
Characterisation of Phage Susceptibility Variation in Salmonellaenterica Serovar Typhimurium DT104 and DT104b


Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains
2.2. Identification of SNPs and Phylogenomics
2.3. Identification of Prophages, Plasmids, and R-M and CRISPR/Cas Systems
3. Results
3.1. Phylogenomics of S. Typhimurium DT104 and DT104b
3.2. Prophages in S. Typhimurium DT104 and DT104b
3.3. R-M Systems S. Typhimurium DT104 and DT104b
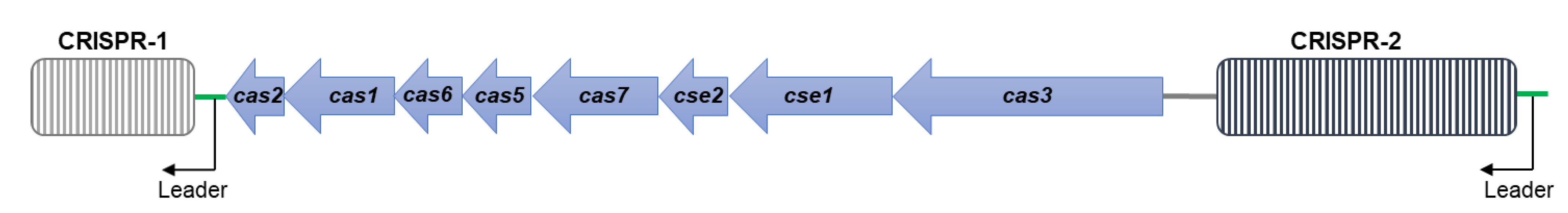
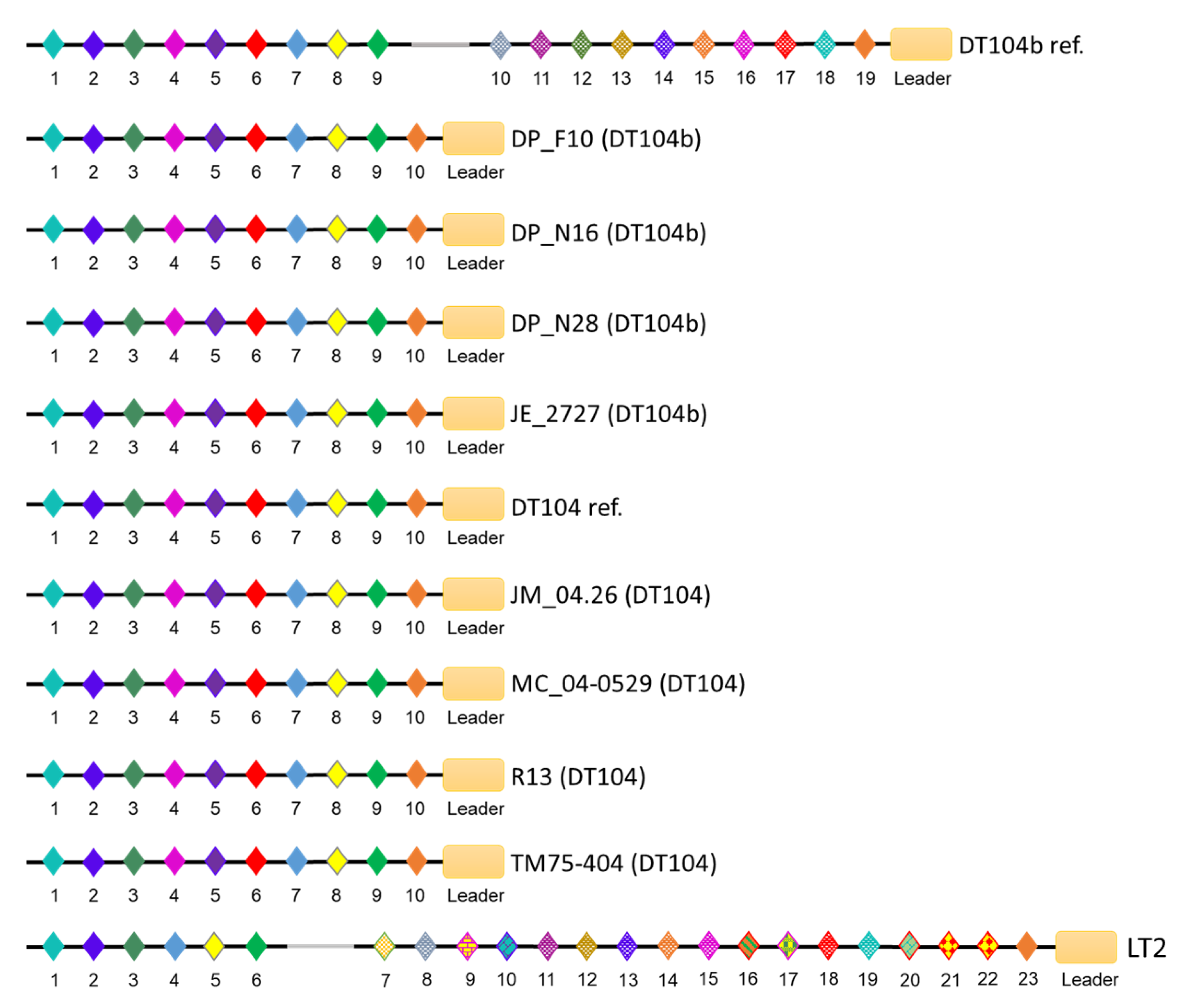
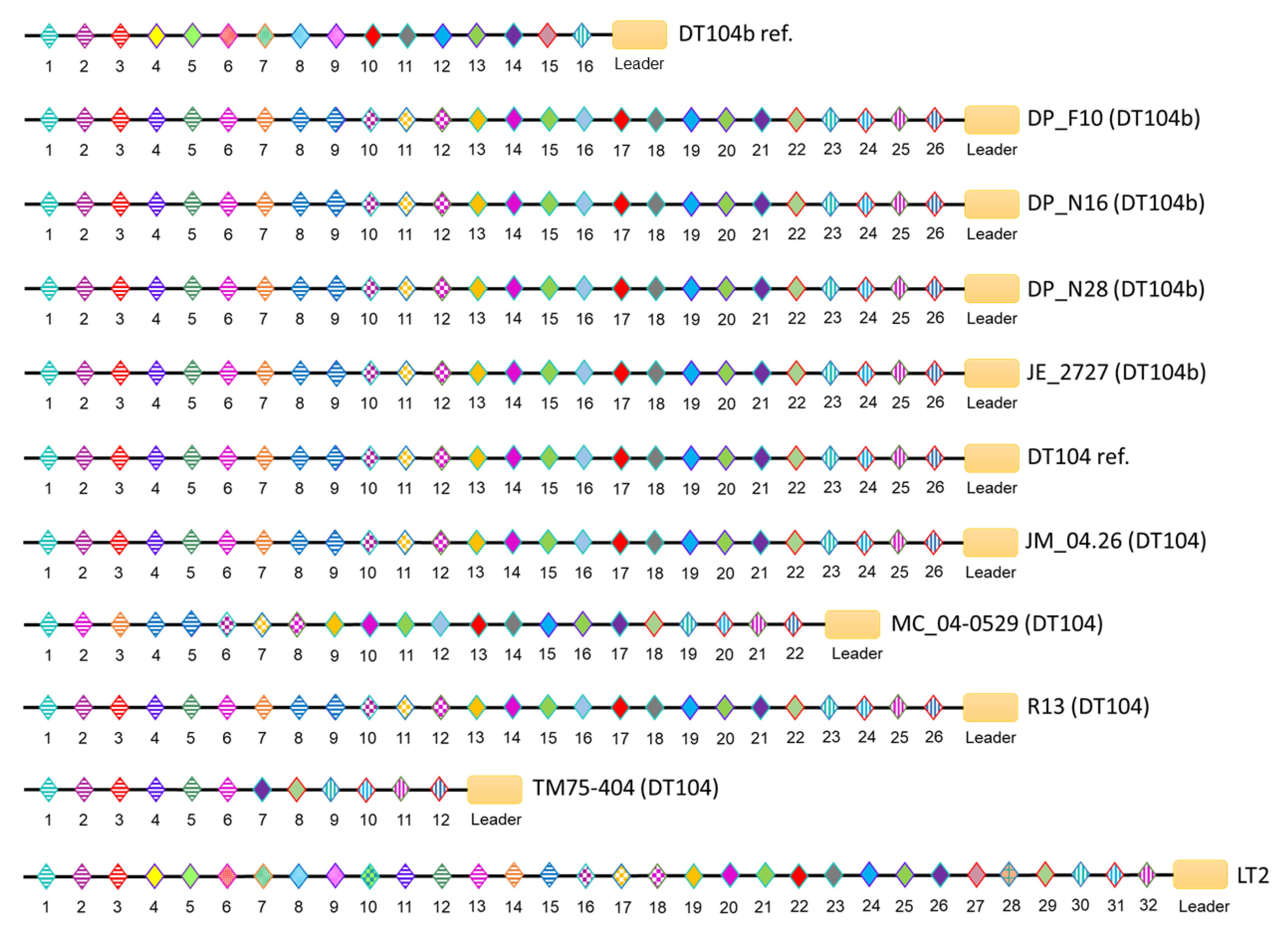
3.4. CRISPR/Cas Systems in S. Typhimurium DT104 and DT104b
3.5. Plasmids in S. Typhimurium DT104 and DT104b
Potential Phage Receptors
3.6. WGS-Based Identification of Antimicrobial Resistance Determinants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mohammed, M.; Le Hello, S.; Leekitcharoenphon, P.; Hendriksen, R. The invasome of Salmonella Dublin as revealed by whole genome sequencing. BMC Infect. Dis. 2017, 17, 544. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, M.; Thapa, S. Evaluation of WGS-subtyping methods for epidemiological surveillance of foodborne salmonellosis. One Health Outlook 2020, 2, 13. [Google Scholar] [CrossRef]
- Rajashekara, G.; Haverly, E.; Halvorson, D.A.; Ferris, K.E.; Lauer, D.C.; Nagaraja, K.V. Multidrug-Resistant Salmonella Typhimurium DT104 in Poultry. J. Food Prot. 2000, 63, 155–161. [Google Scholar] [CrossRef]
