
Comparative Genomics Analysis of Keratin-Degrading Chryseobacterium Species Reveals Their Keratinolytic Potential for Secondary Metabolite Production

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. DNA Preparation
2.2. Genome Sequencing, Assembling, and Functional Annotation
2.3. Whole-Genome Phylogenetic Analysis
2.4. Secondary Metabolite Gene Cluster Detection and Annotation
2.5. Metabolic Networks Construction and Protease Families Prediction
3. Results and Discussion
3.1. Genome Feature Comparison of Four Keratinolytic Chryseobacterium Species
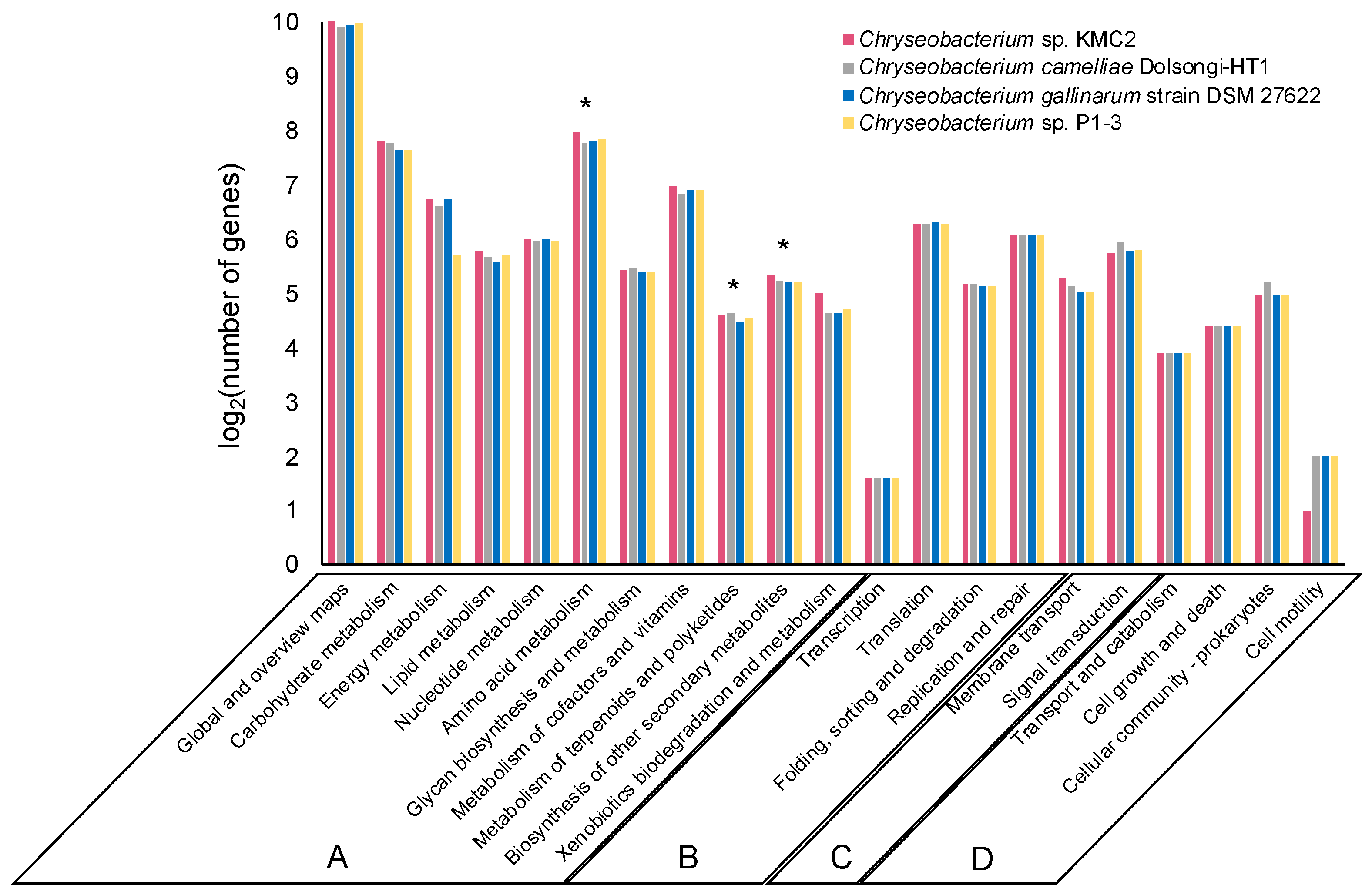
3.2. Metabolic Potential Comparison of Four Keratinolytic Chryseobacterium Genomes
3.3. Mining and Comparing Secondary Metabolite Gene Clusters
3.4. Synteny Analysis and Features of Secondary Metabolite Gene Clusters
3.4.1. Flexirubin-Type Pigment
3.4.2. Microviridin
3.4.3. Siderophore
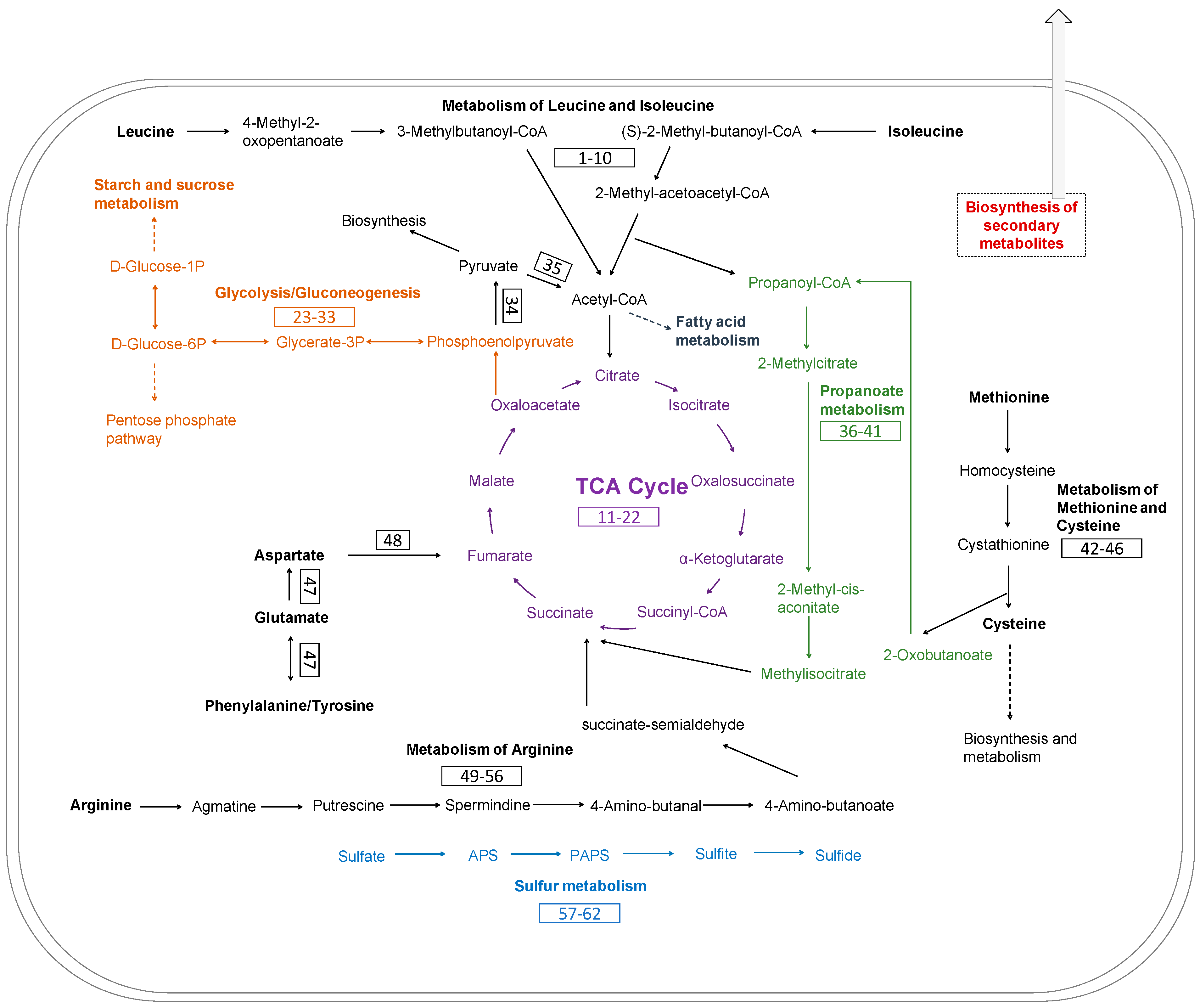
3.5. Metabolic Pathways of Keratin Utilization in Chryseobacterium sp. KMC2 Genome
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, B.; Yang, W.; McKittrick, J.; Meyers, M.A. Keratin: Structure, mechanical properties, occurrence in biological organisms, and efforts at bioinspiration. Prog. Mater. Sci. 2016, 76, 229–318. [Google Scholar] [CrossRef]
- Coulombe, P.A.; Omary, M.B. ‘Hard’and ‘soft’principles defining the structure, function and regulation of keratin intermediate filaments. Curr. Opin. Cell Biol. 2002, 14, 110–122. [Google Scholar] [CrossRef]
- Korniłłowicz-Kowalska, T.; Bohacz, J. Biodegradation of keratin waste: Theory and practical aspects. Waste Manag. 2011, 31, 1689–1701. [Google Scholar] [CrossRef] [PubMed]
- Khosa, M.; Ullah, A. A sustainable role of keratin biopolymer in green chemistry: A review. J. Food Process. Beverages 2013, 1, 8. [Google Scholar]
- Rajabi, M.; Ali, A.; McConnell, M.; Cabral, J. Keratinous materials: Structures and functions in biomedical applications. Mater. Sci. Eng. C 2020, 110, 110612. [Google Scholar] [CrossRef]
- Holkar, C.R.; Jain, S.S.; Jadhav, A.J.; Pinjari, D.V. Valorization of keratin based waste. Process. Saf. Environ. Prot. 2018, 115, 85–98. [Google Scholar] [CrossRef]
- Guo, L.; Lu, L.; Yin, M.; Yang, R.; Zhang, Z.; Zhao, W. Valorization of refractory keratinous waste using a new and sustainable bio-catalysis. Chem. Eng. J. 2020, 397, 125420. [Google Scholar] [CrossRef]
- Dios, D. Fishmeal replacement with feather-enzymatic hydrolyzates co-extruded with soya-bean meal in practical diets for the Pacific white shrimp (Litopenaeus vannamei). Aquac. Nutr. 2001, 7, 143–151. [Google Scholar]
