoniae. Infect. Immun. 2019, 87, e00570-19. [Google Scholar] [CrossRef]
- Lawlor, M.S.; O’Connor, C.; Miller, V.L. Yersiniabactin Is a Virulence Factor for Klebsiella Pneumoniae during Pulmonary Infection. Infect. Immun. 2007, 75, 1463–1472. [Google Scholar] [CrossRef]
- Yoshida, K.; Matsumoto, T.; Tateda, K.; Uchida, K.; Tsujimoto, S.; Yamaguchi, K. Role of Bacterial Capsule in Local and Systemic Inflammatory Responses of Mice during Pulmonary Infection with Klebsiella Pneumoniae. J. Med. Microbiol. 2000, 49, 1003–1010. [Google Scholar] [CrossRef]
- Walker, K.A.; Miner, T.A.; Palacios, M.; Trzilova, D.; Frederick, D.R.; Broberg, C.A.; Sepúlveda, V.E.; Quinn, J.D.; Miller, V.L. A Klebsiella Pneumoniae Regulatory Mutant Has Reduced Capsule Expression but Retains Hypermucoviscosity. mBio 2019, 10, 16. [Google Scholar] [CrossRef]
- Paczosa, M.K.; Silver, R.J.; McCabe, A.L.; Tai, A.K.; McLeish, C.H.; Lazinski, D.W.; Mecsas, J. Transposon Mutagenesis Screen of Klebsiella Pneumoniae Identifies Multiple Genes Important for Resisting Antimicrobial Activities of Neutrophils in Mice. Infect. Immun. 2020, 88, e00034-20. [Google Scholar] [CrossRef]
- Brown, R.L.; Sequeira, R.P.; Clarke, T.B. The Microbiota Protects against Respiratory Infection via GM-CSF Signaling. Nat. Commun. 2017, 8, 1512. [Google Scholar] [CrossRef]
- Clarke, T.B. Early Innate Immunity to Bacterial Infection in the Lung Is Regulated Systemically by the Commensal Microbiota via Nod-Like Receptor Ligands. Infect. Immun. 2014, 82, 4596–4606. [Google Scholar] [CrossRef] [PubMed]
- Wolff, N.S.; Jacobs, M.C.; Haak, B.W.; Roelofs, J.J.T.H.; de Vos, A.F.; Hugenholtz, F.; Wiersinga, W.J. Vendor Effects on Murine Gut Microbiota and Its Influence on Lipopolysaccharide-Induced Lung Inflammation and Gram-Negative Pneumonia. Intensive Care Med. Exp. 2020, 8, 47. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Gan, C.; Li, R.; Ye, Y.; Zhang, S.; Wu, X.; Yang, Y.Y.; Fan, W.; Wu, M. A Novel Chemosynthetic Peptide with β-Sheet Motif Efficiently Kills Klebsiella Pneumoniae in a Mouse Model. Int. J. Nanomed. 2015, 10, 1045–1059. [Google Scholar] [CrossRef]
- Geller, B.L.; Li, L.; Martinez, F.; Sully, E.; Sturge, C.R.; Daly, S.M.; Pybus, C.; Greenberg, D.E. Morpholino Oligomers Tested In Vitro, in Biofilm and In Vivo against Multidrug-Resistant Klebsiella Pneumoniae. J. Antimicrob. Chemother. 2018, 73, 1611–1619. [Google Scholar] [CrossRef]
- Chhibber, S.; Kaur, S.; Kumari, S. Therapeutic Potential of Bacteriophage in Treating Klebsiella Pneumoniae B5055-Mediated Lobar Pneumonia in Mice. J. Med. Microbiol. 2008, 57, 1508–1513. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Wang, X.; Wang, L.; Li, Z.; Che, J.; Wang, L.; Li, X.; Cao, Z.; Zhang, J.; Jin, L.; et al. Evaluation of the Efficacy of a Bacteriophage in the Treatment of Pneumonia Induced by Multidrug Resistance Klebsiella Pneumoniae in Mice. BioMed Res. Int. 2015, 2015, 1–9. [Google Scholar] [CrossRef]