- Khan, A.; Nawaz, M.; Khan, S.; Cerniglia, C. Detection of multidrug-resistant Salmonella typhimurium DT104 by multiplex polymerase chain reaction. FEMS Microbiol. Lett. 2000, 182, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, M.; Cormican, M. Whole genome sequencing provides possible explanations for the difference in phage susceptibility among two Salmonella Typhimurium phage types (DT8 and DT30) associated with a single foodborne outbreak. BMC Res. Notes 2015, 8, 728. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, M. Who Fights Whom? Understanding the Complex Dynamics of Bacteria Phage Interaction Using Anderson Phage Typing System. J. Infect. Dis. Ther. 2018, 6, 3. [Google Scholar] [CrossRef]
- Food Standards Agency 2016. Research Project: Systematic and Critical Review on the Potential Use of Bacteriophages on Foods. Food Standards Agency. Available online: https://www.food.gov.uk/research/research-projects/systematic-and-critical-review-on-the-potential-use-of-bacteriophages-on-foods (accessed on 6 May 2020).
- Orzechowska, B.; Mohammed, M. The War between Bacteria and Bacteriophages. In Growing and Handling of Bacterial Cultures; Mishra, M., Ed.; IntechOpen: London, UK, 2019. [Google Scholar]
- Andres, D.; Hanke, C.; Baxa, U.; Seul, A.; Barbirz, S.; Seckler, R. Tailspike interactions with lipopolysaccharide effect DNA ejection from phage P22 particles in vitro. J. Biol. Chem. 2010, 285, 36768–36775. [Google Scholar] [CrossRef]
