
Mining Synergistic Microbial Interactions: A Roadmap on How to Integrate Multi-Omics Data

 and
and
Abstract
1. Introduction
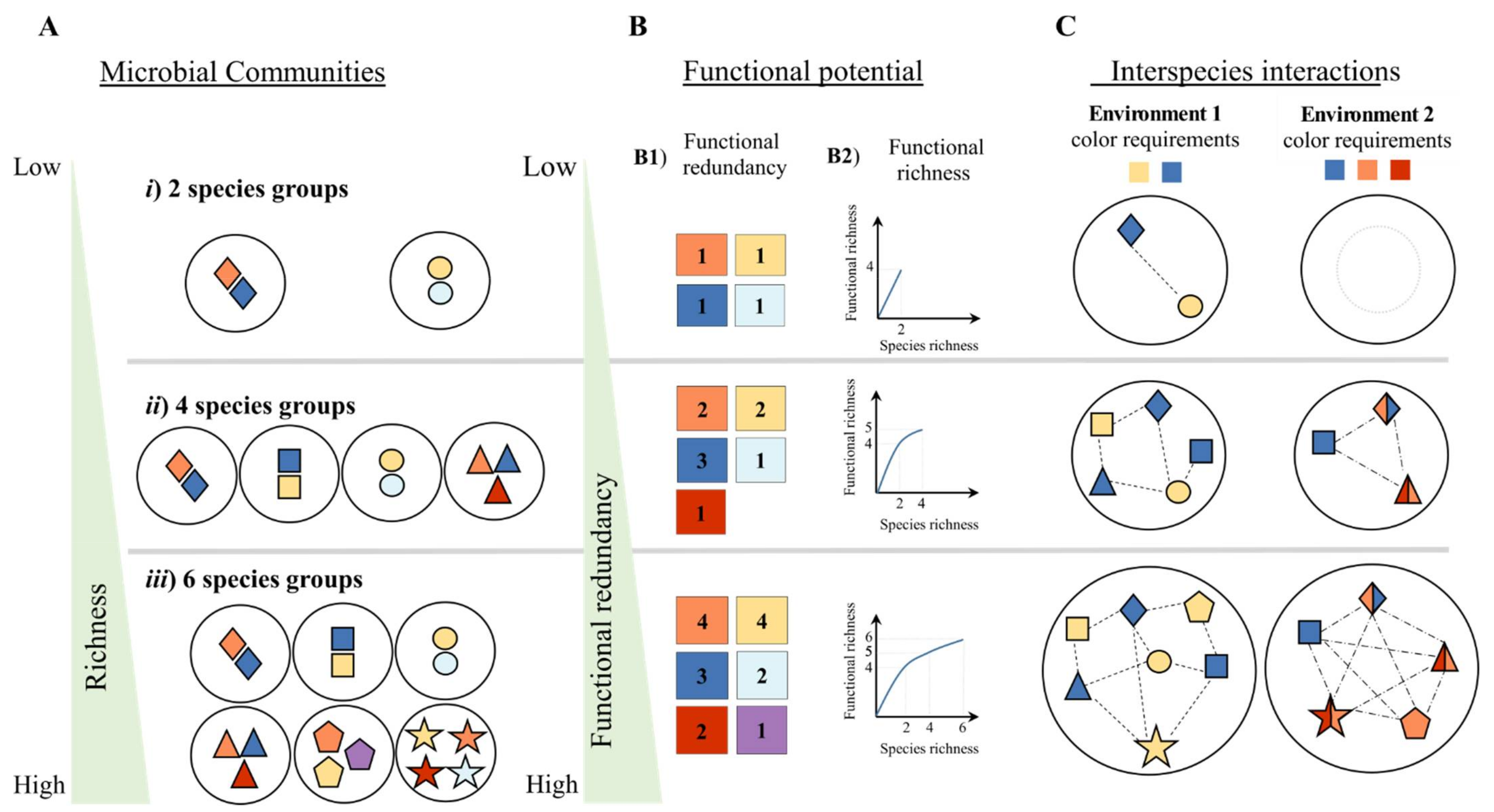
2. Synergistic Interspecies Interactions Drive Ecosystem Processes
From Where Will One Extract the Genomes to Explore Microbial Interactions?
3. Current Approaches to Predict Microbial Community Functional Profiles and Interspecies Interactions
3.1. The Supra-Organism Approach
3.2. The Population-Based Approach
3.3. The Guild-Based Approach
3.4. Advantages and Limitation of Current Approaches Mining Microbial Interactions
- Identification of all species in a community;
- Incomplete functional annotation of genomes;
- Data integration and experimental validation; and
- Exponential increase of search space with relatively small increase of number of species or pathway size.