- Ichida, J.M.; Krizova, L.; LeFevre, C.A.; Keener, H.M.; Elwell, D.L.; Burtt, E.H., Jr. Bacterial inoculum enhances keratin degradation and biofilm formation in poultry compost. J. Microbiol. Methods 2001, 47, 199–208. [Google Scholar] [CrossRef]
- Donadio, S.; Monciardini, P.; Alduina, R.; Mazza, P.; Chiocchini, C.; Cavaletti, L.; Sosio, M.; Puglia, A.M. Microbial technologies for the discovery of novel bioactive metabolites. J. Biotechnol. 2002, 99, 187–198. [Google Scholar] [CrossRef]
- Smitha, M.; Singh, S.; Singh, R. Microbial biotransformation: A process for chemical alterations. J. Bacteriol. Mycol. Open Access 2017, 4, 85. [Google Scholar]
- Ledesma-Amaro, R.; Nicaud, J.-M. Metabolic engineering for expanding the substrate range of Yarrowia lipolytica. Trends Biotechnol. 2016, 34, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Zhang, Y.; Li, Y. Production of fuels and chemicals from renewable resources using engineered Escherichia coli. Biotechnol. Adv. 2019, 37, 107402. [Google Scholar] [CrossRef]
- Chen, C.; Lin, J.; Wang, W.; Huang, H.; Li, S. Cost-effective production of surfactin from xylose-rich corncob hydrolysate using Bacillus subtilis BS-37. Waste Biomass Valorization 2019, 10, 341–347. [Google Scholar] [CrossRef]
- Straub, C.T.; Bing, R.G.; Wang, J.P.; Chiang, V.L.; Adams, M.W.; Kelly, R.M. Use of the lignocellulose-degrading bacterium Caldicellulosiruptor bescii to assess recalcitrance and conversion of wild-type and transgenic poplar. Biotechnol. Biofuels 2020, 13, 1–10. [Google Scholar] [CrossRef]
- Sgobba, E.; Blöbaum, L.; Wendisch, V.F. Production of food and feed additives from non-food-competing feedstocks: Valorizing N-acetylmuramic acid for amino acid and carotenoid fermentation with Corynebacterium glutamicum. Front. Microbiol. 2018, 9, 2046. [Google Scholar] [CrossRef]
- Zhu, X.; Zhou, Y.; Wang, Y.; Wu, T.; Li, X.; Li, D.; Tao, Y. Production of high-concentration n-caproic acid from lactate through fermentation using a newly isolated Ruminococcaceae bacterium CPB6. Biotechnol. Biofuels 2017, 10, 102. [Google Scholar] [CrossRef]
- Li, Q. Progress in microbial degradation of feather waste. Front. Microbiol. 2019, 10, 2717. [Google Scholar] [CrossRef]
- Kang, D.; Huang, Y.; Nesme, J.; Herschend, J.; Jacquiod, S.; Kot, W.; Hansen, L.H.; Lange, L.; Sørensen, S.J. Metagenomic analysis of a keratin-degrading bacterial consortium provides insight into the keratinolytic mechanisms. Sci. Total Environ. 2020, 143281. [Google Scholar] [CrossRef]
- Huang, Y.; Łężyk, M.; Herbst, F.-A.; Busk, P.K.; Lange, L. Novel keratinolytic enzymes, discovered from a talented and efficient bacterial keratin degrader. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef] [PubMed]