- Bondy-Denomy, J.; Qian, J.; Westra, E.; Buckling, A.; Guttman, D.S.; Davidson, A.R.; Maxwell, K.L. Prophages mediate defense against phage infection through diverse mechanisms. ISME J. 2016, 10, 2854–2866. [Google Scholar] [CrossRef]
- Labrie, S.; Samson, J.; Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 2010, 8, 317. [Google Scholar] [CrossRef]
- Wang, C.; Nie, T.; Lin, F.; Connerton, I.F.; Lu, Z.; Zhou, S.; Hang, H. Resistance mechanisms adopted by a Salmonella Typhimurium mutant against bacteriophage. Virus Res. 2019, 273, 197759. [Google Scholar] [CrossRef]
- Samson, J.; Magadán, A.; Sabri, M.; Moineau, S. Revenge of the phages: Defeating bacterial defences. Nat. Rev. Microbiol. 2013, 11, 675–687. [Google Scholar] [CrossRef]
- Anderson, E.S.; Ward, L.R.; Saxe, M.J.; de Sa, J.D. Bacteriophage-typing designations of Salmonella typhimurium. Epidemiol. Infect. 1977, 78, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef]
- Kaas, R.S.; Leekitcharoenphon, P.; Aarestrup, F.M.; Lund, O. Solving the Problem of Comparing Whole Bacterial Genomes across Different Sequencing Platforms. PLoS ONE 2014, 9, e104984. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- CGE 2015. Restriction-ModificationFinder 1.1. Center for Genomic Epidemiology. Available online: https://cge.cbs.dtu.dk/services/Restriction-ModificationFinder/ (accessed on 20 February 2020).
