
4E Interacting Protein as a Potential Novel Drug Target for Nucleoside Analogues in Trypanosoma brucei

 , ,
, ,  , , , , and
, , , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animals and Parasites
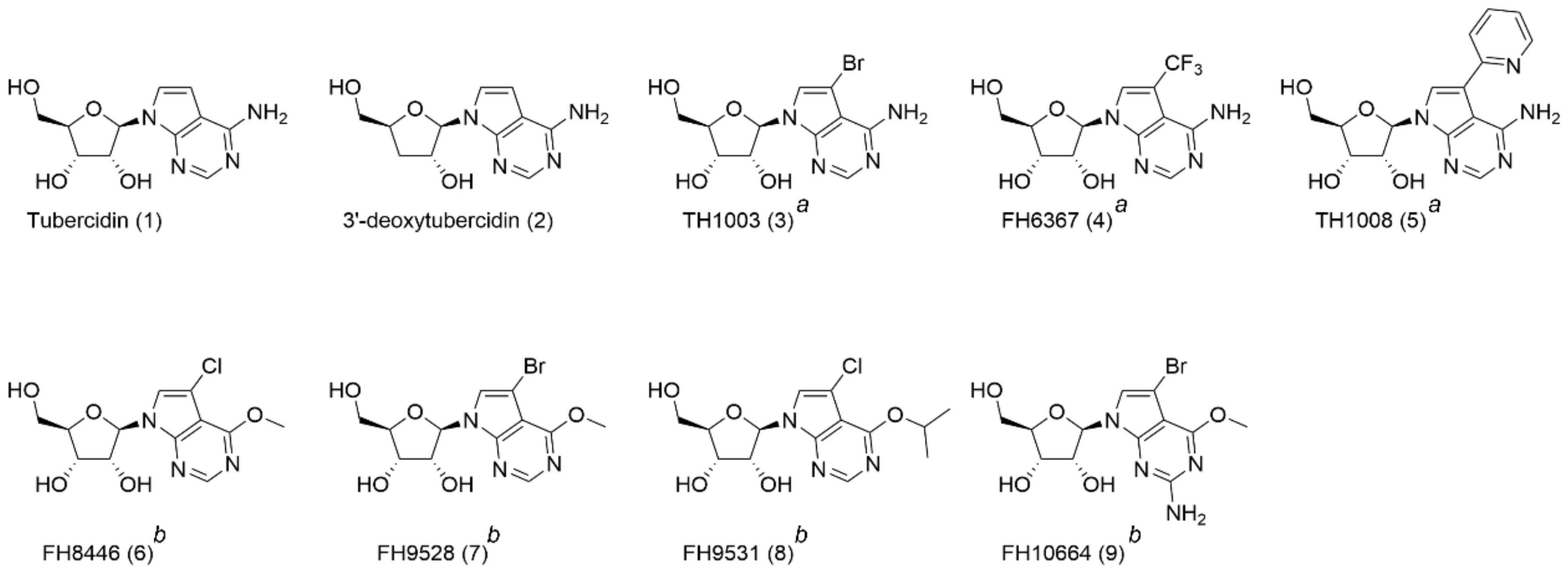
2.3. Compounds
2.4. In Vitro Cytotoxicity Assay
2.5. Resazurin-Based Susceptibility Assay
2.6. RNAi Library Screening
2.7. RNAi Insert Identification
2.8. Independent Confirmation of the RNAi Phenotype
2.9. Efficiency of the RNAi Silencing
2.10. In Vitro Growth
2.11. Cell Cycle Analysis and RNA Quantification
2.12. Western Blot Analysis of PAD1
2.13. In Vivo Infectivity
2.14. Tsetse Fly Infections
2.15. Graphs and Statistical Analyses
3. Results
3.1. 7-Deaza Adenosine Analogues Act Independently of DNA and RNA Synthesis Inhibition
3.2. Involvement of Adenosine Kinase and 4E-Interacting Protein in the Antitrypanosomal Activity
3.3. 7-Deaza Adenosine Analogues Show a Variable Dependence on ADKIN and 4EIP
3.4. 4 EIP but Not ADKIN Is Essential for In Vivo Infectivity in Mice
3.5. 5 Does Not Specifically Impact Parasite Differentiation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kennedy, P.G. Clinical features, diagnosis, and treatment of human African trypanosomiasis (sleeping sickness). Lancet Neurol. 2013, 12, 186–194. [Google Scholar] [CrossRef]
- Tirados, I.; Esterhuizen, J.; Kovacic, V.; Mangwiro, T.N.; Vale, G.A.; Hastings, I.; Solano, P.; Lehane, M.J.; Torr, S.J. Tsetse Control and Gambian Sleeping Sickness; Implications for Control Strategy. PLoS Negl. Trop Dis. 2015, 9, e0003822. [Google Scholar] [CrossRef] [PubMed]
- Malvy, D.; Chappuis, F. Sleeping sickness. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2011, 17, 986–995. [Google Scholar] [CrossRef]
- Babokhov, P.; Sanyaolu, A.O.; Oyibo, W.A.; Fagbenro-Beyioku, A.F.; Iriemenam, N.C. A current analysis of chemotherapy strategies for the treatment of human African trypanosomiasis. Pathog. Glob. Health 2013, 107, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Eperon, G.; Balasegaram, M.; Potet, J.; Mowbray, C.; Valverde, O.; Chappuis, F. Treatment options for second-stage gambiense human African trypanosomiasis. Expert Rev. Anti Infect. Ther. 2014, 12, 1407–1417. [Google Scholar] [CrossRef]