- Anand, T. Phage Therapy for Treatment of Virulent Klebsiella Pneumoniae Infection in a Mouse Model. J. Glob. Antimicrob. Resist. 2020, 8. [Google Scholar] [CrossRef]
- Yadav, V.; Sharma, S.; Harjai, K.; Mohan, H.; Chhibber, S. Lipopolysaccharide-Mediated Protection against Klebsiella Pneumoniae-Induced Lobar Pneumonia: Intranasal vs. Intramuscular Route of Immunization. Folia Microbiol. 2005, 50, 83–86. [Google Scholar] [CrossRef]
- Yoshida, K.; Matsumoto, T.; Tateda, K.; Uchida, K.; Tsujimoto, S.; Yamaguchi, K. Induction of Interleukin-10 and down-Regulation of Cytokine Production by Klebsiella Pneumoniae Capsule in Mice with Pulmonary Infection. J. Med. Microbiol. 2001, 50, 456–461. [Google Scholar] [CrossRef]
- Domenico, P.; Johanson, W.G.; Straus, D.C. Lobar Pneumonia in Rats Produced by Clinical Isolates of Klebsiella Pneumoniae. Infect. Immun. 1982, 37, 327–335. [Google Scholar] [CrossRef]
- Broug-Holub, E.; Toews, G.B.; van Iwaarden, J.F.; Strieter, R.M.; Kunkel, S.L.; Paine, R.; Standiford, T.J. Alveolar Macrophages Are Required for Protective Pulmonary Defenses in Murine Klebsiella Pneumonia: Elimination of Alveolar Macrophages Increases Neutrophil Recruitment but Decreases Bacterial Clearance and Survival. Infect. Immun. 1997, 65, 1139–1146. [Google Scholar] [CrossRef]
- Deng, J.C.; Zeng, X.; Newstead, M.; Moore, T.A.; Tsai, W.C.; Thannickal, V.J.; Standiford, T.J. STAT4 Is a Critical Mediator of Early Innate Immune Responses against Pulmonary Klebsiella Infection. J. Immunol. 2004, 173, 4075–4083. [Google Scholar] [CrossRef]
- Lau, H.Y.; Clegg, S.; Moore, T.A. Identification of Klebsiella Pneumoniae Genes Uniquely Expressed in a Strain Virulent Using a Murine Model of Bacterial Pneumonia. Microb. Pathog. 2007, 42, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.A.; Hilliard, J.K.; Tiemann, K.M.; Todd, E.M.; Morley, S.C.; Hunstad, D.A. Klebsiella Pneumoniae FimK Promotes Virulence in Murine Pneumonia. J. Infect. Dis. 2016, 213, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.A.; Twentyman, J.; Hunstad, D.A. High Levels of Cyclic Di-GMP in Klebsiella Pneumoniae Attenuate Virulence in the Lung. Infect. Immun. 2017, 86, e00647-17. [Google Scholar] [CrossRef] [PubMed]
- Cortés, G.; Borrell, N.; de Astorza, B.; Gómez, C.; Sauleda, J.; Albertí, S. Molecular Analysis of the Contribution of the Capsular Polysaccharide and the Lipopolysaccharide O Side Chain to the Virulence of Klebsiella Pneumoniae in a Murine Model of Pneumonia. Infect. Immun. 2002, 70, 2583–2590. [Google Scholar] [CrossRef]
- Padilla, E.; Llobet, E.; Doménech-Sánchez, A.; Martínez-Martínez, L.; Bengoechea, J.A.; Albertí, S. Klebsiella Pneumoniae AcrAB Efflux Pump Contributes to Antimicrobial Resistance and Virulence. Antimicrob. Agents Chemother. 2010, 54, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Bachman, M.A.; Breen, P.; Deornellas, V.; Mu, Q.; Zhao, L.; Wu, W.; Cavalcoli, J.D.; Mobley, H.L.T. Genome-Wide Identification of Klebsiella Pneumoniae Fitness Genes during Lung Infection. mBio 2015, 6, e00775-15. [Google Scholar] [CrossRef] [PubMed]