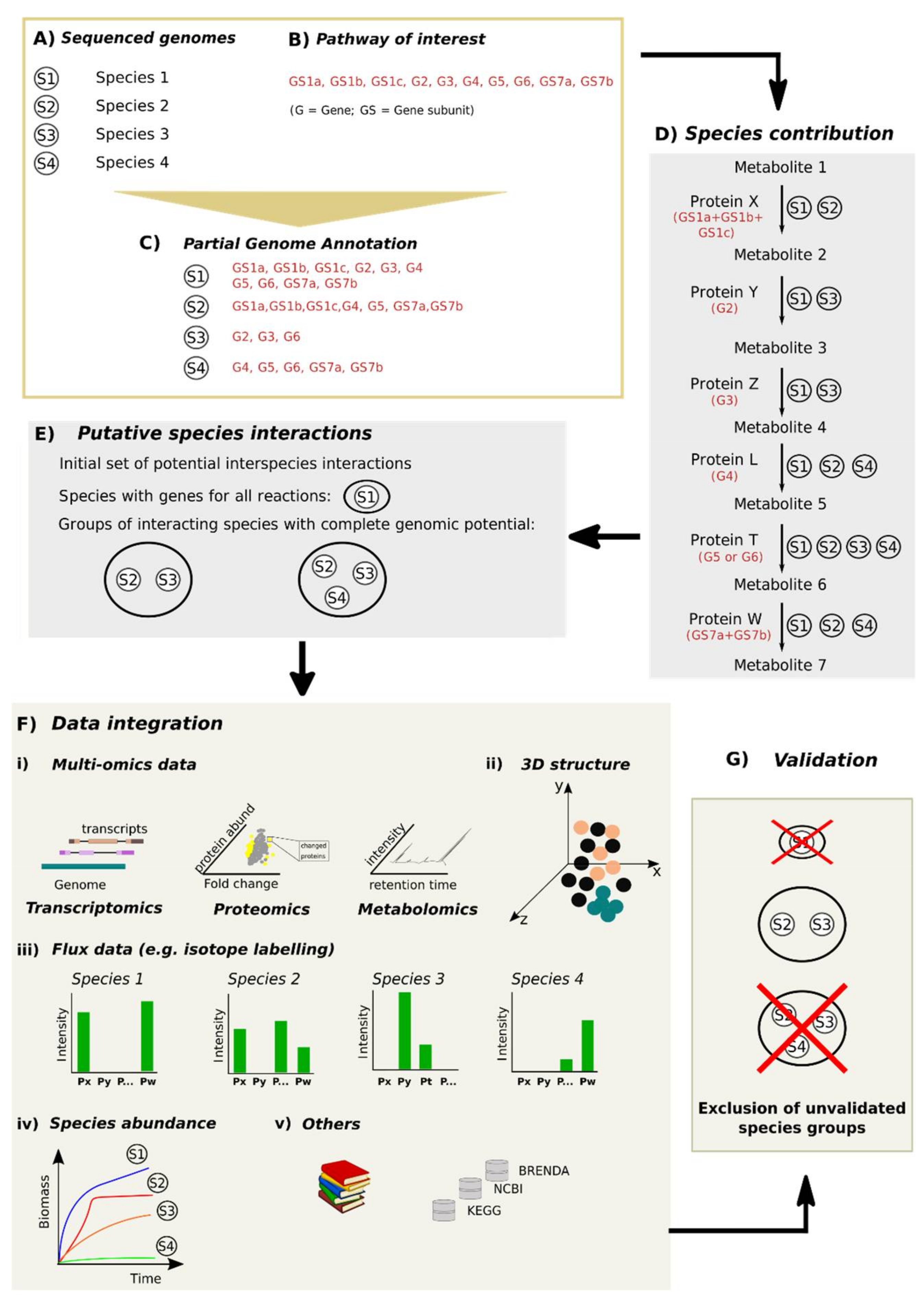
4. Beyond Genetic Potential: Drawing a Strategy to Mine and Validate Microbial Interactions
4.1. Validation of Putative Microbial Interaction through Integration of Different Data Sources
4.1.1. Assumption that Gene Presence is Directly Linked to Function
4.1.2. Spatial (Three Dimensional) Structure of Microbial Communities
4.1.3. Different Levels of Protein Activity within Species or Populations
4.1.4. Temporal Variability
4.2. From Mining to Validation: A Workflow to Identify Mechanisms Underlying Microbial Interactions
4.3. Assembling a Workflow to Determine Microbial Interactions

{kind=link}
{kind=link}
| Outcome | Limitations | Methods | Environment | Validation | Ref. a | |||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |||||
| Improvement in the identification of microbial community species. | Lack of mechanistic understanding of species interactions. | Combination of MALDI-TOF MS b analysis and high-throughput sequencing 16S rRNA c. | Kimchi | ✓ | O | O | ✓ | [78] |
| 16S rRNA gene sequencing. | Human oral environments | O | ✓ | O | O | [79] | ||
| Demonstration of the influence of abiotic factors on microbial community dynamics. | High computational and data requirements for reconstruction of individual metabolic models. | Metagenomics, metabolic network reconstruction and FBA d. | Anaerobic digestion microbiomes | ✓ | O | ✓ | O | [80] |
| Lack of mechanistic understanding of species interactions. | PLS-PM e | Rice soil rhizosphere | O | ✓ | O | ✓ | [81] | |
| 16S rRNA gene sequencing. | Urban and forest park soil litter layers | O | ✓ | O | ✓ | [82] | ||
| In vivo experiment of meadow steppe soil under different precipitation regimes. | Topsoil | ✓ | ✓ | O | ✓ | [83] | ||
| High computational and data requirements for reconstruction of individual metabolic models and complex wet-lab experiments required for validation. | Metabolic network reconstruction, EFM f and FBA. | Acid-sulfate-chloride springs | ✓ | O | ✓ | O | [84] | |
| Demonstration of the influence of interspecies interactions on microbial community dynamics. | Lack of mechanistic understanding of species interactions. | Co-culture of isolates, RNA-Seq g and RT-qPCR h. | Wine fermentation | ✓ | O | O | ✓ | [85] |
| qPCRi and 16S rRNA gene sequencing. | Mixed bacterial consortia | ✓ | O | O | ✓ | [86] | ||
| Improved mechanistic understanding of interspecies interactions. | Complex wet-lab experiments required for validation. | SIP j and Metagenomics. | Continuous up-flow anaerobic sludge blanket reactors | ✓ | O | ✓ | ✓ | [87] |
| Pure and co-cultures and cyclic voltammetry analysis. | Palm oil mill effluent | O | O | ✓ | ✓ | [88] | ||
| High computational and data requirements for reconstruction of individual metabolic models. | Mono- and co-culture, metabolic network reconstruction, bipartite graphs, HPLC k, CGQ l, GC-MS m; SIP. | In silicon experiments with pure and co-culture | ✓ | O | ✓ | ✓ | [89] | |
| Metabolic network reconstruction and cFBA n. | In silicon experiments pure cultures | ✓ | O | ✓ | O | [27] | ||
| Metabolic network reconstruction, evolutionary game theory and FBA. | In silicon experiments pure cultures | ✓ | O | O | O | [90] | ||
| Metagenomics, Metatranscriptomics. | Synthetic human gut | ✓ | ✓ | O | O | [5] | ||
4.3.1. Identifying Microbial Species and Their Genetic Potential
4.3.2. Defining an Ecosystem Process and Links between Genes, Enzymes and Reactions for a Given Ecosystem Process
4.3.3. Mining Putative Species Interactions
4.3.4. Validating Microbial Interactions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Currie, W.S. Units of Nature or Processes across Scales? The Ecosystem Concept at Age 75. New Phytol. 2011, 190, 21–34. [Google Scholar] [CrossRef]
- Song, H.-S.; Cannon, W.; Beliaev, A.; Konopka, A. Mathematical Modeling of Microbial Community Dynamics: A Methodological Review. Processes 2014, 2, 711–752. [Google Scholar] [CrossRef]
- Gentry, T.J.; Pepper, I.L.; Pierson, L.S. Chapter 19—Microbial Diversity and Interactions in Natural Ecosystems. In Environmental Microbiology, 3rd ed.; Pepper, I.L., Gerba, C.P., Gentry, T.J., Eds.; Academic Press: San Diego, CA, USA, 2015; pp. 441–460. ISBN 978-0-12-394626-3. [Google Scholar]