- Weber, T.; Kim, H.U. The secondary metabolite bioinformatics portal: Computational tools to facilitate synthetic biology of secondary metabolite production. Synth. Syst. Biotechnol. 2016, 1, 69–79. [Google Scholar] [CrossRef]
- Zheng, Y.; Saitou, A.; Wang, C.-M.; Toyoda, A.; Minakuchi, Y.; Sekiguchi, Y.; Ueda, K.; Takano, H.; Sakai, Y.; Abe, K. Genome features and secondary metabolites biosynthetic potential of the class Ktedonobacteria. Front. Microbiol. 2019, 10, 893. [Google Scholar] [CrossRef] [PubMed]
- Kjærbølling, I.; Vesth, T.C.; Frisvad, J.C.; Nybo, J.L.; Theobald, S.; Kuo, A.; Bowyer, P.; Matsuda, Y.; Mondo, S.; Lyhne, E.K. Linking secondary metabolites to gene clusters through genome sequencing of six diverse Aspergillus species. Proc. Natl. Acad. Sci. USA 2018, 115, E753–E761. [Google Scholar] [CrossRef] [PubMed]
- Fontoura, R.; Daroit, D.J.; Corrêa, A.P.F.; Moresco, K.S.; Santi, L.; Beys-da-Silva, W.O.; Yates III, J.R.; Moreira, J.C.F.; Brandelli, A. Characterization of a novel antioxidant peptide from feather keratin hydrolysates. New Biotechnol. 2019, 49, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Kshetri, P.; Roy, S.S.; Sharma, S.K.; Singh, T.S.; Ansari, M.A.; Prakash, N.; Ngachan, S. Transforming chicken feather waste into feather protein hydrolysate using a newly isolated multifaceted keratinolytic bacterium Chryseobacterium sediminis RCM-SSR-7. Waste Biomass Valorization 2019, 10, 1–11. [Google Scholar] [CrossRef]
- Kang, D.; Jacquiod, S.; Herschend, J.; Wei, S.; Nesme, J.; Sørensen, S.J. Construction of simplified microbial consortia to degrade recalcitrant materials based on enrichment and dilution-to-extinction cultures. Front. Microbiol. 2020, 10, 3010. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Herschend, J.; Al-Soud, W.A.; Mortensen, M.S.; Gonzalo, M.; Jacquiod, S.; Sørensen, S.J. Enrichment and characterization of an environmental microbial consortium displaying efficient keratinolytic activity. Bioresour. Technol. 2018, 270, 303–310. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [PubMed]
- Bertels, F.; Silander, O.K.; Pachkov, M.; Rainey, P.B.; van Nimwegen, E. Automated reconstruction of whole-genome phylogenies from short-sequence reads. Mol. Biol. Evol. 2014, 31, 1077–1088. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef]
- Avram, O.; Rapoport, D.; Portugez, S.; Pupko, T. M1CR0B1AL1Z3R—a user-friendly web server for the analysis of large-scale microbial genomics data. Nucleic Acids Res. 2019, 47, W88–W92. [Google Scholar] [CrossRef]