- Mesu, V.; Kalonji, W.M.; Bardonneau, C.; Mordt, O.V.; Blesson, S.; Simon, F.; Delhomme, S.; Bernhard, S.; Kuziena, W.; Lubaki, J.F.; et al. Oral fexinidazole for late-stage African Trypanosoma brucei gambiense trypanosomiasis: A pivotal multicentre, randomised, non-inferiority trial. Lancet 2018, 391, 144–154. [Google Scholar] [CrossRef]
- Dickie, E.A.; Giordani, F.; Gould, M.K.; Maser, P.; Burri, C.; Mottram, J.C.; Rao, S.P.S.; Barrett, M.P. New Drugs for Human African Trypanosomiasis: A Twenty First Century Success Story. Trop. Med. Infect. Dis. 2020, 5, 29. [Google Scholar] [CrossRef]
- Baker, N.; de Koning, H.P.; Maser, P.; Horn, D. Drug resistance in African trypanosomiasis: The melarsoprol and pentamidine story. Trends Parasitol. 2013, 29, 110–118. [Google Scholar] [CrossRef]
- Sokolova, A.Y.; Wyllie, S.; Patterson, S.; Oza, S.L.; Read, K.D.; Fairlamb, A.H. Cross-resistance to nitro drugs and implications for treatment of human African trypanosomiasis. Antimicrob. Agents Chemother. 2010, 54, 2893–2900. [Google Scholar] [CrossRef]
- Wyllie, S.; Foth, B.J.; Kelner, A.; Sokolova, A.Y.; Berriman, M.; Fairlamb, A.H. Nitroheterocyclic drug resistance mechanisms in Trypanosoma brucei. J. Antimicrob. Chemother. 2016, 71, 625–634. [Google Scholar] [CrossRef]
- Berg, M.; Van der Veken, P.; Goeminne, A.; Haemers, A.; Augustyns, K. Inhibitors of the Purine Salvage Pathway: A Valuable Approach for Antiprotozoal Chemotherapy? Curr. Med. Chem. 2010, 17, 2456–2481. [Google Scholar] [CrossRef]
- Vodnala, S.K.; Ferella, M.; Lundén-Miguel, H.; Betha, E.; van Reet, N.; Amin, D.N.; Öberg, B.; Andersson, B.; Kristensson, K.; Wigzell, H.; et al. Preclinical Assessment of the Treatment of Second-Stage African Trypanosomiasis with Cordycepin and Deoxycoformycin. PLoS Negl. Trop Dis. 2009, 3, e495. [Google Scholar] [CrossRef]
- Vodnala, S.K.; Lundbäck, T.; Yeheskieli, E.; Sjöberg, B.; Gustavsson, A.-L.; Svensson, R.; Olivera, G.C.; Eze, A.A.; de Koning, H.P.; Hammarström, L.G.J.; et al. Structure–Activity Relationships of Synthetic Cordycepin Analogues as Experimental Therapeutics for African Trypanosomiasis. J. Med. Chem. 2013, 56, 9861–9873. [Google Scholar] [CrossRef] [PubMed]
- Drew, M.E.; Morris, J.C.; Wang, Z.; Wells, L.; Sanchez, M.; Landfear, S.M.; Englund, P.T. The Adenosine Analog Tubercidin Inhibits Glycolysis in Trypanosoma brucei as Revealed by an RNA Interference Library. J. Biol. Chem. 2003, 278, 46596–46600. [Google Scholar] [CrossRef] [PubMed]