- Holden, V.I.; Breen, P.; Houle, S.; Dozois, C.M.; Bachman, M.A. Klebsiella Pneumoniae Siderophores Induce Inflammation, Bacterial Dissemination, and HIF-1α Stabilization during Pneumonia. mBio 2016, 7, e01397-16. [Google Scholar] [CrossRef] [PubMed]
- Willsey, G.G.; Ventrone, S.; Schutz, K.C.; Wallace, A.M.; Ribis, J.W.; Suratt, B.T.; Wargo, M.J. Pulmonary Surfactant Promotes Virulence Gene Expression and Biofilm Formation in Klebsiella Pneumoniae. Infect. Immun. 2018, 86, e00135-18. [Google Scholar] [CrossRef] [PubMed]
- Chhibber, S.; Aggarwal, S.; Yadav, V. Contribution of Capsular and Lipopolysaccharide Antigens to the Pathogenesis of Klebsiella Pneumoniae Respiratory Tract Infection. Folia Microbiol. 2003, 48, 699–702. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, J.; Li, J.; Wu, X.; Xiao, L.; Liu, X.; Yang, X.; Yang, L.; Zou, Q.; Huang, W. AmpR Increases the Virulence of Carbapenem-Resistant Klebsiella Pneumoniae by Regulating the Initial Step of Capsule Synthesis. Infect. Drug Resist. 2020, 13, 3431–3441. [Google Scholar] [CrossRef]
- Bachman, M.A.; Oyler, J.E.; Burns, S.H.; Caza, M.; Lépine, F.; Dozois, C.M.; Weiser, J.N. Klebsiella Pneumoniae Yersiniabactin Promotes Respiratory Tract Infection through Evasion of Lipocalin 2. Infect. Immun. 2011, 79, 3309–3316. [Google Scholar] [CrossRef] [PubMed]
- Bachman, M.A.; Lenio, S.; Schmidt, L.; Oyler, J.E.; Weiser, J.N. Interaction of Lipocalin 2, Transferrin, and Siderophores Determines the Replicative Niche of Klebsiella Pneumoniae during Pneumonia. mBio 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Morinaga, Y.; Take, Y.; Sasaki, D.; Ota, K.; Kaku, N.; Uno, N.; Sakamoto, K.; Kosai, K.; Miyazaki, T.; Hasegawa, H.; et al. Exploring the Microbiota of Upper Respiratory Tract during the Development of Pneumonia in a Mouse Model. PLoS ONE 2019, 14, e0222589. [Google Scholar] [CrossRef]
- Fagundes, C.T.; Amaral, F.A.; Vieira, A.T.; Soares, A.C.; Pinho, V.; Nicoli, J.R.; Vieira, L.Q.; Teixeira, M.M.; Souza, D.G. Transient TLR Activation Restores Inflammatory Response and Ability to Control Pulmonary Bacterial Infection in Germfree Mice. J. Immunol. 2012, 188, 1411–1420. [Google Scholar] [CrossRef]
- Vieira, A.T.; Rocha, V.M.; Tavares, L.; Garcia, C.C.; Teixeira, M.M.; Oliveira, S.C.; Cassali, G.D.; Gamba, C.; Martins, F.S.; Nicoli, J.R. Control of Klebsiella Pneumoniae Pulmonary Infection and Immunomodulation by Oral Treatment with the Commensal Probiotic Bifidobacterium Longum 51A. Microbes Infect. 2016, 18, 180–189. [Google Scholar] [CrossRef]
- van der Weide, H.; Cossío, U.; Gracia, R.; te Welscher, Y.M.; ten Kate, M.T.; van der Meijden, A.; Marradi, M.; Ritsema, J.A.S.; Vermeulen-de Jongh, D.M.C.; Storm, G.; et al. Therapeutic Efficacy of Novel Antimicrobial Peptide AA139-Nanomedicines in a Multidrug-Resistant Klebsiella Pneumoniae Pneumonia-Septicemia Model in Rats. Antimicrob. Agents Chemother. 2020, 64, e00517-20. [Google Scholar] [CrossRef]