- Cohen, O.; Ashkenazy, H.; Belinky, F.; Huchon, D.; Pupko, T. GLOOME: Gain loss mapping engine. Bioinformatics 2010, 26, 2914–2915. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Kim, Y.O.; Park, S.-C.; Chun, J. OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 2016, 66, 1100–1103. [Google Scholar] [CrossRef]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef]
- Sievers, F.; Higgins, D.G. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 2018, 27, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, M.C.F.; Nielsen, M. Seq2Logo: A method for construction and visualization of amino acid binding motifs and sequence profiles including sequence weighting, pseudo counts and two-sided representation of amino acid enrichment and depletion. Nucleic Acids Res. 2012, 40, W281–W287. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2007, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Caspi, R.; Altman, T.; Billington, R.; Dreher, K.; Foerster, H.; Fulcher, C.A.; Holland, T.A.; Keseler, I.M.; Kothari, A.; Kubo, A. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2014, 42, D459–D471. [Google Scholar] [CrossRef] [PubMed]
- Busk, P.K.; Lange, L. Function-based classification of carbohydrate-active enzymes by recognition of short, conserved peptide motifs. Appl. Environ. Microbiol. 2013, 79, 3380–3391. [Google Scholar] [CrossRef] [PubMed]
- Busk, P.K.; Pilgaard, B.; Lezyk, M.J.; Meyer, A.S.; Lange, L. Homology to peptide pattern for annotation of carbohydrate-active enzymes and prediction of function. BMC Bioinform. 2017, 18, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Armenteros, J.J.A.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.-M.; Hwang, K.H.; Park, J.-S. Complete Genome Sequence of Chryseobacterium camelliae Dolsongi-HT1, a Green Tea Isolate with Keratinolytic Activity. Genome Announc. 2018, 6. [Google Scholar] [CrossRef]
- Park, G.-S.; Hong, S.-J.; Jung, B.K.; Khan, A.R.; Park, Y.-J.; Park, C.E.; Lee, A.; Kwak, Y.; Lee, Y.-J.; Lee, D.-W. Complete genome sequence of a keratin-degrading bacterium Chryseobacterium gallinarum strain DSM 27622T isolated from chicken. J. Biotechnol. 2015, 211, 66–67. [Google Scholar] [CrossRef] [PubMed]
- Park, G.-S.; Hong, S.-J.; Lee, C.-H.; Khan, A.R.; Ullah, I.; Jung, B.K.; Choi, J.; Kwak, Y.; Back, C.-G.; Jung, H.-Y. Draft genome sequence of Chryseobacterium sp. strain P1-3, a keratinolytic bacterium isolated from poultry waste. Genome Announc. 2014, 2. [Google Scholar] [CrossRef]
- Lin, P.-C.; Pakrasi, H.B. Engineering cyanobacteria for production of terpenoids. Planta 2019, 249, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Barajas, J.F.; Blake-Hedges, J.M.; Bailey, C.B.; Curran, S.; Keasling, J.D. Engineered polyketides: Synergy between protein and host level engineering. Synth. Syst. Biotechnol. 2017, 2, 147–166. [Google Scholar] [CrossRef]