- Stams, A.J.M.; Bok, F.A.M.D.; Plugge, C.M.; Eekert, M.H.A.V.; Dolfing, J.; Schraa, G. Exocellular Electron Transfer in Anaerobic Microbial Communities. Environ. Microbiol. 2006, 8, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Franzosa, E.A.; McIver, L.J.; Rahnavard, G.; Thompson, L.R.; Schirmer, M.; Weingart, G.; Lipson, K.S.; Knight, R.; Caporaso, J.G.; Segata, N.; et al. Species-Level Functional Profiling of Metagenomes and Metatranscriptomes. Nat. Methods 2018, 15, 962–968. [Google Scholar] [CrossRef]
- Hall, E.K.; Bernhardt, E.S.; Bier, R.L.; Bradford, M.A.; Boot, C.M.; Cotner, J.B.; del Giorgio, P.A.; Evans, S.E.; Graham, E.B.; Jones, S.E.; et al. Understanding How Microbiomes Influence the Systems They Inhabit. Nat. Microbiol. 2018, 3, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Baas Becking, L.G.M.; Nicolai, E. On the Ecology of a Sphagnum Bog. Blumea Biodivers. Evol. Biogeogr. Plants 1934, 1, 10–45. [Google Scholar]
- Girguis, P. Microbial Ecology: Here, There and Everywhere. Nat. Microbiol. 2016, 1, 16123. [Google Scholar] [CrossRef]
- Bell, T.; Newman, J.A.; Silverman, B.W.; Turner, S.L.; Lilley, A.K. The Contribution of Species Richness and Composition to Bacterial Services. Nature 2005, 436, 1157–1160. [Google Scholar] [CrossRef]
- Oh, Y.K.; Palsson, B.O.; Park, S.M.; Schilling, C.H.; Mahadevan, R. Genome-Scale Reconstruction of Metabolic Network in Bacillus Subtilis Based on High-Throughput Phenotyping and Gene Essentiality Data. J. Biol. Chem. 2007, 282, 28791–28799. [Google Scholar] [CrossRef] [PubMed]
- Huddleston, J.R. Horizontal Gene Transfer in the Human Gastrointestinal Tract: Potential Spread of Antibiotic Resistance Genes. Infect. Drug Resist. 2014, 7, 167–176. [Google Scholar] [CrossRef]
- Tyc, O.; Song, C.; Dickschat, J.S.; Vos, M.; Garbeva, P. The Ecological Role of Volatile and Soluble Secondary Metabolites Produced by Soil Bacteria. Trends Microbiol. 2017, 25, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Seth, E.C.; Taga, M.E. Nutrient Cross-Feeding in the Microbial World. Front. Microbiol. 2014, 5, 350. [Google Scholar] [CrossRef] [PubMed]
- Fetzer, I.; Johst, K.; Schäwe, R.; Banitz, T.; Harms, H.; Chatzinotas, A. The Extent of Functional Redundancy Changes as Species’ Roles Shift in Different Environments. Proc. Natl. Acad. Sci. USA 2015, 112, 14888–14893. [Google Scholar] [CrossRef]
- Gawad, C.; Koh, W.; Quake, S.R. Single-Cell Genome Sequencing: Current State of the Science. Nat. Rev. Genet. 2016, 17, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Rinke, C.; Chuvochina, M.; Chaumeil, P.-A.; Woodcroft, B.J.; Evans, P.N.; Hugenholtz, P.; Tyson, G.W. Recovery of Nearly 8000 Metagenome-Assembled Genomes Substantially Expands the Tree of Life. Nat. Microbiol. 2017, 2, 1533–1542. [Google Scholar] [CrossRef] [PubMed]
- Nayfach, S.; Roux, S.; Seshadri, R.; Udwary, D.; Varghese, N.; Schulz, F.; Wu, D.; Paez-Espino, D.; Chen, I.-M.; Huntemann, M.; et al. A Genomic Catalog of Earth’s Microbiomes. Nat. Biotechnol. 2020. [Google Scholar] [CrossRef]
- Da Rocha, U.N.; Cadillo-Quiroz, H.; Karaoz, U.; Rajeev, L.; Klitgord, N.; Dunn, S.; Truong, V.; Buenrostro, M.; Bowen, B.P.; Garcia-Pichel, F.; et al. Isolation of a Significant Fraction of Non-Phototroph Diversity from a Desert Biological Soil Crust. Front. Microbiol. 2015, 6, 277. [Google Scholar] [CrossRef]
- Gloor, G.B.; Macklaim, J.M.; Pawlowsky-Glahn, V.; Egozcue, J.J. Microbiome Datasets Are Compositional: And This Is Not Optional. Front. Microbiol. 2017, 8, 2224. [Google Scholar] [CrossRef]
- Clingenpeel, S.; Clum, A.; Schwientek, P.; Rinke, C.; Woyke, T. Reconstructing Each Cell’s Genome within Complex Microbial Communities-Dream or Reality? Front. Microbiol. 2014, 5, 771. [Google Scholar] [CrossRef]
- Velsko, I.M.; Frantz, L.A.F.; Herbig, A.; Larson, G.; Warinner, C. Selection of Appropriate Metagenome Taxonomic Classifiers for Ancient Microbiome Research. mSystems 2018, 3. [Google Scholar] [CrossRef]
- Frigg, R.; Hartmann, S. Models in Science. 2006. Available online: https://seop.illc.uva.nl/entries/models-science/ (accessed on 26 January 2021).