- Chhibber, S.; Wadhwa, S.; Yadav, V. Protective Role of Liposome Incorporated Lipopolysaccharide Antigen of Klebsiella Pneumoniae in a Rat Model of Lobar Pneumonia. Jpn. J. Infect. Dis. 2004, 57, 150–155. [Google Scholar]
- Sale, L., Jr.; Wood, W.B., Jr. Studies on the Mechanism of Recovery in Pneumonia Due to Friedländer’s Bacillus: I. The Pathogenesis of Experimental Friedländer’s Bacillus Pneumonia. J. Exp. Med. 1947, 86, 239–248. [Google Scholar] [CrossRef]
- Sale, L.; Smith, M.R.; Wood, W.B. Studies on the Mechanism of Recovery in Pneumonia Due to Friedländer’s Bacillus: II. The Effect of Sulfonamide Chemotherapy upon the Pulmonary Lesion of Experimental Friedländer’s Bacillus Pneumonia. J. Exp. Med. 1947, 86, 249–256. [Google Scholar] [CrossRef]
- Struve, C.; Bojer, M.; Krogfelt, K.A. Identification of a Conserved Chromosomal Region Encoding Klebsiella Pneumoniae Type 1 and Type 3 Fimbriae and Assessment of the Role of Fimbriae in Pathogenicity. Infect. Immun. 2009, 77, 5016–5024. [Google Scholar] [CrossRef]
- Struve, C.; Krogfelt, K.A. Role of Capsule in Klebsiella Pneumoniae Virulence: Lack of Correlation between In Vitro and In Vivo Studies. FEMS Microbiol. Lett. 2003, 218, 149–154. [Google Scholar] [CrossRef]
- Camprubi, S.; Merino, S.; BenedÃ, V.-J.; Tomás, J.M. The Role of the O-Antigen Lipopolysaccharide and Capsule on an Experimental Klebsiella Pneumoniae Infection of the Rat Urinary Tract. FEMS Microbiol. Lett. 1993, 111, 9–13. [Google Scholar] [CrossRef]
- Fader, R.C.; Davis, C.P. Effect of Piliation on Klebsiella Pneumoniae Infection in Rat Bladders. Infect. Immun. 1980, 30, 554–561. [Google Scholar] [CrossRef]
- Maayan, M.C.; Ofek, I.; Medalia, O.; Aronson, M. Population Shift in Mannose-Specific Fimbriated Phase of Klebsiella Pneumoniae during Experimental Urinary Tract Infection in Mice. Infect. Immun. 1985, 49, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.A.; Pinkner, J.S.; Walker, J.N.; Elam, J.S.; Jones, J.M.; Hultgren, S.J. Molecular Variations in Klebsiella Pneumoniae and Escherichia Coli FimH Affect Function and Pathogenesis in the Urinary Tract. Infect. Immun. 2008, 76, 3346–3356. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.A.; Pinkner, J.S.; Jones, J.M.; Walker, J.N.; Clegg, S.; Hultgren, S.J. Utilization of an Intracellular Bacterial Community Pathway in Klebsiella Pneumoniae Urinary Tract Infection and the Effects of FimK on Type 1 Pilus Expression. Infect. Immun. 2008, 76, 3337–3345. [Google Scholar] [CrossRef]
- Rosen, D.A.; Hung, C.-S.; Kline, K.A.; Hultgren, S.J. Streptozocin-Induced Diabetic Mouse Model of Urinary Tract Infection. Infect. Immun. 2008, 76, 4290–4298. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.É.I.; Pacheco, T.; da Silva dos Santos, C.; Pereira, J.A.; Ribeiro, M.L.; Darrieux, M.; Ferraz, L.F.C. Functional Insights From KpfR, a New Transcriptional Regulator of Fimbrial Expression That Is Crucial for Klebsiella Pneumoniae Pathogenicity. Front. Microbiol. 2021, 11, 601921. [Google Scholar] [CrossRef]
