
Use of High-Throughput Sequencing and Two RNA Input Methods to Identify Viruses Infecting Tomato Crops

 , ,
, ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Nucleic Acid Extraction, Library Construction and Sequencing
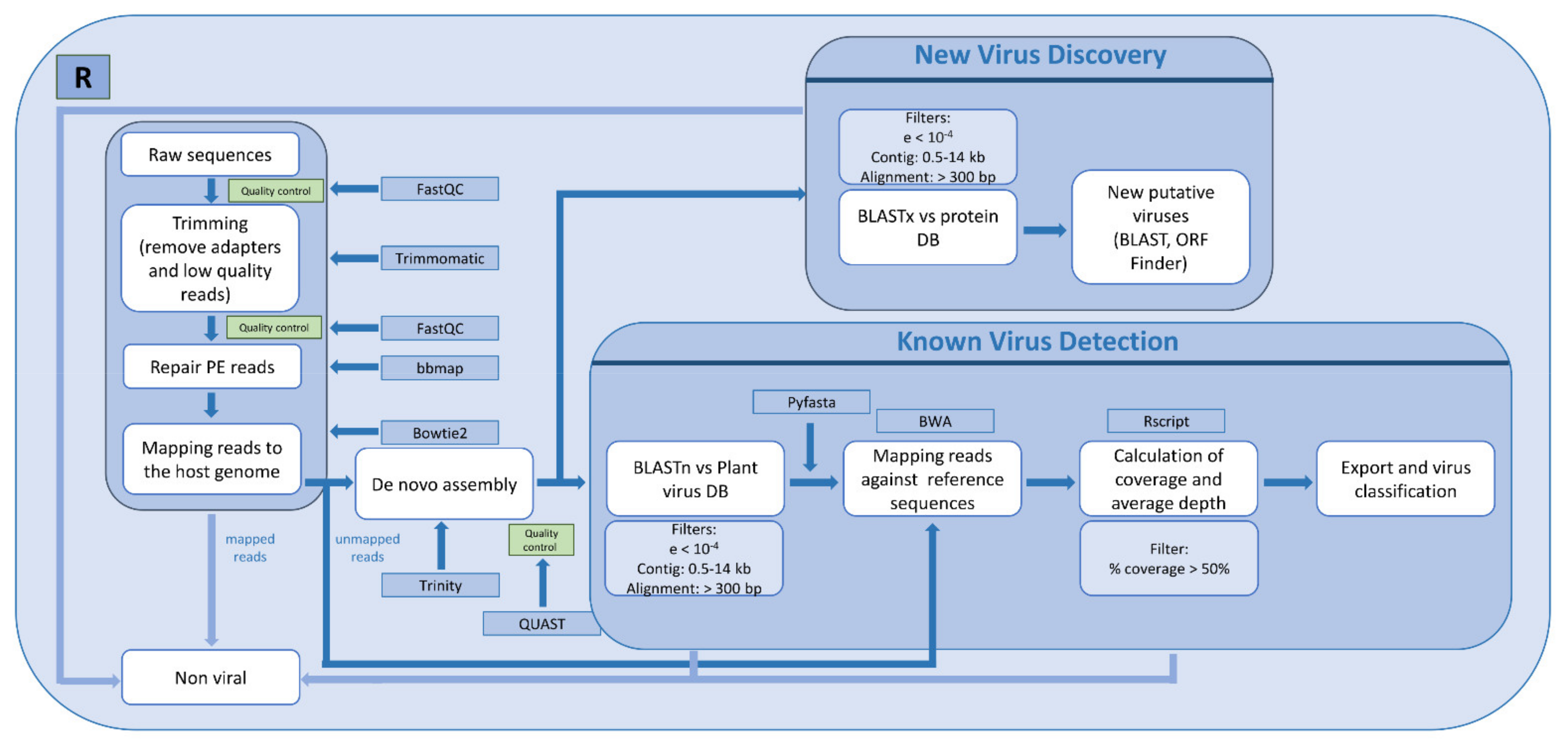
2.3. Bioinformatic Analysis
2.4. Conventional RT-PCR and qRT-PCR
3. Results
3.1. HTS Using Two Different RNA Inputs: Total RNA and dsRNA
3.2. Comparison of Viral Species Found in Two Technical Replicates Using Total RNA as the Input
3.3. Viral Species Found Using Total RNA or dsRNA as the Input
3.4. In Silico Analysis of the Minimum Sequencing Depth Needed to Detect the Less Abundant Viruses
3.5. Viruses Already Reported to Infect Tomato Plants in Spain
3.6. Viruses Not Previously Reported to Infect Tomato Plants in Spain
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanssen, I.M.; Lapidot, M. Major tomato viruses in the Mediterranean basin. In Advances in Virus Research; Elsevier: Amsterdam, The Netherlands, 2012; Volume 84, pp. 31–66. ISBN 0065-3527. [Google Scholar]
- Woolhouse, M.E.J.; Gowtage-Sequeria, S. Host range and emerging and reemerging pathogens. Emerg. Infect. Dis. 2005, 11, 1842. [Google Scholar] [CrossRef] [PubMed]
- Roossinck, M.J.; Martin, D.P.; Roumagnac, P. Plant virus metagenomics: Advances in virus discovery. Phytopathology 2015, 105, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jia, A.; Qiao, Y.; Xiang, J.; Zhang, Y.; Wang, W. Virome analysis of lily plants reveals a new potyvirus. Arch. Virol. 2018, 163, 1079–1082. [Google Scholar] [CrossRef]
- Jo, Y.; Choi, H.; Kim, S.-M.; Kim, S.-L.; Lee, B.C.; Cho, W.K. The pepper virome: Natural co-infection of diverse viruses and their quasispecies. BMC Genom. 2017, 18, 1–12. [Google Scholar] [CrossRef]
- Jo, Y.; Cho, W.K. RNA viromes of the oriental hybrid lily cultivar “Sorbonne”. BMC Genom. 2018, 19, 748. [Google Scholar] [CrossRef] [PubMed]
- Mutuku, J.M.; Wamonje, F.O.; Mukeshimana, G.; Njuguna, J.; Wamalwa, M.; Choi, S.-K.; Tungadi, T.; Djikeng, A.; Kelly, K.; Domelevo Entfellner, J.-B. Metagenomic analysis of plant virus occurrence in common bean (Phaseolus vulgaris) in Central Kenya. Front. Microbiol. 2018, 9, 2939. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Sun, X.; Taylor, A.; Jiao, C.; Xu, Y.; Cai, X.; Wang, X.; Ge, C.; Pan, G.; Wang, Q. Diversity, distribution, and evolution of tomato viruses in China uncovered by small RNA sequencing. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Ma, Y.; Marais, A.; Lefebvre, M.; Faure, C.; Candresse, T. Metagenomic analysis of virome cross-talk between cultivated Solanum lycopersicum and wild Solanum nigrum. Virology 2020, 540, 38–44. [Google Scholar] [CrossRef]
