
Comparative Analysis of the Apple Root Transcriptome as Affected by Rootstock Genotype and Brassicaceae Seed Meal Soil Amendment: Implications for Plant Health


Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Design
2.2. Treatment Effects on Plant Biomass and P. penetrans Root Density
2.3. Total RNA Isolation and High-Throughput Transcriptome Sequencing
2.4. RNA-Seq Analysis
2.5. Validation of DEG Expression Patterns by Real-Time RT-qPCR
2.6. Quantification of Microbial Density in the Rhizosphere
2.7. 16S rRNA Sequencing of Rhizosphere DNA
3. Results
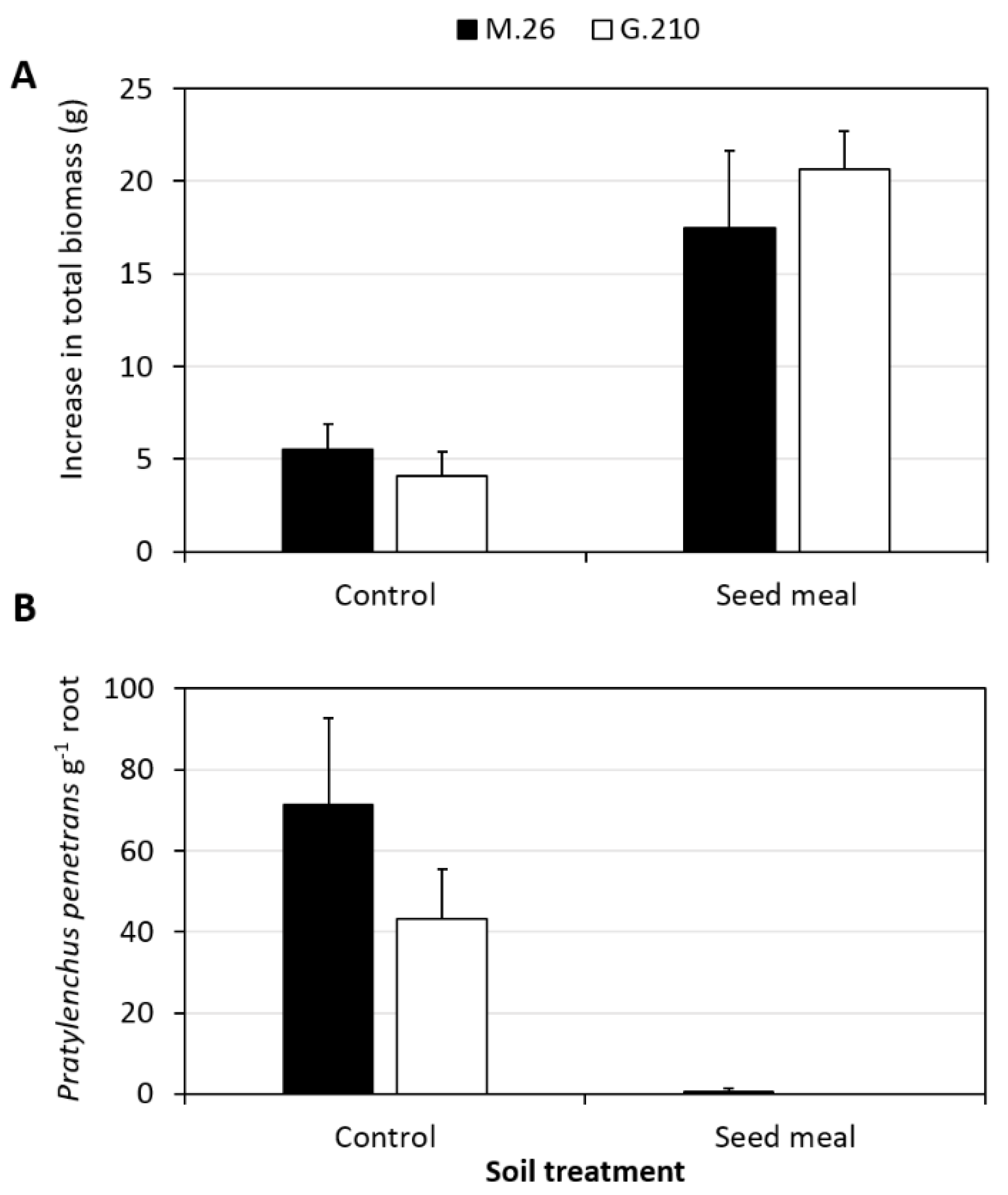
3.1. Influence of Brassicaceae SM Amendment on Plant Growth and P. penetrans Root Density
3.2. Mapping Statistics and Differential Gene Expression