- Merino, S.; Altarriba, M.; Izquierdo, L.; Nogueras, M.M.; Regué, M.; Tomás, J.M. Cloning and Sequencing of the Klebsiella Pneumoniae O5wb Gene Cluster and Its Role in Pathogenesis. Infect. Immun. 2000, 68, 2435–2440. [Google Scholar] [CrossRef]
- Murphy, C.N.; Mortensen, M.S.; Krogfelt, K.A.; Clegg, S. Role of Klebsiella Pneumoniae Type 1 and Type 3 Fimbriae in Colonizing Silicone Tubes Implanted into the Bladders of Mice as a Model of Catheter-Associated Urinary Tract Infections. Infect. Immun. 2013, 81, 3009–3017. [Google Scholar] [CrossRef] [PubMed]
- Fung, C.-P.; Lin, Y.-T.; Lin, J.-C.; Chen, T.-L.; Yeh, K.-M.; Chang, F.-Y.; Chuang, H.-C.; Wu, H.-S.; Tseng, C.-P.; Siu, L.K. Klebsiella Pneumoniae in Gastrointestinal Tract and Pyogenic Liver Abscess. Emerg. Infect. Dis. 2012, 18, 1322–1325. [Google Scholar] [CrossRef] [PubMed]
- Buffie, C.G.; Pamer, E.G. Microbiota-Mediated Colonization Resistance against Intestinal Pathogens. Nat. Rev. Immunol. 2014, 13, 790–801. [Google Scholar] [CrossRef] [PubMed]
- Van der Waaij, D.; Berghuis-de Vries, J.M.; Lekkerkerk-van der Wees, J.E.C. Colonization Resistance of the Digestive Tract in Conventional and Antibiotic-Treated Mice. Epidemiol. Infect. 1971, 69, 405–411. [Google Scholar] [CrossRef]
- Miller, C.P.; Bohnhoff, M. Changes in the Mouse’s Enteric Microflora Associated with Enhanced Susceptibility to Salmonella Infection Following Streptomycin Treatment. J. Infect. Dis. 1963, 113, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Honma, K.; Mishima, E.; Inagaki, S.; Sharma, A. The OxyR Homologue in Tannerella Forsythia Regulates Expression of Oxidative Stress Responses and Biofilm Formation. Microbiology 2009, 155, 1912–1922. [Google Scholar] [CrossRef]
- Johnson, J.R.; Clabots, C.; Rosen, H. Effect of Inactivation of the Global Oxidative Stress Regulator OxyR on the Colonization Ability of Escherichia Coli O1:K1:H7 in a Mouse Model of Ascending Urinary Tract Infection. Infect. Immun. 2006, 74, 461–468. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chung, C.-H.; Fen, S.; Yu, S.-C.; Wong, H. Influence of OxyR on Growth, Biofilm Formation, and Mobility of Vibrio Parahaemolyticus. Appl. Environ. Microbiol. 2016, 82, 788–796. [Google Scholar] [CrossRef]
- Sana, T.G.; Flaugnatti, N.; Lugo, K.A.; Lam, L.H.; Jacobson, A.; Baylot, V.; Durand, E.; Journet, L.; Cascales, E.; Monack, D.M. Salmonella Typhimurium Utilizes a T6SS-Mediated Antibacterial Weapon to Establish in the Host Gut. Proc. Natl. Acad. Sci. USA 2016, 113, E5044–E5051. [Google Scholar] [CrossRef]
- Chen, Y.-T.; Chang, H.-Y.; Lai, Y.-C.; Pan, C.-C.; Tsai, S.-F.; Peng, H.-L. Sequencing and Analysis of the Large Virulence Plasmid PLVPK of Klebsiella Pneumoniae CG43. Gene 2004, 337, 189–198. [Google Scholar] [CrossRef]
- Cortés, G.; de Astorza, B.; Benedí, V.J.; Albertí, S. Role of the HtrA Gene in Klebsiella Pneumoniae Virulence. Infect. Immun. 2002, 70, 4772–4776. [Google Scholar] [CrossRef] [PubMed]