- Kutnjak, D.; Rupar, M.; Gutierrez-Aguirre, I.; Curk, T.; Kreuze, J.F.; Ravnikar, M. Deep sequencing of virus-derived small interfering RNAs and RNA from viral particles shows highly similar mutational landscapes of a plant virus population. J. Virol. 2015, 89, 4760–4769. [Google Scholar] [CrossRef]
- Pecman, A.; Kutnjak, D.; Gutiérrez-Aguirre, I.; Adams, I.; Fox, A.; Boonham, N.; Ravnikar, M. Next generation sequencing for detection and discovery of plant viruses and viroids: Comparison of two approaches. Front. Microbiol. 2017, 8, 1998. [Google Scholar] [CrossRef]
- Ma, Y.; Marais, A.; Lefebvre, M.; Theil, S.; Svanella-Dumas, L.; Faure, C.; Candresse, T. Phytovirome analysis of wild plant populations: Comparison of double-stranded RNA and virion-associated nucleic acid metagenomic approaches. J. Virol. 2019, 94, e0146219. [Google Scholar] [CrossRef]
- Valverde, R.A.; Nameth, S.T.; Jordan, R.L. Analysis of double-stranded RNA for plant virus diagnosis. Plant Dis. 1990, 74, 255–258. [Google Scholar]
- Marais, A.; Faure, C.; Bergey, B.; Candresse, T. Viral double-stranded RNAs (dsRNAs) from plants: Alternative nucleic acid substrates for high-throughput sequencing. In Viral Metagenomics; Springer: Berlin/Heidelberg, Germany, 2018; pp. 45–53. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner; Lawrence Berkeley National Lab.(LBNL): Berkeley, CA, USA, 2014. [Google Scholar]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Camacho, C.; Madded, T.; Tao, T.; Agarwala, R.; Morgulis, A. BLAST® Command Line Applications User Manual; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2018. [Google Scholar]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Li, H. Seqtk: A Fast and Lightweight Tool for Processing Sequences; Broad Inst.: Cambridge, MA, USA, 2016. [Google Scholar]
- Gutiérrez-Aguirre, I.; Mehle, N.; Delić, D.; Gruden, K.; Mumford, R.; Ravnikar, M. Real-time quantitative PCR based sensitive detection and genotype discrimination of Pepino mosaic virus. J. Virol. Methods 2009, 162, 46–55. [Google Scholar] [CrossRef]
- Torre, C.; Donaire, L.; Gómez-Aix, C.; Juárez, M.; Peterschmitt, M.; Urbino, C.; Hernando, Y.; Agüero, J.; Aranda, M.A. Characterization of begomoviruses sampled during severe epidemics in tomato cultivars carrying the Ty-1 Gene. Int. J. Mol. Sci. 2018, 19, 2614. [Google Scholar] [CrossRef] [PubMed]