- Roberts, R.J.; Vincze, T.; Posfai, J.; Macelis, D. REBASE-a database for DNA restriction and modification: Enzymes, genes and genomes. Nucleic Acids Res. 2015, 43, D298–D299. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007, 35, W52–W57. [Google Scholar] [CrossRef]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef]
- Galata, V.; Fehlmann, T.; Backes, C.; Keller, A. PLSDB: A resource of complete bacterial plasmids. Nucleic Acids Res. 2018, 47, D195–D202. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, M.; Delappe, N.; O’Connor, J.; McKeown, P.; Garvey, P.; Cormican, M. Whole genome sequencing provides an unambiguous link between Salmonella Dublin outbreak strain and a historical isolate. Epidemiol. Infect. 2016, 144, 576–581. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Authority, E.F.S. The European Union Summary Report on Antimicrobial Resistance in zoonotic and indicator bacteria from humans, animals and food in 2017/2018. EFSA J. 2020, 18, e06007. [Google Scholar]
- Sillankorva, S.M.; Oliveira, H.; Azeredo, J. Bacteriophages and Their Role in Food Safety. Int. J. Microbiol. 2012, 2012, 863945. [Google Scholar] [CrossRef]
- Cooper, I.R. A review of current methods using bacteriophages in live animals, food and animal products intended for human consumption. J. Microbiol. Methods 2016, 130, 38–47. [Google Scholar] [CrossRef]
- Baggesen, D.L.; Sørensen, G.; Nielsen, E.M.; Wegener, H.C. Phage typing of Salmonella Typhimurium-is it still a useful tool for surveillance and outbreak investigation? Eurosurveillance 2010, 15, 12–14. [Google Scholar] [CrossRef]
- Ashton, P.M.; Peters, T.; Ameh, L.; McAleer, R.; Petrie, S.; Nair, S.; Muscat, I.; de Pinna, E.; Dallman, T. Whole Genome Sequencing for the Retrospective Investigation of an Outbreak of Salmonella Typhimurium DT 8. PLoS Curr. 2015, 10, 7. [Google Scholar] [CrossRef]
- Kwong, J.C.; Mccallum, N.; Sintchenko, V.; Howden, B.P. Whole genome sequencing in clinical and public health microbiology. Pathology 2015, 47, 199–210. [Google Scholar] [CrossRef]
- Rychlik, I.; Gregorova, D.; Hradecka, H. Distribution and function of plasmids in Salmonella enterica. Vet Microbiol. 2006, 112, 1–10. [Google Scholar] [CrossRef]
- Lang, A.S.; Zhaxybayeva, O.; Beatty, J.T. Gene transfer agents: Phage-like elements of genetic exchange. Nat. Rev. Microbiol. 2012, 10, 472. [Google Scholar] [CrossRef]
- Pang, S.; Octavia, S.; Feng, L.; Liu, B.; Reeves, P.R.; Lan, R.; Wang, L. Genomic diversity and adaptation of Salmonella enterica serovar Typhimurium from analysis of six genomes of different phage types. BMC Genom. 2013, 14, 718. [Google Scholar] [CrossRef]
- Louwen, R.; Staals, R.H.; Endtz, H.P.; van Baarlen, P.; van der Oost, J. The role of CRISPR-Cas systems in virulence of pathogenic bacteria. Microbiol. Mol. Biol. Rev. 2014, 78, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Ramisetty, B.C.M.; Sudhakari, P.A. Bacterial ‘Grounded’ Prophages: Hotspots for Genetic Renovation and Innovation. Front. Genet. 2019, 10, 65. [Google Scholar] [CrossRef]