- Athamna, A.; Ofek, I.; Keisari, Y.; Dutton, G.G.S.; Sharon, N. Lectinophagocytosis of Encapsulated Klebsiella Pneumoniae Mediated by Surface Lectins of Guinea Pig Alveolar Macrophages and Human Monocyte-Derived Macrophages. Infect. Immun. 1991, 59, 10. [Google Scholar] [CrossRef]
- Keisari, Y.; Kabha, K.; Nissimov, L.; Schlepper-Schafer, J.; Ofek, I. Phagocyte-Bacteria Interactions. Adv. Dent. Res. 1997, 11, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Jeng, K.C.; Ping, L.I. Exogenous Cytokine Modulation or Neutralization of Interleukin-10 Enhance Survival in Lipopolysaccharide-Hyporesponsive C3H/HeJ Mice with Klebsiella Infection. Immunology 1999, 98, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Greenberger, M.J.; Strieter, R.M.; Kunkel, S.L.; Danforth, J.M.; Goodman, R.E.; Standiford, T.J. Neutralization of IL-10 Increases Survival in a Murine Model of Klebsiella Pneumonia. J. Immunol. 1995, 155, 722–729. [Google Scholar]
- Ko, W.-C. Community-Acquired Klebsiella Pneumoniae Bacteremia: Global Differences in Clinical Patterns. Emerg. Infect. Dis. 2002, 8, 160–166. [Google Scholar] [CrossRef]
- Rello, J.; Rodriguez, R.; Jubert, P.; Alvarez, B.; Study Group for Severe Community-Acquired Pneumonia. Severe Community-Acquired Pneumonia in the Elderly: Epidemiology and Prognosis. Clin. Infect. Dis. 1996, 23, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Song, J.-H.; Oh, W.S.; Kang, C.-I.; Chung, D.R.; Peck, K.R.; Ko, K.S.; Yeom, J.S.; Kim, C.K.; Kim, S.W.; Chang, H.-H.; et al. Epidemiology and Clinical Outcomes of Community-Acquired Pneumonia in Adult Patients in Asian Countries: A Prospective Study by the Asian Network for Surveillance of Resistant Pathogens. Int. J. Antimicrob. Agents 2008, 31, 107–114. [Google Scholar] [CrossRef]
- Jones, R.N. Microbial Etiologies of Hospital-Acquired Bacterial Pneumonia and Ventilator-Associated Bacterial Pneumonia. Clin. Infect. Dis. 2010, 51, S81–S87. [Google Scholar] [CrossRef]
- Rammaert, B.; Goyet, S.; Beauté, J.; Hem, S.; Te, V.; Try, P.L.; Mayaud, C.; Borand, L.; Buchy, P.; Guillard, B.; et al. Klebsiella Pneumoniae Related Community-Acquired Acute Lower Respiratory Infections in Cambodia: Clinical Characteristics and Treatment. BMC Infect. Dis. 2012, 12, 3. [Google Scholar] [CrossRef] [PubMed]
- Percival, S.L.; Suleman, L.; Vuotto, C.; Donelli, G. Healthcare-Associated Infections, Medical Devices and Biofilms: Risk, Tolerance and Control. J. Med. Microbiol. 2015, 64, 323–334. [Google Scholar] [CrossRef]
- Mizgerd, J.P.; Skerrett, S.J. Animal Models of Human Pneumonia. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294, L387–L398. [Google Scholar] [CrossRef]
- Bielen, K.; ’S Jongers, B.; Malhotra-Kumar, S.; Jorens, P.G.; Goossens, H.; Kumar-Singh, S. Animal Models of Hospital-Acquired Pneumonia: Current Practices and Future Perspectives. Ann. Transl. Med. 2017, 5, 132. [Google Scholar] [CrossRef]