- Ciuffo, M.; Kinoti, W.M.; Tiberini, A.; Forgia, M.; Tomassoli, L.; Constable, F.E.; Turina, M. A new blunervirus infects tomato crops in Italy and Australia. Arch. Virol. 2020, 165, 2379–2384. [Google Scholar] [CrossRef] [PubMed]
- Borodynko, N.; Hasiów-Jaroszewska, B.; Pospieszny, H. Identification and characterization of an Olive latent virus 1 isolate from a new host: Solanum lycopersicum. J. Plant Pathol. 2010, 92, 789–792. [Google Scholar]
- Visser, M.; Bester, R.; Burger, J.T.; Maree, H.J. Next-generation sequencing for virus detection: Covering all the bases. Virol. J. 2016, 13, 1–6. [Google Scholar] [CrossRef] [PubMed]
- García, Y.G.; Montoya, M.M.; Gutiérrez, P.A. Detection of RNA viruses in Cape gooseberry (Physalis peruviana L.) by RNAseq using total RNA and dsRNA inputs. Arch. Phytopathol. Plant Prot. 2020, 53, 395–413. [Google Scholar] [CrossRef]
- Czosnek, H.; Ber, R.; Navot, N.; Antignus, Y.; Cohen, S.; Zamir, D. Tomato yellow leaf curl virus DNA forms in the viral capside, in infected plants and in the insect vector. J. Phytopathol. 1989, 125, 47–54. [Google Scholar] [CrossRef]
- Maree, H.J.; Fox, A.; Al Rwahnih, M.; Boonham, N.; Candresse, T. Application of HTS for routine plant virus diagnostics: State of the art and challenges. Front. Plant Sci. 2018, 9, 1082. [Google Scholar] [CrossRef]
- Al Rwahnih, M.; Daubert, S.; Golino, D.; Islas, C.; Rowhani, A. Comparison of next-generation sequencing versus biological indexing for the optimal detection of viral pathogens in grapevine. Phytopathology 2015, 105, 758–763. [Google Scholar] [CrossRef]
- Candresse, T.; Filloux, D.; Muhire, B.; Julian, C.; Galzi, S. Appearances Can Be Deceptive: Revealing a Hidden Viral Infection with Deep Sequencing. PLoS ONE 2014, 9, e102945. [Google Scholar] [CrossRef] [PubMed]
- Akinyemi, I.A.; Wang, F.; Zhou, B.; Qi, S.; Wu, Q. Ecogenomic survey of plant viruses infecting tobacco by next generation sequencing. Virol. J. 2016, 13, 181. [Google Scholar] [CrossRef]
- Alcalá-Briseño, R.I.; Casarrubias-Castillo, K.; López-Ley, D.; Garrett, K.A.; Silva-Rosales, L. Network analysis of the papaya oroecological regions of Chiapas, Mexicochard virome from two agr. Msystems 2020, 5, e0042319. [Google Scholar] [CrossRef] [PubMed]
- Bernal-Vicente, A.; Donaire, L.; Torre, C.; Gómez-Aix, C.; Sánchez-Pina, M.A.; Juarez, M.; Hernando, Y.; Aranda, M.A. Small RNA-seq to characterize viruses responsible of Lettuce big vein disease in Spain. Front. Microbiol. 2018, 9, 3188. [Google Scholar] [CrossRef] [PubMed]