- Kirwan, L.; Connolly, J.; Finn, J.A.; Brophy, C.; Lüscher, A.; Nyfeler, D.; Sebastià, M.-T. Diversity–Interaction Modeling: Estimating Contributions of Species Identities and Interactions to Ecosystem Function. Ecology 2009, 90, 2032–2038. [Google Scholar] [CrossRef] [PubMed]
- Kreft, J.-U.; Plugge, C.M.; Prats, C.; Leveau, J.H.J.; Zhang, W.; Hellweger, F.L. From Genes to Ecosystems in Microbiology: Modeling Approaches and the Importance of Individuality. Front. Microbiol. 2017, 8, 2299. [Google Scholar] [CrossRef]
- Biggs, M.B.; Medlock, G.L.; Kolling, G.L.; Papin, J.A. Metabolic Network Modeling of Microbial Communities. Wiley Interdiscip. Rev. Syst. Biol. Med. 2015, 7, 317–334. [Google Scholar] [CrossRef]
- Yarygin, K.; Tyakht, A.; Larin, A.; Kostryukova, E.; Kolchenko, S.; Bitner, V.; Alexeev, D. Abundance Profiling of Specific Gene Groups Using Precomputed Gut Metagenomes Yields Novel Biological Hypotheses. PLoS ONE 2017, 12, e0176154. [Google Scholar] [CrossRef]
- Khandelwal, R.A.; Olivier, B.G.; Röling, W.F.M.; Teusink, B.; Bruggeman, F.J. Community Flux Balance Analysis for Microbial Consortia at Balanced Growth. PLoS ONE 2013, 8, e64567. [Google Scholar] [CrossRef]
- Rocha, I.; Förster, J.; Nielsen, J. Design and Application of Genome-Scale Reconstructed Metabolic Models. In Microbial Gene Essentiality: Protocols and Bioinformatics; Methods in Molecular Biology™ Book Series; Humana Press: Totowa, NJ, USA, 2008; Volume 416, pp. 409–431. [Google Scholar]
- Dias, O.; Rocha, M.; Ferreira, E.C.; Rocha, I. Reconstructing Genome-Scale Metabolic Models with Merlin. Nucleic Acids Res. 2015, 43, 3899–3910. [Google Scholar] [CrossRef]
- Gottstein, W.; Olivier, B.G.; Bruggeman, F.J.; Teusink, B. Constraint-Based Stoichiometric Modelling from Single Organisms to Microbial Communities. J. R. Soc. Interface 2016, 13, 20160627. [Google Scholar] [CrossRef]
- Gu, C.; Kim, G.B.; Kim, W.J.; Kim, H.U.; Lee, S.Y. Current Status and Applications of Genome-Scale Metabolic Models. Genome Biol. 2019, 20, 121. [Google Scholar] [CrossRef]
- Blondel, J. Guilds or Functional Groups: Does It Matter? Oikos 2003, 100, 223–231. [Google Scholar] [CrossRef]
- Kettle, H.; Louis, P.; Holtrop, G.; Duncan, S.H.; Flint, H.J. Modelling the Emergent Dynamics and Major Metabolites of the Human Colonic Microbiota. Environ. Microbiol. 2015, 17, 1615–1630. [Google Scholar] [CrossRef] [PubMed]
- Hanemaaijer, M.; Röling, W.F.M.; Olivier, B.G.; Khandelwal, R.A.; Teusink, B.; Bruggeman, F.J. Systems Modeling Approaches for Microbial Community Studies: From Metagenomics to Inference of the Community Structure. Front. Microbiol. 2015, 6, 213. [Google Scholar] [CrossRef] [PubMed]
- Talbot, J.M.; Martin, F.; Kohler, A.; Henrissat, B.; Peay, K.G. Functional Guild Classification Predicts the Enzymatic Role of Fungi in Litter and Soil Biogeochemistry. Soil Biol. Biochem. 2015, 88, 441–456. [Google Scholar] [CrossRef]
- Faust, K.; Raes, J. Microbial Interactions: From Networks to Models. Nat. Rev. Microbiol. 2012, 10, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Thiele, I.; Palsson, B.Ø. A Protocol for Generating a High-Quality Genome-Scale Metabolic Reconstruction. Nat. Protoc. 2010, 5, 93–121. [Google Scholar] [CrossRef]
- Rodríguez, J.; Kleerebezem, R.; Lema, J.M.; van Loosdrecht, M.C.M. Modeling Product Formation in Anaerobic Mixed Culture Fermentations. Biotechnol. Bioeng. 2006, 93, 592–606. [Google Scholar] [CrossRef]
- Tringe, S.G.; von Mering, C.; Kobayashi, A.; Salamov, A.A.; Chen, K.; Chang, H.W.; Podar, M.; Short, J.M.; Mathur, E.J.; Detter, J.C.; et al. Comparative Metagenomics of Microbial Communities. Science 2005, 308, 554–557. [Google Scholar] [CrossRef]
- Hay, M.E.; Parker, J.D.; Burkepile, D.E.; Caudill, C.C.; Wilson, A.E.; Hallinan, Z.P.; Chequer, A.D. Mutualisms and Aquatic Community Structure: The Enemy of My Enemy Is My Friend. Annu. Rev. Ecol. Evol. Syst. 2004, 35, 175–197. [Google Scholar] [CrossRef]
- Boetius, A.; Ravenschlag, K.; Schubert, C.J.; Rickert, D.; Widdel, F.; Gieseke, A.; Amann, R.; Jørgensen, B.B.; Witte, U.; Pfannkuche, O. A Marine Microbial Consortium Apparently Mediating Anaerobic Oxidation of Methane. Nature 2000, 407, 623–626. [Google Scholar] [CrossRef]
- Traore, A.S.; Fardeau, M.-L.; Hatchikian, C.E.; Le Gall, J.; Belaich, J.-P. Energetics of Growth of a Defined Mixed Culture of Desulfovibrio vulgaris and Methanosarcina barkeri: Interspecies Hydrogen Transfer in Batch and Continuous Cultures. Appl. Environ. Microbiol. 1983, 46, 1152–1156. [Google Scholar] [CrossRef]
- Chuang, J.S.; Rivoire, O.; Leibler, S. Simpson’s Paradox in a Synthetic Microbial System. Science 2009, 323, 272–275. [Google Scholar] [CrossRef]
- Wertz, S.; Degrange, V.; Prosser, J.I.; Poly, F.; Commeaux, C.; Guillaumaud, N.; Roux, X.L. Decline of Soil Microbial Diversity Does Not Influence the Resistance and Resilience of Key Soil Microbial Functional Groups Following a Model Disturbance. Environ. Microbiol. 2007, 9, 2211–2219. [Google Scholar] [CrossRef]