- Dickson, R.P.; Erb-Downward, J.R.; Falkowski, N.R.; Hunter, E.M.; Ashley, S.L.; Huffnagle, G.B. The Lung Microbiota of Healthy Mice Are Highly Variable, Cluster by Environment, and Reflect Variation in Baseline Lung Innate Immunity. Am. J. Respir. Crit. Care Med. 2018, 198, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Dumas, A.; Bernard, L.; Poquet, Y.; Lugo-Villarino, G.; Neyrolles, O. The Role of the Lung Microbiota and the Gut–Lung Axis in Respiratory Infectious Diseases. Cell. Microbiol. 2018, 20, e12966. [Google Scholar] [CrossRef] [PubMed]
- Flores-Mireles, A.L.; Walker, J.N.; Caparon, M.; Hultgren, S.J. Urinary Tract Infections: Epidemiology, Mechanisms of Infection and Treatment Options. Nat. Rev. Microbiol. 2015, 13, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Clegg, S.; Murphy, C.N. Epidemiology and Virulence of Klebsiella Pneumoniae. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Langermann, S.; Möllby, R.; Burlein, J.E.; Palaszynski, S.R.; Gale Auguste, C.; DeFusco, A.; Strouse, R.; Schenerman, M.A.; Hultgren, S.J.; Pinkner, J.S.; et al. Vaccination with FimH Adhesin Protects Cynomolgus Monkeys from Colonization and Infection by Uropathogenic Eschevichia Coli. J. Infect. Dis. 2000, 181, 774–778. [Google Scholar] [CrossRef]
- Livrelli, V.; De Champs, C.; Di Martino, P.; Darfeuille-Michaud, A.; Forestier, C.; Joly, B. Adhesive Properties and Antibiotic Resistance of Klebsiella, Enterobacter, and Serratia Clinical Isolates Involved in Nosocomial Infections. J. Clin. Microbiol. 1996, 34, 1963–1969. [Google Scholar] [CrossRef]
- Panthier, J.-J.; Montagutelli, X. Le Collaborative Cross—Un outil révolutionnaire à l’assaut des caractères complexes. Med. Sci. 2012, 28, 103–108. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Model | Inoculation Mode | Read-Out | Technique | Strains | References | |
|---|---|---|---|---|---|---|
| cKp a | hvKp b | |||||
| Gastrointestinal colonization | Intragastric | Colonization levels | CFU counting | ☑ | ☑ | [13,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46] |
| Host status and immune response | Histopathology | ☑ | [27,40,41,47] | |||
| Cytokine quantification | ☑ | [35,40,47] | ||||
| Inflammatory marker measurement | ☑ | [40,41,42,47,48] | ||||
| FISH | ☑ | [26,28,41,42,47] | ||||
| Microscopy | ☑ | [41] | ||||
| Microbiota modification | 16S DNA sequencing | ☑ | [13,26,35,39,41] | |||
| CFU counting | ☑ | [29,42] | ||||
| Systemic dissemination | Intragastric | Colonization levels | CFU counting | ☑ | [49,50,51,52,53] | |
| Host status and immune response | Histopathology | ☑ | [49,51] | |||
| Cytokine quantification | ☑ | [49] | ||||
| Microscopy | ☑ | [54] | ||||
| Lethality | Survival | ☑ | [51,55] | |||
| Intravenous | Colonization levels | CFU counting | ☑ | ☑ | [16,56,57,58] | |
| Host status and immune response | Histopathology | ☑ | [56,58] | |||
| Cytokine quantification | ☑ | [56,57,58] | ||||
| Microscopy | ☑ | [59] | ||||
| Lethality | Survival | ☑ | ☑ | [56,58,59,60] | ||
| Intraperitoneal | Colonization levels | CFU counting | ☑ | ☑ | [53,55,61,62,63,64,65,66] | |