- Jo, Y.; Bae, J.-Y.; Kim, S.-M.; Choi, H.; Lee, B.C.; Cho, W.K. Barley RNA viromes in six different geographical regions in Korea. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Mumo, N.N.; Mamati, G.E.; Ateka, E.M.; Rimberia, F.K.; Asudi, G.O.; Boykin, L.M.; Machuka, E.M.; Njuguna, J.N.; Pelle, R.; Stomeo, F. Metagenomic Analysis of Plant Viruses Associated With Papaya Ringspot Disease in Carica papaya L. in Kenya. Front. Microbiol. 2020, 11, 205. [Google Scholar] [CrossRef]
- Skums, P.; Artyomenko, A.; Glebova, O.; Ramachandran, S.; Campo, D.S.; Dimitrova, Z.; Măndoiu, I.I.; Zelikovsky, A.; Khudyakov, Y. Pooling Strategy for Massive Viral Sequencing. In Computational Methods for Next Generation Sequencing Data Analysis; Măndoiu, I., Zelikovsky., A., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; pp. 57–83. [Google Scholar]
- Agüero, J.; Gómez-Aix, C.; Sempere, R.N.; García-Villalba, J.; García-Núñez, J.; Hernando, Y.; Aranda, M.A. Stable and broad spectrum cross-protection against Pepino mosaic virus attained by mixed infection. Front. Plant Sci. 2018, 9, 1810. [Google Scholar] [CrossRef]
- Gómez, P.; Sempere, R.N.; Elena, S.F.; Aranda, M.A. Mixed infections of Pepino mosaic virus strains modulate the evolutionary dynamics of this emergent virus. J. Virol. 2009, 83, 12378–12387. [Google Scholar] [CrossRef]
- Fiallo-Olivé, E.; Navas-Castillo, J. Tomato chlorosis virus, an emergent plant virus still expanding its geographical and host ranges. Mol. Plant Pathol. 2019, 20, 1307–1320. [Google Scholar] [CrossRef]
- Navas-Castillo, J.; López-Moya, J.J.; Aranda, M.A. Whitefly-Transmitted RNA Viruses that Affect Intensive Vegetable Production. Ann. Appl. Biol. 2014, 165, 155–171. [Google Scholar] [CrossRef]
- Orílio, A.F.; Fortes, I.M.; Navas-Castillo, J. Infectious cDNA clones of the crinivirus Tomato chlorosis virus are competent for systemic plant infection and whitefly-transmission. Virology 2014, 464, 365–374. [Google Scholar] [CrossRef]
- Navas-Castillo, J.; Fiallo-Olivé, E.; Sánchez-Campos, S. Emerging Virus Diseases Transmitted by Whiteflies. Annu. Rev. Phytopathol. 2011, 49, 219–248. [Google Scholar] [CrossRef]
- Elvira-Gonzalez, L.; Carpino, C.; Alfaro Fernández, A.O.; Font San Ambrosio, M.I.; Peiró Barber, R.M.; Rubio MIgélez, L.; Galipienso-Torregrosa, L. A sensitive real-time RT-PCR reveals a high incidence of Southern tomato virus (STV) in Spanish tomato crops. Span. J. Agric. Res. 2018, 16. [Google Scholar] [CrossRef]
- Campbell, R.N.; Lot, H. Lettuce ring necrosis, a viruslike disease of lettuce: Evidence for transmission by Olpidium brassicae. Plant Dis. 1996, 80, 611–615. [Google Scholar] [CrossRef]