- Rottenberg, M.E.; Masocha, W.; Ferella, M.; Petitto-Assis, F.; Goto, H.; Kristensson, K.; McCaffrey, R.; Wigzell, H. Treatment of African trypanosomiasis with cordycepin and adenosine deaminase inhibitors in a mouse model. J. Infect Dis. 2005, 192, 1658–1665. [Google Scholar] [CrossRef]
- Smulson, M.E.; Suhadolnik, R.J. The biosynthesis of the 7-deazaadenine ribonucleoside, tubercidin, by Streptomyces tubercidicus. J. Biol. Chem. 1967, 242, 2872–2876. [Google Scholar] [CrossRef]
- El Kouni, M.H.; Diop, D.; O’Shea, P.; Carlisle, R.; Sommadossi, J.P. Prevention of tubercidin host toxicity by nitrobenzylthioinosine 5’-monophosphate for the treatment of schistosomiasis. Antimicrob. Agents Chemother. 1989, 33, 824–827. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Aoki, J.I.; Coelho, A.C.; Muxel, S.M.; Zampieri, R.A.; Sanchez, E.M.; Nerland, A.H.; Floeter-Winter, L.M.; Cotrim, P.C. Characterization of a Novel Endoplasmic Reticulum Protein Involved in Tubercidin Resistance in Leishmania major. PLoS Negl. Trop Dis. 2016, 10, e0004972. [Google Scholar] [CrossRef]
- Hulpia, F.; Campagnaro, G.D.; Scortichini, M.; Van Hecke, K.; Maes, L.; de Koning, H.P.; Caljon, G.; Van Calenbergh, S. Revisiting tubercidin against kinetoplastid parasites: Aromatic substitutions at position 7 improve activity and reduce toxicity. Eur. J. Med. Chem. 2019, 164, 689–705. [Google Scholar] [CrossRef] [PubMed]
- Hulpia, F.; Mabille, D.; Campagnaro, G.D.; Schumann, G.; Maes, L.; Roditi, I.; Hofer, A.; de Koning, H.P.; Caljon, G.; Van Calenbergh, S. Combining tubercidin and cordycepin scaffolds results in highly active candidates to treat late-stage sleeping sickness. Nat. Commun. 2019, 10, 5564. [Google Scholar] [CrossRef]
- Hulpia, F.; Bouton, J.; Campagnaro, G.D.; Alfayez, I.A.; Mabille, D.; Maes, L.; de Koning, H.P.; Caljon, G.; Van Calenbergh, S. C6-O-alkylated 7-deazainosine nucleoside analogues: Discovery of potent and selective anti-sleeping sickness agents. Eur. J. Med. Chem. 2020, 188, 112018. [Google Scholar] [CrossRef]
- Ho, H.T.B.; Wang, J. The Nucleoside Transporters CNTs and ENTs. In Drug Transporters: Molecular Characterization and Role in Drug Disposition, 2nd ed.; Guofeng You, M.E.M., Ed.; Wiley: Hoboken, NJ, USA, 2014; pp. 107–126. [Google Scholar]
- Target Product Profile Sleeping Sickness, DNDi. Available online: https://dndi.org/diseases/sleeping-sickness/target-product-profile (accessed on 29 January 2021).