- Wang, W.; Li, S.; Li, Z.; Zhang, J.; Fan, K.; Tan, G.; Ai, G.; Lam, S.M.; Shui, G.; Yang, Z. Harnessing the intracellular triacylglycerols for titer improvement of polyketides in Streptomyces. Nat. Biotechnol. 2020, 38, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Adamek, M.; Spohn, M.; Stegmann, E.; Ziemert, N. Mining bacterial genomes for secondary metabolite gene clusters. In Antibiotics; Springer: Berlin/Heidelberg, Germany, 2017; pp. 23–47. [Google Scholar]
- Reichenbach, H.; Kohl, W.; Böttger-Vetter, A.; Achenbach, H. Flexirubin-type pigments in Flavobacterium. Arch. Microbiol. 1980, 126, 291–293. [Google Scholar] [CrossRef]
- Vandamme, P.; Bernardet, J.-F.; Segers, P.; Kersters, K.; Holmes, B. New Perspectives in the Classification of the Flavobacteria: Description of Chryseobacterium gen. nov., Bergeyella gen. nov., and Empedobacter nom. rev. Int. J. Syst. Evol. Microbiol. 1994, 44, 827–831. [Google Scholar] [CrossRef]
- Venil, C.K.; Zakaria, Z.A.; Usha, R.; Ahmad, W.A. Isolation and characterization of flexirubin type pigment from Chryseobacterium sp. UTM-3T. Biocatal. Agric. Biotechnol. 2014, 3, 103–107. [Google Scholar] [CrossRef]
- Javidpour, P.; Deutsch, S.; Mutalik, V.K.; Hillson, N.J.; Petzold, C.J.; Keasling, J.D.; Beller, H.R. Investigation of proposed ladderane biosynthetic genes from anammox bacteria by heterologous expression in E. coli. PLoS ONE 2016, 11, e0151087. [Google Scholar]
- Mercer, J.A.; Cohen, C.M.; Shuken, S.R.; Wagner, A.M.; Smith, M.W.; Moss III, F.R.; Smith, M.D.; Vahala, R.; Gonzalez-Martinez, A.; Boxer, S.G. Chemical synthesis and self-assembly of a ladderane phospholipid. J. Am. Chem. Soc. 2016, 138, 15845–15848. [Google Scholar] [CrossRef]
- Adamek, M.; Alanjary, M.; Sales-Ortells, H.; Goodfellow, M.; Bull, A.T.; Winkler, A.; Wibberg, D.; Kalinowski, J.; Ziemert, N. Comparative genomics reveals phylogenetic distribution patterns of secondary metabolites in Amycolatopsis species. BMC Genom. 2018, 19, 1–15. [Google Scholar] [CrossRef]
- Venil, C.K.; Zakaria, Z.A.; Ahmad, W.A. Bacterial pigments and their applications. Process. Biochem. 2013, 48, 1065–1079. [Google Scholar] [CrossRef]
- Aruldass, C.A.; Dufossé, L.; Ahmad, W.A. Current perspective of yellowish-orange pigments from microorganisms—A review. J. Clean. Prod. 2018, 180, 168–182. [Google Scholar] [CrossRef]
- Philmus, B.; Christiansen, G.; Yoshida, W.Y.; Hemscheidt, T.K. Post-translational modification in microviridin biosynthesis. ChemBioChem 2008, 9, 3066–3073. [Google Scholar] [CrossRef] [PubMed]