- Gallitelli, D.; Savino, V. Olive latent virus-1, an isometric virus with a single RNA species isolated from olive in Apulia, Southern Italy. Ann. Appl. Biol. 1985, 106, 295–303. [Google Scholar] [CrossRef]
- Kanematsu, S.; Yumiko, T.; Morikawa, T. Isolation of Olive latent virus 1 from tulip in Toyama Prefecture. J. Gen. Plant Pathol. 2001, 67, 333–334. [Google Scholar] [CrossRef]
- Martelli, G.P.; Yilmaz, M.A.; Savino, V.; Baloglu, S.; Grieco, F.; Güldür, M.E.; Greco, N.; Lafortezza, R. Properties of a citrus isolate of olive latent virus 1, a new necrovirus. Eur. J. Plant Pathol. 1996, 102, 527–536. [Google Scholar] [CrossRef]
- Quito-Avila, D.F.; Freitas-Astúa, J.; Melzer, M.J. Bluner-, Cile-, and higreviruses (kitaviridae). Encycl. Virol. 2020, 3, 247–251. [Google Scholar]
- Moreno, A.B.; López-Moya, J.J. When viruses play team sports: Mixed infections in plants. Phytopathology 2020, 110, 29–48. [Google Scholar] [CrossRef]
- Massart, S.; Candresse, T.; Gil, J.; Lacomme, C.; Predajna, L.; Ravnikar, M.; Reynard, J.-S.; Rumbou, A.; Saldarelli, P.; Škorić, D. A framework for the evaluation of biosecurity, commercial, regulatory, and scientific impacts of plant viruses and viroids identified by NGS technologies. Front. Microbiol. 2017, 8, 45. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Sample ID | Surveyed | Location | Symptoms | Virus Detected 1 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| OLV1 | TYLCV | ToCV | STV 2 | ToFBV | PepMV-EU | PepMV-CH2 | LRNV | ||||
| R20-01 | March 2020 | Almería | Vein clearing | − | − | − | + | − | ++ | +++ | +++ |
| R19-12 | November 2019 | Almería | Necrotic spots on leaves | − | − | + | + | − | + | +++ | + |
| R19-09 | October 2019 | Almería | Leaf curling, leaf mosaics | − | − | − | + | − | +++ | +++ | − |
| R19-08 | October 2019 | Almería | Leaf curling, leaf mosaics | − | − | − | − | − | +++ | ++ | − |
| R19-07 | September 2019 | Almería | Chlorosis, yellow spots on leaves, leaf mosaics | − | − | − | + | ++ | +++ | ++ | − |
| R17-01 | Febuary 2017 | Murcia | Upward curling of leaves, chlorosis on leaves | − | − | − | − | ++ | + | ++ | − |
| H-57 | December 2016 | Murcia | Leaf mosaics | − | − | − | + | − | ++ | ++ | − |
| H-55 | June 2016 | Murcia | Leaf distortion | − | − | + | + | − | − | ++ | − |
| H-54 | May 2016 | Murcia | Leaf distortion | − | − | − | + | − | ++ | ++ | − |
| H-53 | May 2016 | Murcia | Leaf distortion | − | + | + | + | +++ | + | +++ | − |
| H-52 | May 2016 | Murcia | Distortion and mosaic in fruit | − | + | + | + | +++ | − | +++ | − |