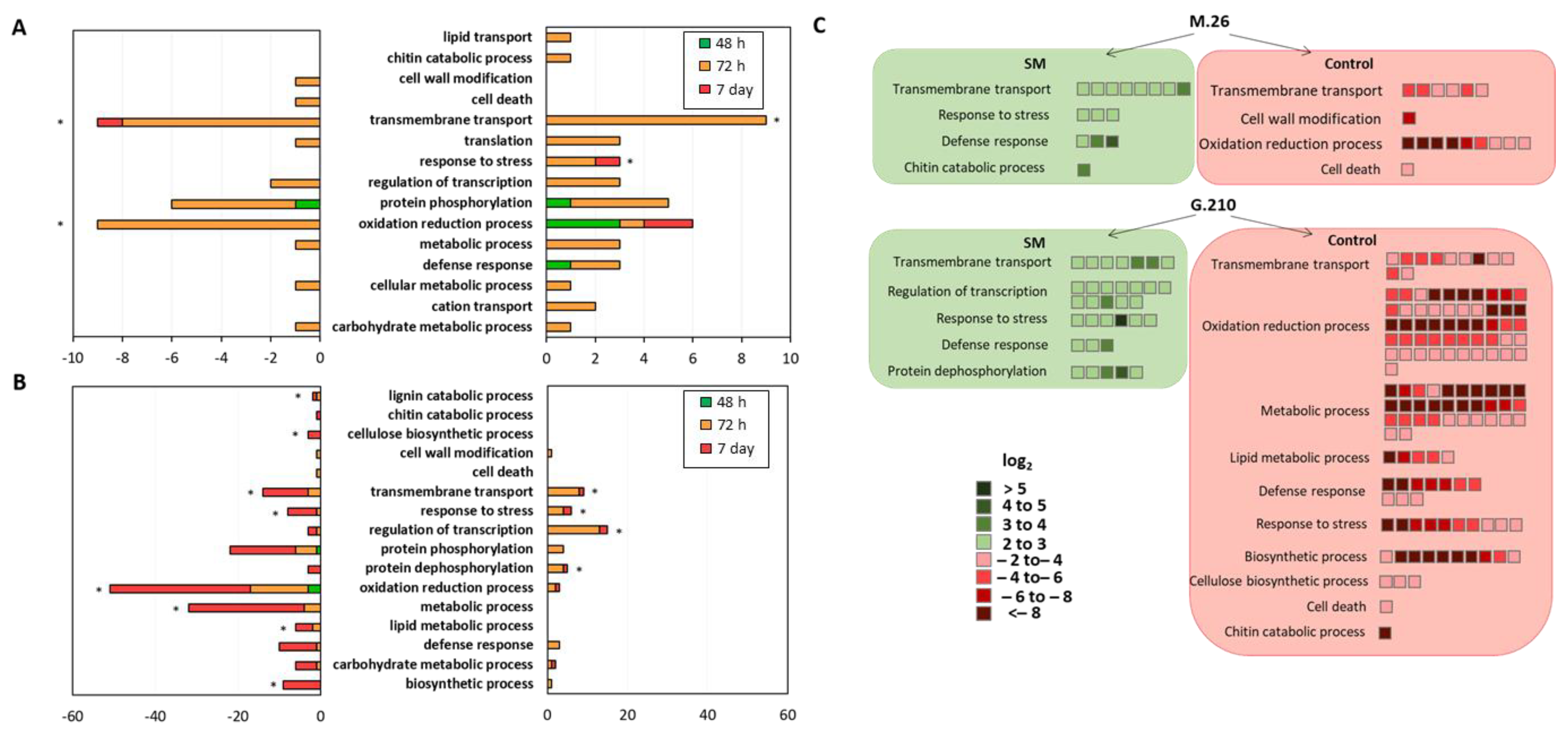
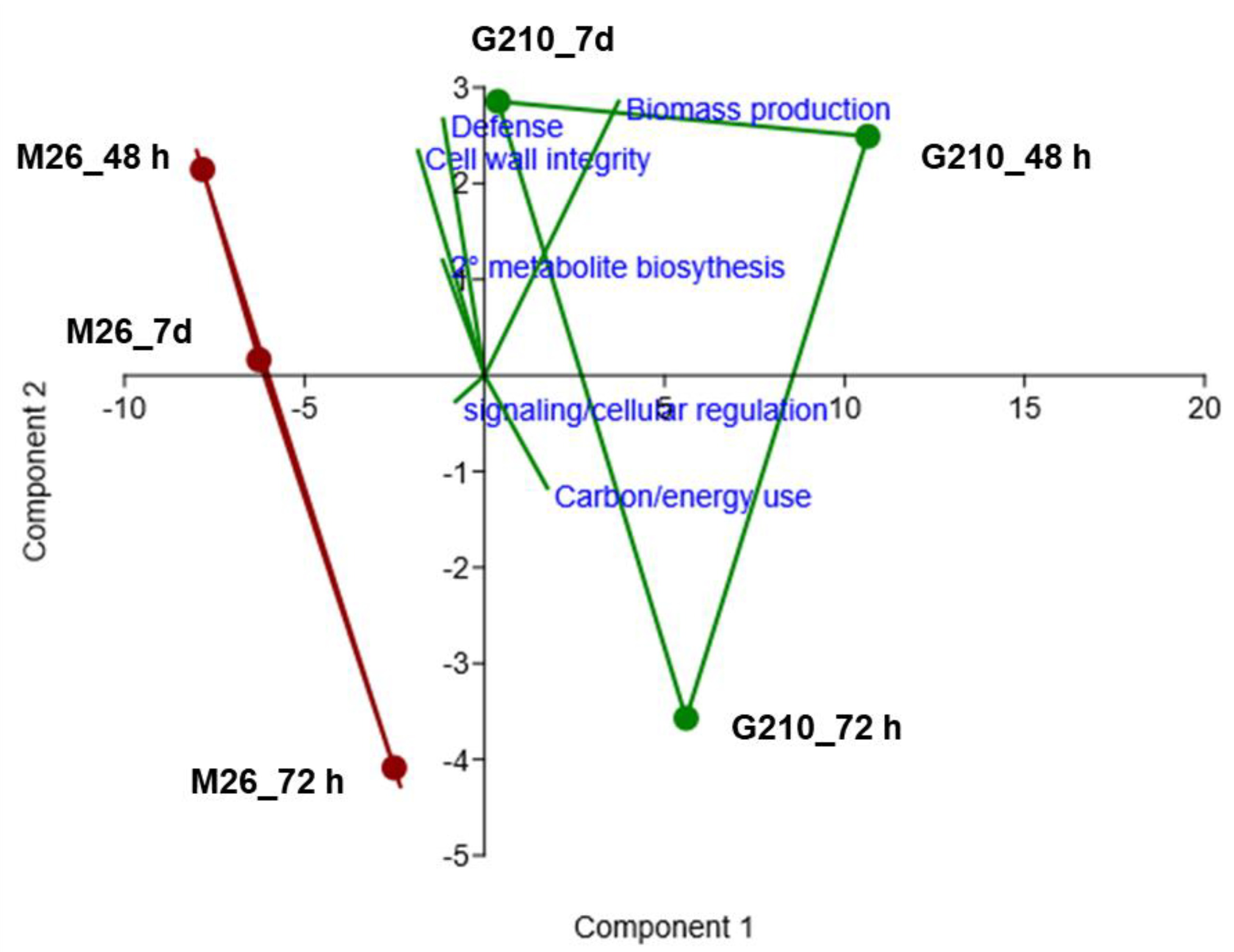
3.3. Gene Ontology (GO) Enrichment Analysis of Biological Processes
3.3.1. ARD Susceptible Genotype M.26
3.3.2. ARD Tolerant Genotype G.210
3.4. KEGG Pathway Analysis and the Role of Secondary Metabolism
3.4.1. ARD Susceptible Genotype M.26
3.4.2. ARD Tolerant Genotype G.210
3.5. DEG Identified in Both Rootstock Genotypes Cultivated in SM Amended Soil
3.6. Validation of the Expression Patterns of Selected DEG by q-PCR
3.7. Effect of SM Treatment on the Root Zone Microbial Density Post-Planting
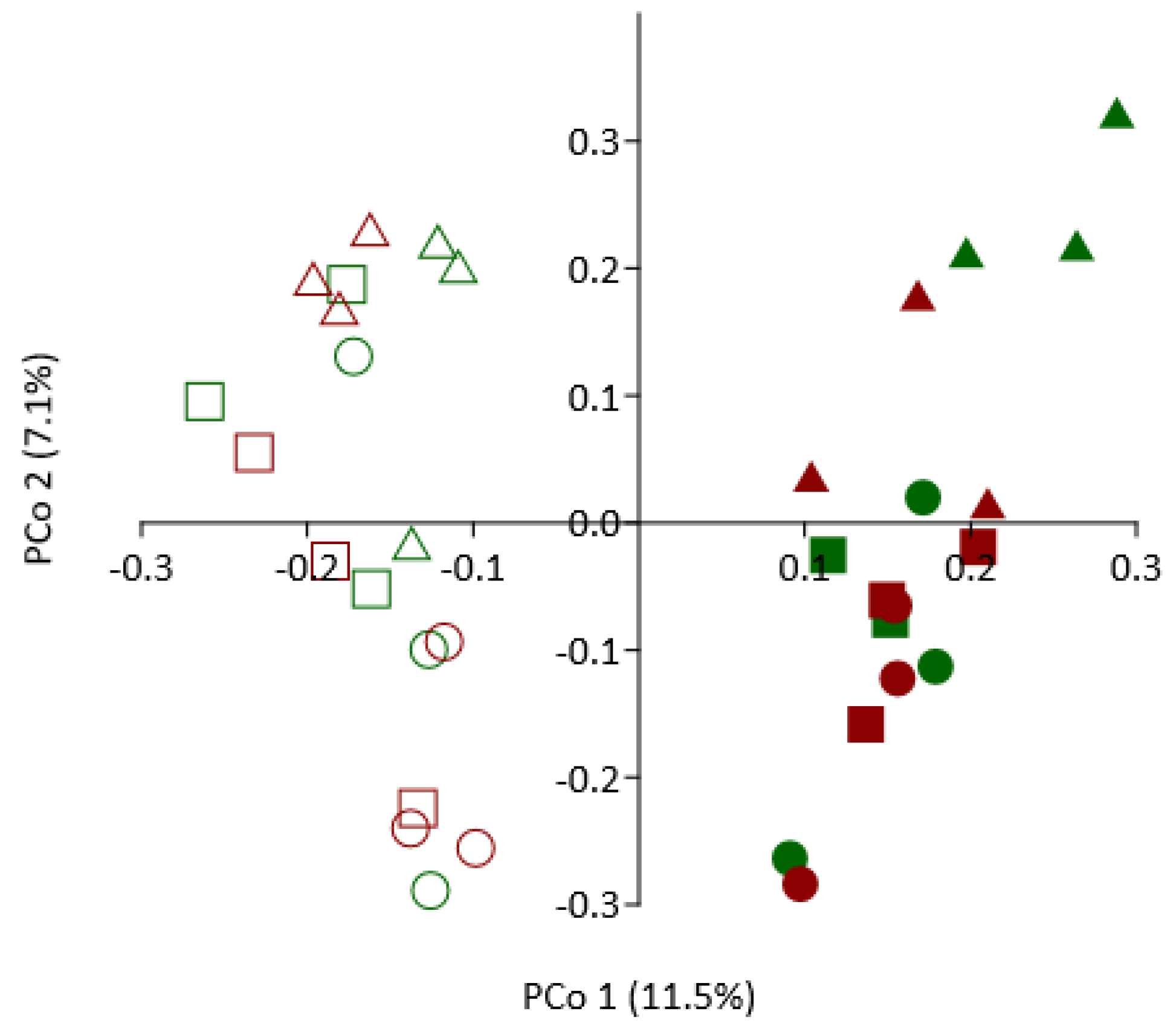
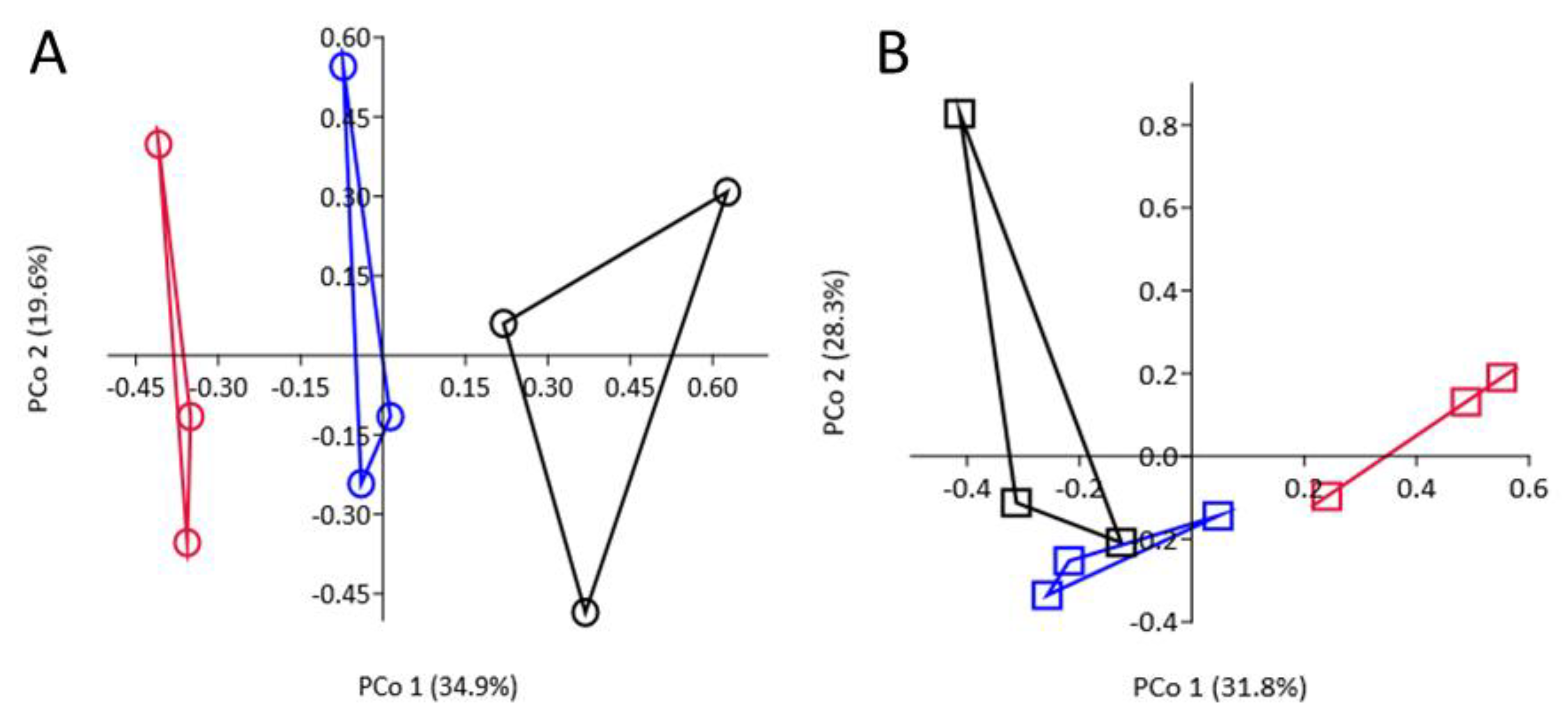
3.8. Effect of SM Amendment and Rootstock Genotype on Bacterial Community Composition