- Penadés, J.R.; Chen, J.; Quiles-Puchalt, N.; Carpena, N.; Novick, R.P. Bacteriophage-mediated spread of bacterial virulence genes. Curr. Opin. Microbiol. 2015, 23, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; DeGiovanni, A.; Jancarik, J.; Adams, P.D.; Yokota, H.; Kim, R.; Kim, S.H. Crystal structure of DNA sequence specificity subunit of a type I restriction-modification enzyme and its functional implications. Proc. Natl. Acad. Sci. USA 2005, 102, 3248–3253. [Google Scholar] [CrossRef] [PubMed]
- Vasu, K.; Nagaraja, V. Diverse Functions of Restriction-Modification Systems in Addition to Cellular Defense. Microbiol. Mol. Biol. Rev. 2013, 77, 53. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Alkhnbashi, O.S.; Costa, F.; Shah, S.A.; Saunders, S.J.; Barrangou, R.; Brouns, S.J.; Charpentier, E.; Haft, D.H.; et al. An updated evolutionary classification of CRISPR–Cas systems. Nat. Rev. Microbiol. 2015, 13, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Hille, F.; Charpentier, E. CRISPR-Cas: Biology, mechanisms and relevance. Philos Trans. R. Soc. Lond. B Biol. Sci. 2016, 371, 20150496. [Google Scholar] [CrossRef] [PubMed]
- Shariat, N.; Timme, R.E.; Pettengill, J.B.; Barrangou, R.; Dudley, E.G. Characterization and evolution of Salmonella CRISPR-Cas systems. Microbiology 2015, 161, 374–386. [Google Scholar] [CrossRef]
- Gregorová, D.; Pravcova, M.; Karpíšková, R.; Rychlik, I. Plasmid pC present in Salmonella enterica serovar Enteritidis PT14b strains encodes a restriction modification system. FEMS Microbiol. Lett. 2002, 214, 195–198. [Google Scholar] [CrossRef][Green Version]
- Jalasvuori, M.; Friman, V.P.; Nieminen, A.; Bamford, J.K.; Buckling, A. Bacteriophage selection against a plasmid-encoded sex apparatus leads to the loss of antibiotic-resistance plasmids. Biol. Lett. 2011, 7, 902–905. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zheng, H.; Yang, M.; Xu, Z.; Wang, X.; Wei, L.; Tang, B.; Liu, F.; Zhang, Y.; Ding, Y.; et al. Genome analysis and in vivo virulence of porcine extraintestinal pathogenic Escherichia coli strain PCN033. BMC Genom. 2015, 16, 717. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 | 24 | 25 | 26 | 27 | 28 | 29 | 30 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DT104 | - | - | - | - | - | - | - | - | - | - | CL | CL | - | - | - | - | CL | - | + | - | - | - | - | - | - | ±/− | −/± | - | −/± | +/+++ |
| DT104b | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | CL | - | ± | - | - | - | - | - | - | ± | ± | - | - | + |
| Strain ID | Phage Type | EnteroBase Barcode | Accession Number | Collection Year | Country of Detection | Source |
|---|---|---|---|---|---|---|
| DP_F10 | DT104b | SAL_KA4333AA | ERS3655651 | 2006 | Ireland | Swine |
| DP_N16 | DT104b | SAL_KA4331AA | ERS3655649 | 2006 | Ireland | Environment |
| DP_N28 | DT104b | SAL_KA4341AA | - | 2006 | Ireland | Swine |
| JE_2727 | DT104b | SAL_KA4322AA | - | 2006 | Ireland | Food |
| DT104b ref. | DT104b | - | - | - | - | - |
| JM_04.26 | DT104 | SAL_KA4067AA | - | 2004 | UK | Human |
| MC_04-0529 | DT104 | SAL_KA3878AA | - | 2004 | Ireland | Human |
| R13 | DT104 | SAL_KA3845AA | - | 1999 | Germany | Bovine |
| TM75-404 | DT104 | SAL_EA5197AA | - | 1975 | France | Human |
| DT104 ref. | DT104 | SAL_EA9332AA | HF937208.1 | - | - | - |
| LT2 | DT4 | - | AE006468.2 | - | - | - |