- Gatte-Picchi, D.; Weiz, A.; Ishida, K.; Hertweck, C.; Dittmann, E. Functional analysis of environmental DNA-derived microviridins provides new insights into the diversity of the tricyclic peptide family. Appl. Environ. Microbiol. 2014, 80, 1380–1387. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.A.; Abol-Fotouh, D.; Omer, A.M.; Tamer, T.M.; Abbas, E. Comprehensive insights into microbial keratinases and their implication in various biotechnological and industrial sectors: A review. Int. J. Biol. Macromol. 2020, 154, 567–583. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Singh, P.; Srivastava, A. Synthesis, nature and utility of universal iron chelator–Siderophore: A review. Microbiol. Res. 2018, 212, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Boda, S.K.; Pandit, S.; Garai, A.; Pal, D.; Basu, B. Bacterial siderophore mimicking iron complexes as DNA targeting antimicrobials. RSC Adv. 2016, 6, 39245–39260. [Google Scholar] [CrossRef]
- Nakouti, I.; Sihanonth, P.; Palaga, T.; Hobbs, G. Effect of a siderophore producer on animal cell apoptosis: A possible role as anti-cancer agent. Int. J. Pharma Med. Biol. Sci. 2013, 2, 1–5. [Google Scholar]
- Remington, S.J. Structure and mechanism of citrate synthase. In Current Topics in Cellular Regulation; Elsevier: Amsterdam, The Netherlands, 1992; Volume 33, pp. 209–229. [Google Scholar]
- Cronan, J.E.; Thomas, J. Bacterial fatty acid synthesis and its relationships with polyketide synthetic pathways. Methods Enzymol. 2009, 459, 395–433. [Google Scholar]
- Park, Y.-K.; Dulermo, T.; Ledesma-Amaro, R.; Nicaud, J.-M. Optimization of odd chain fatty acid production by Yarrowia lipolytica. Biotechnol. Biofuels 2018, 11, 1–12. [Google Scholar] [CrossRef]
- Park, Y.-k.; Ledesma-Amaro, R.; Nicaud, J.-M. De novo biosynthesis of odd-chain fatty acids in Yarrowia lipolytica enabled by modular pathway engineering. Front. Bioeng. Biotechnol. 2020, 7, 484. [Google Scholar] [CrossRef]
- Nasipuri, P.; Herschend, J.; Brejnrod, A.D.; Madsen, J.S.; Espersen, R.; Svensson, B.; Burmølle, M.; Jacquiod, S.; Sørensen, S.J. Community-intrinsic properties enhance keratin degradation from bacterial consortia. PLoS ONE 2020, 15, e0228108. [Google Scholar] [CrossRef]
- Ramnani, P.; Gupta, R. Keratinases vis-à-vis conventional proteases and feather degradation. World J. Microbiol. Biotechnol. 2007, 23, 1537–1540. [Google Scholar] [CrossRef]
- Navone, L.; Speight, R. Understanding the dynamics of keratin weakening and hydrolysis by proteases. PLoS ONE 2018, 13, e0202608. [Google Scholar] [CrossRef] [PubMed]