| H-50 | April 2016 | Murcia | Leaf distortion | − | − | − | + | − | + | +++ | − |
| H-43 | December 2015 | Granada | No clear symptoms | − | − | − | + | − | − | + | − |
| H-42 | December 2015 | Granada | Leaf curling | − | ++ | − | − | − | + | + | − |
| H-31 | October 2015 | Almería | Yellow mosaic | − | ++ | + | + | − | − | − | − |
| H-20 | April 2015 | Portugal 3 | No clear symptoms | − | − | + | + | ++ | − | +++ | − |
| H-13 | Aprli 2015 | Portugal 3 | No clear symptoms | − | + | − | − | − | − | +++ | − |
| H-11 | April 2015 | Portugal 3 | No clear symptoms | ++ | − | − | + | − | − | ++ | − |
| H-10 | April 2015 | Almería | Necrosis, yellow mosaic and distortion of leaves | − | + | ++ | + | − | + | ++ | + |
| H-09 | April 2015 | Almería | Necrosis, yellow mosaic and distortion of leaves | + | + | ++ | + | - | + | +++ | + |
| Tom1 | Tom2 | TomDS | ||||
|---|---|---|---|---|---|---|
| Reads | % | Reads | % | Reads | % | |
| Raw reads | 86,284,538 | 84,739,174 | 64,540,826 | |||
| Clean reads | 85,026,574 | 98.54 | 83,516,356 | 98.56 | 63,715,596 | 98.72 |
| Host mappings | 40,905,042 | 48.11 | 39,992,206 | 47.89 | 16,395,174 | 25.73 |
| Filter reads | 44,121,532 | 51.89 | 43,524,150 | 52.11 | 47,320,422 | 74.27 |
| Viral contigs | 63 | 51 | 55 | |||
| Unique viruses | 7 | 7 | 6 | |||
| Viral reads | 6,790,296 | 7.99 | 7,159,776 | 8.57 | 20,491,882 | 32.16 |
| Virus | Accession | Genome | Segment | Ref. Length | Tom1 | Tom2 | TomDS | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reads | AD | PC | Reads | AD | PC | Reads | AD | PC | |||||
| OLV1 | DQ083996 | (+)ssRNA | 3702 | 390 | 9 | 97.97 | 364 | 9 | 89.68 | 678 | 590 | 0.62 | |
| TYLCV | HF548826 | (+)ssDNA | 2787 | 1694 | 65 | 100 | 1596 | 60 | 99.64 | 1344 | 69 | 90.17 | |
| ToCV | KF018280 | (+)ssRNA | RNA1 | 8596 | 1106 | 14 | 96.92 | 1076 | 15 | 94.16 | 222,112 | 3441 | 98.15 |
| KJ815045 | RNA2 | 8249 | 2736 | 42 | 99.33 | 2794 | 43 | 99.52 | 235,672 | 3759 | 99.79 | ||
| STV | KT438549 | dsRNA | 3463 | 1782 | 63 | 98.84 | 1812 | 64 | 98.64 | 3,459,440 | 7319 | 99.19 | |
| ToFBV | MK517477 | (+)ssRNA | RNA1 | 5811 | 39,452 | 878 | 99.78 | 41,498 | 930 | 99.78 | 255,152 | 5665 | 99.88 |
| MK517478 | RNA2 | 3643 | 17,810 | 626 | 99.75 | 17,760 | 628 | 99.45 | 79,414 | 2892 | 99.56 | ||
| MK517479 | RNA3 | 2872 | 72,830 | 2096 | 99.51 | 81,684 | 2417 | 99.65 | 500,460 | 6360 | 99.93 | ||
| MK517480 | RNA4 | 1946 | 47,102 | 2938 | 100 | 51,060 | 3158 | 100 | 317,660 | 7309 | 100 | ||
| PepMV | NC_004067 | (+)ssRNA | 6450 | 6,431,722 | 7687 | 100 | 6,809,266 | 7686 | 100 | 15,351,220 | 7831 | 100 | |