- Gill, A.S.; Purnell, K.; Palmer, M.I.; Stein, J.; McGuire, K.L. Microbial Composition and Functional Diversity Differ across Urban Green Infrastructure Types. Front. Microbiol. 2020, 11, 912. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, D.J.; Chaves-Moreno, D.; van Elsas, J.D. Unveiling the Metabolic Potential of Two Soil-Derived Microbial Consortia Selected on Wheat Straw. Sci. Rep. 2015, 5, 13845. [Google Scholar] [CrossRef] [PubMed]
- Jeffries, T.C.; Rayu, S.; Nielsen, U.N.; Lai, K.; Ijaz, A.; Nazaries, L.; Singh, B.K. Metagenomic Functional Potential Predicts Degradation Rates of a Model Organophosphorus Xenobiotic in Pesticide Contaminated Soils. Front. Microbiol. 2018, 9, 147. [Google Scholar] [CrossRef] [PubMed]
- Rondon, M.R.; August, P.R.; Bettermann, A.D.; Brady, S.F.; Grossman, T.H.; Liles, M.R.; Loiacono, K.A.; Lynch, B.A.; MacNeil, I.A.; Minor, C.; et al. Cloning the Soil Metagenome: A Strategy for Accessing the Genetic and Functional Diversity of Uncultured Microorganisms. Appl. Environ. Microbiol. 2000, 66, 2541–2547. [Google Scholar] [CrossRef] [PubMed]
- Van Rossum, T.; Ferretti, P.; Maistrenko, O.M.; Bork, P. Diversity within Species: Interpreting Strains in Microbiomes. Nat. Rev. Microbiol. 2020, 18, 491–506. [Google Scholar] [CrossRef]
- Prosser, J.I. Dispersing Misconceptions and Identifying Opportunities for the Use of “omics” in Soil Microbial Ecology. Nat. Rev. Microbiol. 2015, 13, 439–446. [Google Scholar] [CrossRef]
- Ancel Meyers, L.; Ancel, F.D.; Lachmann, M. Evolution of Genetic Potential. PLoS Comput. Biol. 2005, 1, e32. [Google Scholar] [CrossRef]
- Garcia-Pichel, F.; Johnson, S.L.; Youngkin, D.; Belnap, J. Small-Scale Vertical Distribution of Bacterial Biomass and Diversity in Biological Soil Crusts from Arid Lands in the Colorado Plateau. Microb. Ecol. 2003, 46, 312–321. [Google Scholar] [CrossRef]
- Quarton, T.; Kang, T.; Papakis, V.; Nguyen, K.; Nowak, C.; Li, Y.; Bleris, L. Uncoupling Gene Expression Noise along the Central Dogma Using Genome Engineered Human Cell Lines. Nucleic Acids Res. 2020, 48, 9406–9413. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Studying Gene Expression and Function. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Doolittle, W.F.; Zhaxybayeva, O. Metagenomics and the Units of Biological Organization. BioScience 2010, 60, 102–112. [Google Scholar] [CrossRef]
- Jansson, J.K.; Hofmockel, K.S. The Soil Microbiome-from Metagenomics to Metaphenomics. Curr. Opin. Microbiol. 2018, 43, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.J.; Ceri, H.; Yerly, J.; Stremick, C.A.; Hu, Y.; Martinuzzi, R.; Turner, R.J. The Use of Microscopy and Three-Dimensional Visualization to Evaluate the Structure of Microbial Biofilms Cultivated in the Calgary Biofilm Device. Biol. Proced. Online 2006, 8, 194–215. [Google Scholar] [CrossRef] [PubMed]
- Stubbendieck, R.M.; Vargas-Bautista, C.; Straight, P.D. Bacterial Communities: Interactions to Scale. Front. Microbiol. 2016, 7, 1234. [Google Scholar] [CrossRef] [PubMed]
- Natale, A.D.; Mele, B.H.; Cennamo, P.; Mondo, A.D.; Petraretti, M.; Pollio, A. Microbial Biofilm Community Structure and Composition on the Lithic Substrates of Herculaneum Suburban Baths. PLoS ONE 2020, 15, e0232512. [Google Scholar] [CrossRef]
- Gonzalez-Gil, G.; Thomas, L.; Emwas, A.-H.; Lens, P.N.L.; Saikaly, P.E. NMR and MALDI-TOF MS Based Characterization of Exopolysaccharides in Anaerobic Microbial Aggregates from Full-Scale Reactors. Sci. Rep. 2015, 5, 14316. [Google Scholar] [CrossRef]
- Ruppé, E.; Ghozlane, A.; Tap, J.; Pons, N.; Alvarez, A.-S.; Maziers, N.; Cuesta, T.; Hernando-Amado, S.; Clares, I.; Martínez, J.L.; et al. Prediction of the Intestinal Resistome by a Three-Dimensional Structure-Based Method. Nat. Microbiol. 2019, 4, 112–123. [Google Scholar] [CrossRef]
- Starr, E.P.; Shi, S.; Blazewicz, S.J.; Probst, A.J.; Herman, D.J.; Firestone, M.K.; Banfield, J.F. Stable Isotope Informed Genome-Resolved Metagenomics Reveals That Saccharibacteria Utilize Microbially-Processed Plant-Derived Carbon. Microbiome 2018, 6, 122. [Google Scholar] [CrossRef] [PubMed]
- Fortunato, C.S.; Huber, J.A. Coupled RNA-SIP and Metatranscriptomics of Active Chemolithoautotrophic Communities at a Deep-Sea Hydrothermal Vent. ISME J. 2016, 10, 1925–1938. [Google Scholar] [CrossRef]
- Kong, Y.; Kuzyakov, Y.; Ruan, Y.; Zhang, J.; Wang, T.; Wang, M.; Guo, S.; Shen, Q.; Ling, N. DNA Stable-Isotope Probing Delineates Carbon Flows from Rice Residues into Soil Microbial Communities Depending on Fertilization. Appl. Environ. Microbiol. 2020, 86, e02151-19. [Google Scholar] [CrossRef]
- Seifert, J.; Taubert, M.; Jehmlich, N.; Schmidt, F.; Völker, U.; Vogt, C.; Richnow, H.-H.; von Bergen, M. Protein-Based Stable Isotope Probing (Protein-SIP) in Functional Metaproteomics. Mass Spectrom. Rev. 2012, 31, 683–697. [Google Scholar] [CrossRef] [PubMed]