- Cedrola, S.M.L.; de Melo, A.C.N.; Mazotto, A.M.; Lins, U.; Zingali, R.B.; Rosado, A.S.; Peixoto, R.S.; Vermelho, A.B. Keratinases and sulfide from Bacillus subtilis SLC to recycle feather waste. World J. Microbiol. Biotechnol. 2012, 28, 1259–1269. [Google Scholar] [CrossRef] [PubMed]
- Brandelli, A.; Daroit, D.J.; Riffel, A. Biochemical features of microbial keratinases and their production and applications. Appl. Microbiol. Biotechnol. 2010, 85, 1735–1750. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Busk, P.K.; Herbst, F.-A.; Lange, L. Genome and secretome analyses provide insights into keratin decomposition by novel proteases from the non-pathogenic fungus Onygena corvina. Appl. Microbiol. Biotechnol. 2015, 99, 9635–9649. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-W.; Liang, S.; Ke, Y.; Deng, J.-J.; Zhang, M.-S.; Lu, D.-L.; Li, J.-Z.; Luo, X.-C. The feather degradation mechanisms of a new Streptomyces sp. isolate SCUT-3. Commun. Biol. 2020, 3, 1–13. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Chryseobacterium sp. KMC2 | Chryseobacterium camelliae Dolsongi-HT1 | Chryseobacterium gallinarum strain DSM 27622 | Chryseobacterium sp. P1-3 |
|---|---|---|---|---|
| Total length (bp) | 5.276.159 | 4.376.354 | 4.633.632 | 4.628.764 |
| Contigs | 63 | 1 | 1 | 45 |
| N50 (bp) | 231.784 | 4.376.354 | 4.633.632 | 342.512 |
| GC content (%) | 36.33 | 41.80 | 37.30 | 37.02 |
| Gene | 4.773 | 4.012 | 4.161 | 4.906 |
| CDS | 4.692 | 4.009 | 4.151 | 4.939 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, D.; Shoaie, S.; Jacquiod, S.; Sørensen, S.J.; Ledesma-Amaro, R. Comparative Genomics Analysis of Keratin-Degrading Chryseobacterium Species Reveals Their Keratinolytic Potential for Secondary Metabolite Production. Microorganisms 2021, 9, 1042. https://doi.org/10.3390/microorganisms9051042
Kang D, Shoaie S, Jacquiod S, Sørensen SJ, Ledesma-Amaro R. Comparative Genomics Analysis of Keratin-Degrading Chryseobacterium Species Reveals Their Keratinolytic Potential for Secondary Metabolite Production. Microorganisms. 2021; 9(5):1042. https://doi.org/10.3390/microorganisms9051042
Chicago/Turabian StyleKang, Dingrong, Saeed Shoaie, Samuel Jacquiod, Søren J. Sørensen, and Rodrigo Ledesma-Amaro. 2021. "Comparative Genomics Analysis of Keratin-Degrading Chryseobacterium Species Reveals Their Keratinolytic Potential for Secondary Metabolite Production" Microorganisms 9, no. 5: 1042. https://doi.org/10.3390/microorganisms9051042
APA StyleKang, D., Shoaie, S., Jacquiod, S., Sørensen, S. J., & Ledesma-Amaro, R. (2021). Comparative Genomics Analysis of Keratin-Degrading Chryseobacterium Species Reveals Their Keratinolytic Potential for Secondary Metabolite Production. Microorganisms, 9(5), 1042. https://doi.org/10.3390/microorganisms9051042