| DP_F10 | DP_N16 | DP_N28 | JE_2727 | DT104b ref. | JM_04.26 | MC_04-0529 | R13 | TM75-404 | DT104 ref. | LT2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| DP_F10 | 0 | 59 | 60 | 53 | 1095 | 72 | 76 | 59 | 139 | 88 | 655 |
| DP_N16 | 59 | 0 | 5 | 50 | 1088 | 69 | 65 | 52 | 134 | 77 | 652 |
| DP_N28 | 60 | 5 | 0 | 49 | 1087 | 70 | 66 | 53 | 133 | 78 | 651 |
| JE_2727 | 53 | 50 | 49 | 0 | 1082 | 57 | 61 | 44 | 122 | 71 | 644 |
| DT104b ref. | 1095 | 1088 | 1087 | 1082 | 0 | 1075 | 1065 | 1064 | 1072 | 1059 | 862 |
| JM_04.26 | 72 | 69 | 70 | 57 | 1075 | 0 | 58 | 39 | 117 | 66 | 639 |
| MC_04-0529 | 76 | 65 | 66 | 61 | 1065 | 58 | 0 | 27 | 123 | 62 | 627 |
| R13 | 59 | 52 | 53 | 44 | 1064 | 39 | 27 | 0 | 104 | 51 | 626 |
| TM75-404 | 139 | 134 | 133 | 122 | 1072 | 117 | 123 | 104 | 0 | 105 | 636 |
| DT104 ref. | 88 | 77 | 78 | 71 | 1059 | 66 | 62 | 51 | 105 | 0 | 617 |
| LT2 | 655 | 652 | 651 | 644 | 862 | 639 | 627 | 626 | 636 | 617 | 0 |
| Strain ID | Phage Type | Prophages, Accession Number | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Salmon_118970_sal3 NC_031940 | Salmon_ST64B NC_004313 | Gifsy−2 NC_010393 | Gifsy−1 NC_010392 | Entero_ST104 NC_005841 | Salmon_SP_004 NC_021774 | Entero_lato NC_001422 | Fels−1 NC_010391 | Fels−2 NC_010463 | ||
| DP_F10 | DT104b | + | + | + | + | + | − | − | − | − |
| DP_N16 | DT104b | + | + | + | + | + | − | − | − | − |
| DP_N28 | DT104b | + | + | + | + | + | − | − | − | − |
| JE_2727 | DT104b | + | + | + | + | + | − | − | − | − |
| DT104b ref. | DT104b | + | + | + | + | − | + | − | − | − |
| JM_04.26 | DT104 | + | + | + | + | + | − | − | − | − |
| MC_04−0529 | DT104 | + | + | + | + | + | − | − | − | − |
| R13 | DT104 | + | + | + | + | + | − | − | − | − |
| TM75−404 | DT104 | + | + | + | + | + | − | + | − | − |
| DT104 ref. | DT104 | + | + | + | + | + | − | − | − | − |
| LT2 | DT4 | − | − | + | + | − | − | − | + | + |
| Type I Restriction Modification System | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Function | Recognition Sequence | DP_F10 (DT104b) | DP_N16 (DT104b) | DP_N28 (DT104b) | JE_2727 (DT104b) | DT104b ref. (DT104b) | JM_04.26 (DT104) | MC_04-0529 (DT104) | R13 (DT104) | TM75-404 (DT104) | DT104 ref. (DT104) | LT2 (DT4) |
| StyUK1IIP | Restriction enzyme | GAGNNNNNNRTAYG | + | + | + | + | + | + | + | + | + | + | − |
| SenLT2IIP | Restriction enzyme | GAGNNNNNNRTAYG | − | − | − | − | − | − | − | − | − | − | + |
| M.SenTFII | Methyltransferase | GAGNNNNNNRTAYG | + | + | + | + | − | + | + | + | + | + | + |
| M.Sen1899II | Methyltransferase | GAGNNNNNNRTAYG | − | − | − | − | + | − | − | − | − | − | − |
| S.StyUK1II | Specificity subunit | GAGNNNNNNRTAYG | + | + | + | + | + | + | + | + | + | + | + |
| Type II Restriction Modification System | |||||||||||||
| Sty13348III Type IIG | Restriction enzyme/Methyltransferase | GATCAG | + | + | + | + | − | + | + | + | + | + | − |
| M.SenAboDcm | Methyltransferase | CCWGG | + | + | + | + | + | + | + | + | + | + | + |
| M.Sen641III | Methyltransferase | ATGCAT | + | + | + | + | + | + | + | + | + | + | + |
| M.StyUK1V | Methyltransferase | * | + | + | + | + | + | + | + | + | + | + | − |
| StyUK1IV Type IIG | Restriction enzyme/Methyltransferase | GATCAG | − | − | − | − | + | − | − | − | − | − | + |