- Chokkathukalam, A.; Kim, D.-H.; Barrett, M.P.; Breitling, R.; Creek, D.J. Stable Isotope-Labeling Studies in Metabolomics: New Insights into Structure and Dynamics of Metabolic Networks. Bioanalysis 2014, 6, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Musat, N.; Musat, F.; Weber, P.K.; Pett-Ridge, J. Tracking Microbial Interactions with NanoSIMS. Curr. Opin. Biotechnol. 2016, 41, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.R.; Sanders, J.G.; McDonald, D.; Amir, A.; Ladau, J.; Locey, K.J.; Prill, R.J.; Tripathi, A.; Gibbons, S.M.; Ackermann, G.; et al. A Communal Catalogue Reveals Earth’s Multiscale Microbial Diversity. Nature 2017, 551, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The Human Microbiome Project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Sunagawa, S.; Acinas, S.G.; Bork, P.; Bowler, C.; Eveillard, D.; Gorsky, G.; Guidi, L.; Iudicone, D.; Karsenti, E.; Lombard, F.; et al. Tara Oceans: Towards Global Ocean Ecosystems Biology. Nat. Rev. Microbiol. 2020, 18, 428–445. [Google Scholar] [CrossRef] [PubMed]
- Manzoni, C.; Kia, D.A.; Vandrovcova, J.; Hardy, J.; Wood, N.W.; Lewis, P.A.; Ferrari, R. Genome, Transcriptome and Proteome: The Rise of Omics Data and Their Integration in Biomedical Sciences. Brief. Bioinform. 2018, 19, 286–302. [Google Scholar] [CrossRef]
- Lauber, C.L.; Ramirez, K.S.; Aanderud, Z.; Lennon, J.; Fierer, N. Temporal Variability in Soil Microbial Communities across Land-Use Types. ISME J. 2013, 7, 1641–1650. [Google Scholar] [CrossRef]
- Hannula, S.E.; Kielak, A.M.; Steinauer, K.; Huberty, M.; Jongen, R.; Long, J.R.D.; Heinen, R.; Bezemer, T.M. Time after Time: Temporal Variation in the Effects of Grass and Forb Species on Soil Bacterial and Fungal Communities. mBio 2019, 10, e02635-19. [Google Scholar] [CrossRef]
- Ladau, J.; Eloe-Fadrosh, E.A. Spatial, Temporal, and Phylogenetic Scales of Microbial Ecology. Trends Microbiol. 2019, 27, 662–669. [Google Scholar] [CrossRef]
- Karakoç, C.; Clark, A.T.; Chatzinotas, A. Diversity and Coexistence Are Influenced by Time-Dependent Species Interactions in a Predator–Prey System. Ecol. Lett. 2020, 23, 983–993. [Google Scholar] [CrossRef]
- Bar-Joseph, Z.; Gitter, A.; Simon, I. Studying and Modelling Dynamic Biological Processes Using Time-Series Gene Expression Data. Nat. Rev. Genet. 2012, 13, 552–564. [Google Scholar] [CrossRef]
- Da Rocha, U.N.; van Elsas, J.D.; van Overbeek, L.S. Real-Time PCR Detection of Holophagae (Acidobacteria) and Verrucomicrobia Subdivision 1 Groups in Bulk and Leek (Allium porrum) Rhizosphere Soils. J. Microbiol. Methods 2010, 83, 141–148. [Google Scholar] [CrossRef]
- Kim, E.; Cho, E.-J.; Yang, S.-M.; Kim, M.-J.; Kim, H.-Y. Novel Approaches for the Identification of Microbial Communities in Kimchi: MALDI-TOF MS Analysis and High-Throughput Sequencing. Food Microbiol. 2021, 94, 103641. [Google Scholar] [CrossRef]
- Moraes, L.C.; Lang, P.M.; Arcanjo, R.A.; Rampelotto, P.H.; Fatturi-Parolo, C.C.; Ferreira, M.B.C.; Montagner, F. Microbial Ecology and Predicted Metabolic Pathways in Various Oral Environments from Patients with Acute Endodontic Infections. Int. Endod. J. 2020, 53, 1603–1617. [Google Scholar] [CrossRef]
- Basile, A.; Campanaro, S.; Kovalovszki, A.; Zampieri, G.; Rossi, A.; Angelidaki, I.; Valle, G.; Treu, L. Revealing Metabolic Mechanisms of Interaction in the Anaerobic Digestion Microbiome by Flux Balance Analysis. Metab. Eng. 2020, 62, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Padhy, S.R.; Das, R.R.; Shahid, M.; Dash, P.K.; Senapati, A.; Panneerselvam, P.; Kumar, U.; Chatterjee, D.; Adak, T.; et al. Elucidating Relationship between Nitrous Oxide Emission and Functional Soil Microbes from Tropical Lowland Rice Soil Exposed to Elevated CO2: A Path Modelling Approach. Agric. Ecosyst. Environ. 2021, 308, 107268. [Google Scholar] [CrossRef]
- Yan, B.; Lu, Q.; He, J.; Qi, Y.; Fu, G.; Xiao, N.; Li, J. Composition and Interaction Frequencies in Soil Bacterial Communities Change in Association with Urban Park Age in Beijing. Pedobiologia 2021, 84, 150699. [Google Scholar] [CrossRef]
- Yang, X.; Zhu, K.; Loik, M.E.; Sun, W. Differential Responses of Soil Bacteria and Fungi to Altered Precipitation in a Meadow Steppe. Geoderma 2021, 384, 114812. [Google Scholar] [CrossRef]
- Hunt, K.A.; Jennings, R.M.; Inskeep, W.P.; Carlson, R.P. Multiscale Analysis of Autotroph-Heterotroph Interactions in a High-Temperature Microbial Community. PLoS Comput. Biol. 2018, 14, e1006431. [Google Scholar] [CrossRef] [PubMed]
- Mencher, A.; Morales, P.; Curiel, J.A.; Gonzalez, R.; Tronchoni, J. Metschnikowia pulcherrima Represses Aerobic Respiration in Saccharomyces cerevisiae Suggesting a Direct Response to Co-Cultivation. Food Microbiol. 2021, 94, 103670. [Google Scholar] [CrossRef]
- Yuan, H.; Huang, S.; Yuan, J.; You, Y.; Zhang, Y. Characteristics of Microbial Denitrification under Different Aeration Intensities: Performance, Mechanism, and Co-Occurrence Network. Sci. Total Environ. 2021, 754, 141965. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Yin, Q.; Shi, Y.; Wu, G. Microbial Physiology and Interactions in Anammox Systems with the Intermittent Addition of Organic Carbons. Bioresour. Technol. 2021, 319, 124226. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.A.; Karim, A.; Mishra, P.; Dubowski, J.J.; Yousuf, A.; Sarmin, S.; Khan, M.M.R. Microbial Synergistic Interactions Enhanced Power Generation in Co-Culture Driven Microbial Fuel Cell. Sci. Total Environ. 2020, 738, 140138. [Google Scholar] [CrossRef]