| Host status and immune response | Histopathology | ☑ | ☑ | [62,66,67,68] | ||
| Cytokine quantification | ☑ | ☑ | [63,64,66,69,70] | |||
| Inflammatory marker measurement | ☑ | ☑ | [65,66,70] | |||
| Microscopy | ☑ | ☑ | [61] | |||
| Intraperitoneal | Microbiota modification | 16S Sequencing | ☑ | ☑ | [66] | |
| Lethality | Survival | ☑ | ☑ | [16,55,61,62,63,64,65,66,68,69,70,71,72,73] | ||
| Subcutaneous | Colonization levels | CFU counting | ☑ | ☑ | [74,75] | |
| Host status and immune response | Liver abscess measurement | ☑ | ☑ | [74] | ||
| Lethality | Survival | ☑ | ☑ | [72,75,76,77] | ||
| Pulmonary infection | Intranasal | Colonization levels | CFU counting | ☑ | ☑ | [78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97] |
| Host status and immune response | Cytokine quantification | ☑ | ☑ | [79,83,84,86,89,90,91,92,95,98] | ||
| Histopathology | ☑ | ☑ | [78,79,80,81,82,86,91,92,95,96,97] | |||
| Inflammatory marker measurement | ☑ | ☑ | [79,82,84,86,88,92] | |||
| Microscopy | ☑ | [92] | ||||
| Lethality | Survival | ☑ | ☑ | [78,81,85,86,93,95,98] | ||
| Intratracheal | Colonization levels | CFU counting | ☑ | ☑ | [75,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118] | |
| Host status and immune response | Cytokine quantification | ☑ | ☑ | [100,101,102,103,108,115,116] | ||
| Histopathology | ☑ | ☑ | [99,100,101,103,104,105,110,113,116,119,120] | |||
| Inflammatory marker measurement | ☑ | ☑ | [100,101,102,103,109,114] | |||
| Microscopy | ☑ | [105,113] | ||||
| Lethality | Survival | ☑ | ☑ | [75,77,100,101,111,112,115,116,117,118,119,120] | ||
| UTIc | Intraurethral | Colonization levels | CFU counting | ☑ | [31,46,121,122,123,124,125,126,127,128,129,130] | |
| Host status and immune response | Histology | ☑ | [127,128,129] | |||
| Microscopy | ☑ | [124,127,128] | ||||
| CAUTId | Intraurethral | Colonization levels | CFU counting | ☑ | [131] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joseph, L.; Merciecca, T.; Forestier, C.; Balestrino, D.; Miquel, S. From Klebsiella pneumoniae Colonization to Dissemination: An Overview of Studies Implementing Murine Models. Microorganisms 2021, 9, 1282. https://doi.org/10.3390/microorganisms9061282
Joseph L, Merciecca T, Forestier C, Balestrino D, Miquel S. From Klebsiella pneumoniae Colonization to Dissemination: An Overview of Studies Implementing Murine Models. Microorganisms. 2021; 9(6):1282. https://doi.org/10.3390/microorganisms9061282
Chicago/Turabian StyleJoseph, Laura, Thomas Merciecca, Christiane Forestier, Damien Balestrino, and Sylvie Miquel. 2021. "From Klebsiella pneumoniae Colonization to Dissemination: An Overview of Studies Implementing Murine Models" Microorganisms 9, no. 6: 1282. https://doi.org/10.3390/microorganisms9061282
APA StyleJoseph, L., Merciecca, T., Forestier, C., Balestrino, D., & Miquel, S. (2021). From Klebsiella pneumoniae Colonization to Dissemination: An Overview of Studies Implementing Murine Models. Microorganisms, 9(6), 1282. https://doi.org/10.3390/microorganisms9061282