| LRNV | NC_006051 | (−)ssRNA | RNA 1 | 7651 | 13,116 | 223 | 99.76 | 10,716 | 183 | 99.48 | 14,738 | 258 | 99.12 |
| NC_006052 | RNA 2 | 1830 | 17,546 | 1258 | 99.89 | 15,668 | 1124 | 99.95 | 10,512 | 758 | 99.89 | ||
| NC_006053 | RNA 3 | 1527 | 108,412 | 6655 | 98.76 | 95,402 | 6507 | 99.41 | 38,700 | 3458 | 97.12 | ||
| NC_006054 | RNA 4 | 1417 | 34,598 | 3226 | 99.86 | 29,080 | 2749 | 98.52 | 4780 | 462 | 96.47 | ||
| Subset 1 (50%) | Subset 2 (37.5%) | Subset 3 (25%) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Tom1 | TomDS | Tom1 | TomDS | Tom1 | TomDS | |||||||
| Reads | % | Reads | % | Reads | % | Reads | % | Reads | % | Reads | % | |
| Subset | 40,000,000 | 30,000,000 | 30,000,000 | 22,500,000 | 20,000,000 | 15,000,000 | ||||||
| Clean reads | 39,416,281 | 98.54 | 29,616,416 | 98.72 | 29,562,358 | 98.54 | 22,212,742 | 98.72 | 19,708,041 | 98.54 | 14,809,024 | 98.73 |
| Host mappings | 19,544,446 | 49.58 | 7,618,386 | 25.72 | 14,223,278 | 48.11 | 5,714,092 | 25.72 | 9,483,289 | 48.12 | 3,808,670 | 25.72 |
| Filter reads | 20,455,554 | 51.90 | 21,998,030 | 74.28 | 15,339,080 | 51.89 | 16,498,650 | 74.28 | 10,224,752 | 51.88 | 11,000,354 | 74.28 |
| Viral contigs | 50 | 56 | 43 | 40 | 36 | 40 | ||||||
| Unique viruses | 7 | 6 | 7 | 6 | 7 | 6 | ||||||
| Viral reads | 3,033,136 | 14.83 | 9,245,404 | 42.03 | 2,275,725 | 14.84 | 6,934,690 | 42.03 | 1,515,690 | 14.82 | 4,622,820 | 42.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maachi, A.; Torre, C.; Sempere, R.N.; Hernando, Y.; Aranda, M.A.; Donaire, L. Use of High-Throughput Sequencing and Two RNA Input Methods to Identify Viruses Infecting Tomato Crops. Microorganisms 2021, 9, 1043. https://doi.org/10.3390/microorganisms9051043
Maachi A, Torre C, Sempere RN, Hernando Y, Aranda MA, Donaire L. Use of High-Throughput Sequencing and Two RNA Input Methods to Identify Viruses Infecting Tomato Crops. Microorganisms. 2021; 9(5):1043. https://doi.org/10.3390/microorganisms9051043
Chicago/Turabian StyleMaachi, Ayoub, Covadonga Torre, Raquel N. Sempere, Yolanda Hernando, Miguel A. Aranda, and Livia Donaire. 2021. "Use of High-Throughput Sequencing and Two RNA Input Methods to Identify Viruses Infecting Tomato Crops" Microorganisms 9, no. 5: 1043. https://doi.org/10.3390/microorganisms9051043
APA StyleMaachi, A., Torre, C., Sempere, R. N., Hernando, Y., Aranda, M. A., & Donaire, L. (2021). Use of High-Throughput Sequencing and Two RNA Input Methods to Identify Viruses Infecting Tomato Crops. Microorganisms, 9(5), 1043. https://doi.org/10.3390/microorganisms9051043