3.9. Differential Gene Expression between Rootstocks in Non-Treated Control Soil
4. Discussion
4.1. Rootstock Transcriptome Responses in SM Treated Soil
4.2. Transformation of the Rhizosphere Microbiome
4.3. Genotype-Specific Transcriptome Response to Cultivation in NTC Replant Orchard Soil
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Matthiessen, J.; Kirkegaard, J. Biofumigation and Enhanced Biodegradation: Opportunity and Challenge in Soilborne Pest and Disease Management. Crit. Rev. Plant Sci. 2006, 25, 235–265. [Google Scholar] [CrossRef]
- Kirkegaard, J.A.; Gardiner, P.A.; Desmarchelier, J.M.; Angus, J.F. Biofumigation using Brassica species to control pests and diseases in horticulture and agriculture. In Proceedings 9th Australian Research Assembly on Brassicas; Wratten, N., Mailer, R.J., Eds.; Wagga Wagga Agricultural Research Institute: Wagga Wagga, New South Wales, Australia, 1993; pp. 77–82. [Google Scholar]
- Ntalli, N.; Caboni, P. A review of isothiocyanates biofumigation activity on plant parasitic nematodes. Phytochem. Rev. 2017, 16, 827–834. [Google Scholar] [CrossRef]
- Brennan, R.J.B.; Glaze-Corcoran, S.; Wick, R.; Hashemi, M. Biofumigation: An alternative strategy for the control of plant parasistic nematodes. J. Integr. Agric. 2020, 19, 1680–1690. [Google Scholar] [CrossRef]
- Weerakoon, M.D.; Reardon, C.L.; Paulitz, T.C.; Izzo, A.D.; Mazzola, M. Long-term suppression of Pythium abappressorium induced by Brassica juncea seed meal amendment is biologically mediated. Soil Biol. Biochem. 2012, 51, 44–52. [Google Scholar] [CrossRef]