- Van den Kerkhof, M.; Sterckx, Y.G.; Leprohon, P.; Maes, L.; Caljon, G. Experimental Strategies to Explore Drug Action and Resistance in Kinetoplastid Parasites. Microorganisms 2020, 8, 950. [Google Scholar] [CrossRef]
- Alsford, S.; Eckert, S.; Baker, N.; Glover, L.; Sanchez-Flores, A.; Leung, K.F.; Turner, D.J.; Field, M.C.; Berriman, M.; Horn, D. High-throughput decoding of antitrypanosomal drug efficacy and resistance. Nature 2012, 482, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.; Alsford, S.; Horn, D. Genome-wide RNAi screens in African trypanosomes identify the nifurtimox activator NTR and the eflornithine transporter AAT6. Mol. Biochem. Parasitol. 2011, 176, 55–57. [Google Scholar] [CrossRef] [PubMed]
- Burkard, G.S.; Jutzi, P.; Roditi, I. Genome-wide RNAi screens in bloodstream form trypanosomes identify drug transporters. Mol. Biochem. Parasitol. 2011, 175, 91–94. [Google Scholar] [CrossRef]
- Moss, C.X.; Brown, E.; Hamilton, A.; Van der Veken, P.; Augustyns, K.; Mottram, J.C. An essential signal peptide peptidase identified in an RNAi screen of serine peptidases of Trypanosoma brucei. PLoS ONE 2015, 10, e0123241. [Google Scholar] [CrossRef]
- Povelones, M.L.; Tiengwe, C.; Gluenz, E.; Gull, K.; Englund, P.T.; Jensen, R.E. Mitochondrial shape and function in trypanosomes requires the outer membrane protein, TbLOK1. Mol. Microbiol. 2013, 87, 713–729. [Google Scholar] [CrossRef] [PubMed]
- Monnerat, S.; Clucas, C.; Brown, E.; Mottram, J.C.; Hammarton, T.C. Searching for novel cell cycle regulators in Trypanosoma brucei with an RNA interference screen. BMC Res. Notes 2009, 2, 46. [Google Scholar] [CrossRef]
- Morris, J.C.; Wang, Z.; Drew, M.E.; Englund, P.T. Glycolysis modulates trypanosome glycoprotein expression as revealed by an RNAi library. EMBO J. 2002, 21, 4429–4438. [Google Scholar] [CrossRef]
- Glover, L.; Alsford, S.; Baker, N.; Turner, D.J.; Sanchez-Flores, A.; Hutchinson, S.; Hertz-Fowler, C.; Berriman, M.; Horn, D. Genome-scale RNAi screens for high-throughput phenotyping in bloodstream-form African trypanosomes. Nat. Protoc. 2015, 10, 106–133. [Google Scholar] [CrossRef]
- Glover, L.; Horn, D. Site-specific DNA double-strand breaks greatly increase stable transformation efficiency in Trypanosoma brucei. Mol. Biochem. Parasitol. 2009, 166, 194–197. [Google Scholar] [CrossRef]
- Burkard, G.; Fragoso, C.M.; Roditi, I. Highly efficient stable transformation of bloodstream forms of Trypanosoma brucei. Mol. Biochem. Parasitol. 2007, 153, 220–223. [Google Scholar] [CrossRef]
- Terrao, M.; Marucha, K.K.; Mugo, E.; Droll, D.; Minia, I.; Egler, F.; Braun, J.; Clayton, C. The supressive cap-binding-complex factor 4EIP is required for normal differentiation. Nucleic Acids Res. 2018, 46, 8993–9010. [Google Scholar] [CrossRef]
- Maes, L.; Cos, P.; Croft, S.L. The Relevance of Susceptibility Tests, Breakpoints, and Markers. In Drug Resistance in Leishmania Parasites: Consequences, Molecular Mechanisms and PossibleTreatments; Ponte-Sucre, A., Diaz, E., Padrón-Nieves, M., Eds.; Springer: Vienna, Austria, 2013; pp. 407–429. [Google Scholar]
- Tritryp Database. Available online: http://tritrypdb.org/tritrypdb (accessed on 11 April 2017).