- Ravikrishnan, A.; Blank, L.M.; Srivastava, S.; Raman, K. Investigating Metabolic Interactions in a Microbial Co-Culture through Integrated Modelling and Experiments. Comput. Struct. Biotechnol. J. 2020, 18, 1249–1258. [Google Scholar] [CrossRef]
- Zomorrodi, A.R.; Segrè, D. Genome-Driven Evolutionary Game Theory Helps Understand the Rise of Metabolic Interdependencies in Microbial Communities. Nat. Commun. 2017, 8, 1563. [Google Scholar] [CrossRef] [PubMed]
- Tláskal, V.; Brabcová, V.; Větrovský, T.; Jomura, M.; López-Mondéjar, R.; Monteiro, L.M.O.; Saraiva, J.P.; Human, Z.R.; Cajthaml, T.; da Rocha, U.N.; et al. Complementary Roles of Wood-Inhabiting Fungi and Bacteria Facilitate Deadwood Decomposition. mSystems 2021, 6, e01078-20. [Google Scholar] [CrossRef]
- Da Rocha, U.N.; Van Overbeek, L.; Elsas, V.; Dirk, J. Exploration of Hitherto-Uncultured Bacteria from the Rhizosphere. FEMS Microbiol. Ecol. 2009, 69, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-X.; Anantharaman, K.; Shaiber, A.; Eren, A.M.; Banfield, J.F. Accurate and Complete Genomes from Metagenomes. Genome Res. 2020, 30, 315–333. [Google Scholar] [CrossRef]
- Kaster, A.-K.; Sobol, M.S. Microbial Single-Cell Omics: The Crux of the Matter. Appl. Microbiol. Biotechnol. 2020, 104, 8209–8220. [Google Scholar] [CrossRef]
- Lagier, J.-C.; Hugon, P.; Khelaifia, S.; Fournier, P.-E.; Scola, B.L.; Raoult, D. The Rebirth of Culture in Microbiology through the Example of Culturomics To Study Human Gut Microbiota. Clin. Microbiol. Rev. 2015, 28, 237–264. [Google Scholar] [CrossRef]
- Karimi, E.; Keller-Costa, T.; Slaby, B.M.; Cox, C.J.; da Rocha, U.N.; Hentschel, U.; Costa, R. Genomic Blueprints of Sponge-Prokaryote Symbiosis Are Shared by Low Abundant and Cultivatable Alphaproteobacteria. Sci. Rep. 2019, 9, 1999. [Google Scholar] [CrossRef]
- NCBI Resource Coordinators. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018, 46, D8–D13. [Google Scholar] [CrossRef]
- Attwood, T.K.; Agit, B.; Ellis, L.B.M. Longevity of Biological Databases. EMBnet. J. 2015, 21, e803. [Google Scholar] [CrossRef]
- Schnoes, A.M.; Brown, S.D.; Dodevski, I.; Babbitt, P.C. Annotation Error in Public Databases: Misannotation of Molecular Function in Enzyme Superfamilies. PLoS Comput. Biol. 2009, 5, e1000605. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.; Jeske, L.; Ulbrich, S.; Hofmann, J.; Koblitz, J.; Schomburg, I.; Neumann-Schaal, M.; Jahn, D.; Schomburg, D. BRENDA, the ELIXIR Core Data Resource in 2021: New Developments and Updates. Nucleic Acids Res. 2021, 49, D498–D508. [Google Scholar] [CrossRef] [PubMed]
- Bairoch, A.; Apweiler, R. The SWISS-PROT Protein Sequence Data Bank and Its New Supplement TREMBL. Nucleic Acids Res. 1996, 24, 21–25. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. UniProt: A Worldwide Hub of Protein Knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG Resource for Deciphering the Genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef]
- Prestat, E.; David, M.M.; Hultman, J.; Taş, N.; Lamendella, R.; Dvornik, J.; Mackelprang, R.; Myrold, D.D.; Jumpponen, A.; Tringe, S.G.; et al. FOAM (Functional Ontology Assignments for Metagenomes): A Hidden Markov Model (HMM) Database with Environmental Focus. Nucleic Acids Res. 2014, 42, e145. [Google Scholar] [CrossRef]
- Huang, L.; Zhang, H.; Wu, P.; Entwistle, S.; Li, X.; Yohe, T.; Yi, H.; Yang, Z.; Yin, Y. DbCAN-Seq: A Database of Carbohydrate-Active Enzyme (CAZyme) Sequence and Annotation. Nucleic Acids Res. 2018, 46, D516–D521. [Google Scholar] [CrossRef]
- Tal, O.; Selvaraj, G.; Medina, S.; Ofaim, S.; Freilich, S. NetMet: A Network-Based Tool for Predicting Metabolic Capacities of Microbial Species and Their Interactions. Microorganisms 2020, 8, 840. [Google Scholar] [CrossRef] [PubMed]
- Rosado, P.M.; Leite, D.C.A.; Duarte, G.A.S.; Chaloub, R.M.; Jospin, G.; da Rocha, U.N.; Saraiva, J.P.; Dini-Andreote, F.; Eisen, J.A.; Bourne, D.G.; et al. Marine Probiotics: Increasing Coral Resistance to Bleaching through Microbiome Manipulation. ISME J. 2019, 13, 921–936. [Google Scholar] [CrossRef]
- Lian, S.; Nikolausz, M.; Nijenhuis, I.; da Rocha, U.N.; Liu, B.; Corrêa, F.B.; Saraiva, J.P.; Richnow, H.H. Biotransformation of Hexachlorocyclohexanes Contaminated Biomass for Energetic Utilization Demonstrated in Continuous Anaerobic Digestion System. J. Hazard. Mater. 2020, 384, 121448. [Google Scholar] [CrossRef]
- Aranda-Díaz, A.; Obadia, B.; Dodge, R.; Thomsen, T.; Hallberg, Z.F.; Güvener, Z.T.; Ludington, W.B.; Huang, K.C. Bacterial Interspecies Interactions Modulate PH-Mediated Antibiotic Tolerance. eLife 2020, 9, e51493. [Google Scholar] [CrossRef] [PubMed]