| Type III Restriction Modification System | |||||||||||||
| M.StyUK1I | Methyltransferase | CAGAG | + | + | + | + | + | + | + | + | + | + | + |
| SenAZII | Restriction enzyme | CAGAG | + | + | + | + | + | + | + | + | + | + | + |
| Type IV Restriction Modification System | |||||||||||||
| StyLT2Mrr | Methyl-directed restriction enzyme | * | + | + | + | + | + | + | + | + | + | + | + |
| Strain ID | Phage Type | Spacer Number | Cas Cluster Subtype | ||
|---|---|---|---|---|---|
| CRISPR-1 | CRISPR-2 | Total | |||
| DP_F10 (DT104b) | DT104b | 10 | 26 | 36 | I-E |
| DP_N16 (DT104b) | DT104b | 10 | 26 | 36 | I-E |
| DP_N28 (DT104b) | DT104b | 10 | 26 | 36 | I-E |
| JE_2727 (DT104b) | DT104b | 10 | 26 | 36 | I-E |
| DT104b ref. (DT104b) | DT104b | 19 | 16 | 35 | I-E |
| JM_04.26 (DT104) | DT104 | 10 | 26 | 36 | I-E |
| MC_04-0529 (DT104) | DT104 | 10 | 22 | 32 | I-E |
| R13 (DT104) | DT104 | 10 | 26 | 36 | I-E |
| TM75-404 (DT104) | DT104 | 10 | 12 | 22 | I-E |
| DT104 ref. (DT104) | DT104 | 10 | 26 | 36 | I-E |
| LT2 (DT4) | DT4 | 23 | 32 | 55 | I-E |
| Plasmid | Accession Number | Length (bp) | DP_F10 (DT104b) | DP_N16 (DT104b) | DP_N28 (DT104b) | JE_2727 (DT104b) | DT104b ref. (DT104b) | JM_04.26 (DT104) | MC_04-0529 (DT104) | R13 (DT104) | TM75-404 (DT104) | DT104 ref. (DT104) | LT2 (DT4) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pSC-09-1 | NZ_CP028319.1 | 94,045 | + | + | + | + | − | + | + | + | + | + | − |
| p1PCN033 | NZ_CP006633.1 | 3319 | + | + | + | + | − | − | − | − | − | − | − |
| unnamed 2 * | NZ_CP043666.1 | 4593 | + | + | + | − | − | − | − | − | − | − | − |
| pSE81-1705-3 | NZ_CP018654.1 | 33,784 | + | + | + | + | + | + | + | + | + | + | + |
| plasmid 3 * | NZ_LN868945.1 | 147,787 | + | + | + | + | + | + | + | + | + | + | + |
| pCERC1 | NC_019070.1 | 6790 | − | + | + | − | − | − | − | − | − | − | − |
| Punnamed 4 * | NZ_CP036207.1 | 4149 | − | − | − | + | − | − | − | − | − | − | − |
| plasmid: 2 ** | NZ_LT855377.1 | 93,862 | − | − | − | − | + | − | − | − | − | − | − |
| pAUSMDU00010534_03 | NZ_CP045935.1 | 57,073 | − | − | − | − | − | − | − | + | − | − | − |
| pSLT | NC_003277.2 | 93,939 | − | − | − | − | − | − | − | − | − | − | + *** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohammed, M.; Orzechowska, B. Characterisation of Phage Susceptibility Variation in Salmonellaenterica Serovar Typhimurium DT104 and DT104b. Microorganisms 2021, 9, 865. https://doi.org/10.3390/microorganisms9040865
Mohammed M, Orzechowska B. Characterisation of Phage Susceptibility Variation in Salmonellaenterica Serovar Typhimurium DT104 and DT104b. Microorganisms. 2021; 9(4):865. https://doi.org/10.3390/microorganisms9040865
Chicago/Turabian StyleMohammed, Manal, and Beata Orzechowska. 2021. "Characterisation of Phage Susceptibility Variation in Salmonellaenterica Serovar Typhimurium DT104 and DT104b" Microorganisms 9, no. 4: 865. https://doi.org/10.3390/microorganisms9040865
APA StyleMohammed, M., & Orzechowska, B. (2021). Characterisation of Phage Susceptibility Variation in Salmonellaenterica Serovar Typhimurium DT104 and DT104b. Microorganisms, 9(4), 865. https://doi.org/10.3390/microorganisms9040865