- ProtParam. Available online: http://web.expasy.org/protparam (accessed on 11 April 2017).
- SignalIP Tool. Available online: www.bds.dtu.dk/services/SignalIP (accessed on 11 April 2017).
- Toolkit. Available online: https://toolkit/tuebingen.mpg.de/hhpred (accessed on 11 April 2017).
- Brenndorfer, M.; Boshart, M. Selection of reference genes for mRNA quantification in Trypanosoma brucei. Mol. Biochem. Parasitol. 2010, 172, 52–55. [Google Scholar] [CrossRef] [PubMed]
- Vanwalleghem, G.; Fontaine, F.; Lecordier, L.; Tebabi, P.; Klewe, K.; Nolan, D.P.; Yamaryo-Botte, Y.; Botte, C.; Kremer, A.; Burkard, G.S.; et al. Coupling of lysosomal and mitochondrial membrane permeabilization in trypanolysis by APOL1. Nat. Commun. 2015, 6, 8078. [Google Scholar] [CrossRef]
- Gonzalez-Andrade, P.; Camara, M.; Ilboudo, H.; Bucheton, B.; Jamonneau, V.; Deborggraeve, S. Diagnosis of trypanosomatid infections: Targeting the spliced leader RNA. J. Mol. Diagn. 2014, 16, 400–404. [Google Scholar] [CrossRef]
- Dean, S.; Marchetti, R.; Kirk, K.; Matthews, K.R. A surface transporter family conveys the trypanosome differentiation signal. Nature 2009, 459, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Acs, G.; Reich, E.; Mori, M. Biological and Biochemical Properties of the Analogue Antibiotic Tubercidin. Proc. Natl. Acad. Sci. USA 1964, 52, 493–501. [Google Scholar] [CrossRef]
- Berriman, M.; Ghedin, E.; Hertz-Fowler, C.; Blandin, G.; Renauld, H.; Bartholomeu, D.C.; Lennard, N.J.; Caler, E.; Hamlin, N.E.; Haas, B.; et al. The genome of the African trypanosome Trypanosoma brucei. Science 2005, 309, 416–422. [Google Scholar] [CrossRef]
- Sun, S.Y.; Wang, C.; Yuan, Y.A.; He, C.Y. An intracellular membrane junction consisting of flagellum adhesion glycoproteins links flagellum biogenesis to cell morphogenesis in Trypanosoma brucei. J. Cell Sci. 2013, 126, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Vodnala, M.; Fijolek, A.; Rofougaran, R.; Mosimann, M.; Maser, P.; Hofer, A. Adenosine kinase mediates high affinity adenosine salvage in Trypanosoma brucei. J. Biol. Chem. 2008, 283, 5380–5388. [Google Scholar] [CrossRef] [PubMed]
- Luscher, A.; Onal, P.; Schweingruber, A.M.; Maser, P. Adenosine kinase of Trypanosoma brucei and its role in susceptibility to adenosine antimetabolites. Antimicrob. Agents Chemother. 2007, 51, 3895–3901. [Google Scholar] [CrossRef] [PubMed]
- Vigueira, P.A.; Paul, K.S. Requirement for acetyl-CoA carboxylase in Trypanosoma brucei is dependent upon the growth environment. Mol. Microbiol. 2011, 80, 117–132. [Google Scholar] [CrossRef] [PubMed]