- Yamazawa, A.; Iikura, T.; Morioka, Y.; Shino, A.; Ogata, Y.; Date, Y.; Kikuchi, J. Cellulose Digestion and Metabolism Induced Biocatalytic Transitions in Anaerobic Microbial Ecosystems. Metabolites 2014, 4, 36–52. [Google Scholar] [CrossRef] [PubMed]
- Herbst, F.-A.; Bahr, A.; Duarte, M.; Pieper, D.H.; Richnow, H.-H.; von Bergen, M.; Seifert, J.; Bombach, P. Elucidation of In Situ Polycyclic Aromatic Hydrocarbon Degradation by Functional Metaproteomics (Protein-SIP). Proteomics 2013, 13, 2910–2920. [Google Scholar] [CrossRef] [PubMed]
- Drigo, B.; Pijl, A.S.; Duyts, H.; Kielak, A.M.; Gamper, H.A.; Houtekamer, M.J.; Boschker, H.T.S.; Bodelier, P.L.E.; Whiteley, A.S.; van Veen, J.A.; et al. Shifting Carbon Flow from Roots into Associated Microbial Communities in Response to Elevated Atmospheric CO2. Proc. Natl. Acad. Sci. USA 2010, 107, 10938–10942. [Google Scholar] [CrossRef]
- Biswas, S.; Mcdonald, M.; Lundberg, D.S.; Dangl, J.L.; Jojic, V. Learning Microbial Interaction Networks from Metagenomic Count Data. J. Comput. Biol. 2016, 23, 526–535. [Google Scholar] [CrossRef]
- Khammar, N.; Malhautier, L.; Degrange, V.; Lensi, R.; Godon, J.-J.; Fanlo, J.-L. Link between Spatial Structure of Microbial Communities and Degradation of a Complex Mixture of Volatile Organic Compounds in Peat Biofilters. J. Appl. Microbiol. 2005, 98, 476–490. [Google Scholar] [CrossRef]
- Van der Waals, M.J.; Atashgahi, S.; da Rocha, U.N.; van der Zaan, B.M.; Smidt, H.; Gerritse, J. Benzene Degradation in a Denitrifying Biofilm Reactor: Activity and Microbial Community Composition. Appl. Microbiol. Biotechnol. 2017, 101, 5175–5188. [Google Scholar] [CrossRef] [PubMed]
- Karaoz, U.; Couradeau, E.; da Rocha, U.N.; Lim, H.-C.; Northen, T.; Garcia-Pichel, F.; Brodie, E.L. Large Blooms of Bacillales (Firmicutes) Underlie the Response to Wetting of Cyanobacterial Biocrusts at Various Stages of Maturity. mBio 2018, 9, e01366-16. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Gao, Y.; Jia, X.; Wang, M.; Ding, J.; Cheng, L.; Bao, F.; Wu, B. Network Analysis Reveals the Strengthening of Microbial Interaction in Biological Soil Crust Development in the Mu Us Sandy Land, Northwestern China. Soil Biol. Biochem. 2020, 144, 107782. [Google Scholar] [CrossRef]
- Wegener, G.; Kellermann, M.Y.; Elvert, M. Tracking Activity and Function of Microorganisms by Stable Isotope Probing of Membrane Lipids. Curr. Opin. Biotechnol. 2016, 41, 43–52. [Google Scholar] [CrossRef]
- Shi, S.; Nuccio, E.; Herman, D.J.; Rijkers, R.; Estera, K.; Li, J.; da Rocha, U.N.; He, Z.; Pett-Ridge, J.; Brodie, E.L.; et al. Successional Trajectories of Rhizosphere Bacterial Communities over Consecutive Seasons. mBio 2015, 6, e00746-15. [Google Scholar] [CrossRef]


| Approach | Pros | Cons | Environments | References |
|---|---|---|---|---|
| Supra-organism | Global reaction network is possible and allows for prediction of shifts in pathway activity by measuring gene relative abundance. | Genetic potential of individual species not determined. | Anaerobic mixed culture fermentations | [38] |
| Contribution of individual species to shifts in pathway activity not determined since interactions are based on genes/reactions. | Agricultural soil and seep sea “whale fall” carcasses | [39] | ||
| Population-based | Species boundaries explicitly defined. Individual species functional potential can be determined. Allows determining direct metabolic interactions between species. | High computational and manual curation efforts since full genome-scale metabolic models for each species is required. | Corals | [40] |
| Anoxic sediments | [41] | |||
| Batch and Continuous cultures | [42] | |||
| Synthetic microbial systems | [43] | |||
| Guild-based | Less complex models since grouping of species is based on their known functional traits. | Requires previous knowledge on functional traits. Individual contribution of species to ecosystem processes is unknown. | Soil | [44,45] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saraiva, J.P.; Worrich, A.; Karakoç, C.; Kallies, R.; Chatzinotas, A.; Centler, F.; Nunes da Rocha, U. Mining Synergistic Microbial Interactions: A Roadmap on How to Integrate Multi-Omics Data. Microorganisms 2021, 9, 840. https://doi.org/10.3390/microorganisms9040840
Saraiva JP, Worrich A, Karakoç C, Kallies R, Chatzinotas A, Centler F, Nunes da Rocha U. Mining Synergistic Microbial Interactions: A Roadmap on How to Integrate Multi-Omics Data. Microorganisms. 2021; 9(4):840. https://doi.org/10.3390/microorganisms9040840
Chicago/Turabian StyleSaraiva, Joao Pedro, Anja Worrich, Canan Karakoç, Rene Kallies, Antonis Chatzinotas, Florian Centler, and Ulisses Nunes da Rocha. 2021. "Mining Synergistic Microbial Interactions: A Roadmap on How to Integrate Multi-Omics Data" Microorganisms 9, no. 4: 840. https://doi.org/10.3390/microorganisms9040840
APA StyleSaraiva, J. P., Worrich, A., Karakoç, C., Kallies, R., Chatzinotas, A., Centler, F., & Nunes da Rocha, U. (2021). Mining Synergistic Microbial Interactions: A Roadmap on How to Integrate Multi-Omics Data. Microorganisms, 9(4), 840. https://doi.org/10.3390/microorganisms9040840