- Ong, H.B.; Sienkiewicz, N.; Wyllie, S.; Patterson, S.; Fairlamb, A.H. Trypanosoma brucei (UMP synthase null mutants) are avirulent in mice, but recover virulence upon prolonged culture in vitro while retaining pyrimidine auxotrophy. Mol. Microbiol. 2013, 90, 443–455. [Google Scholar] [CrossRef]
- Meleppattu, S.; Arthanari, H.; Zinoviev, A.; Boeszoermenyi, A.; Wagner, G.; Shapira, M.; Leger-Abraham, M. Structural basis for LeishIF4E-1 modulation by an interacting protein in the human parasite Leishmania major. Nucleic Acids Res. 2018, 46, 3791–3801. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 ± SEM (µM) | CC50 ± SEM (µM) | SI 1 |
|---|---|---|---|
| Tubercidin (1) | 0.12 ± 0.003 | 2.23 ± 0.68 | 18 |
| 3 | 0.38 ± 0.064 | 33.3 ± 12.6 | 88 |
| 4 | 0.38 ± 0.017 | 19.2 ± 15.2 | 50 |
| 5 | 0.15 ± 0.005 | 15.1 ± 4.1 | 103 |
| 6 | 0.05 ± 0.003 | 48 ± 16 | 960 |
| 7 | 0.08 ± 0.001 | >64 | 800 |
| 8 | 0.10 ± 0.008 | >64 | 640 |
| 9 | 0.10 ± 0.001 | 3.24 ± 0.01 | 32 |
| % Knock-Down | ||
|---|---|---|
| Target | Clone A 1 | Clone B 1 |
| FLABP1 | 44.1 ± 1.7 | 42.8 ± 0.6 |
| EndoG | 33.2 ± 0.9 | 31.0 ± 0.9 |
| ADKIN | 81.2 ± 0.2 | 83.4 ± 0.5 |
| 4EIP | 85.0 ± 0.7 | 85.1 ± 0.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mabille, D.; Cardoso Santos, C.; Hendrickx, R.; Claes, M.; Takac, P.; Clayton, C.; Hendrickx, S.; Hulpia, F.; Maes, L.; Van Calenbergh, S.; et al. 4E Interacting Protein as a Potential Novel Drug Target for Nucleoside Analogues in Trypanosoma brucei. Microorganisms 2021, 9, 826. https://doi.org/10.3390/microorganisms9040826
Mabille D, Cardoso Santos C, Hendrickx R, Claes M, Takac P, Clayton C, Hendrickx S, Hulpia F, Maes L, Van Calenbergh S, et al. 4E Interacting Protein as a Potential Novel Drug Target for Nucleoside Analogues in Trypanosoma brucei. Microorganisms. 2021; 9(4):826. https://doi.org/10.3390/microorganisms9040826
Chicago/Turabian StyleMabille, Dorien, Camila Cardoso Santos, Rik Hendrickx, Mathieu Claes, Peter Takac, Christine Clayton, Sarah Hendrickx, Fabian Hulpia, Louis Maes, Serge Van Calenbergh, and et al. 2021. "4E Interacting Protein as a Potential Novel Drug Target for Nucleoside Analogues in Trypanosoma brucei" Microorganisms 9, no. 4: 826. https://doi.org/10.3390/microorganisms9040826
APA StyleMabille, D., Cardoso Santos, C., Hendrickx, R., Claes, M., Takac, P., Clayton, C., Hendrickx, S., Hulpia, F., Maes, L., Van Calenbergh, S., & Caljon, G. (2021). 4E Interacting Protein as a Potential Novel Drug Target for Nucleoside Analogues in Trypanosoma brucei. Microorganisms, 9(4), 826. https://doi.org/10.3390/microorganisms9040826