- Cohen, M.F.; Yamasaki, H.; Mazzola, M. Brassica napus seed meal soil amendment modifies microbial community structure, nitric oxide production and incidence of Rhizoctonia root rot. Soil Biol. Biochem. 2005, 37, 1215–1227. [Google Scholar] [CrossRef]
- Mazzola, M.; Granatstein, D.M.; Elfving, D.C.; Mullinix, K. Suppression of specific apple root pathogens by Brassica napus seed meal amendment regardless of glucosinolate content. Phytopathology 2001, 91, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.F.; Mazzola, M. Impact of resident bacteria, nitric oxide emission and particle size on root infection by Pythium spp. and R. solani AG-5 in Brassica napus seed meal amended soils. Plant Soil 2006, 286, 75–86. [Google Scholar] [CrossRef]
- Somera, T.S.; Freilich, S.; Mazzola, M. Comprehensive analysis of the apple rhizobiome as influenced by different Brassica seed meal and rootstocks in the same soil/plant system. Appl. Soil Ecol. 2021. [Google Scholar] [CrossRef]
- Bastian, F.; Bouziri, L.; Nicolardot, B.; Ranjard, L. Impact of wheat straw decomposition on successional patterns of soil microbial community structure. Soil Biol. Biochem. 2009, 41, 262–275. [Google Scholar] [CrossRef]
- Blaud, A.; Diouf, F.; Herrmann, A.M.; Lerch, T.Z. Analyzing the effect of soil organic matter on bacterial communities using T-RFLP fingerprinting: Different methods, different stories? Biol. Fert. Soils 2015, 51, 959–971. [Google Scholar] [CrossRef][Green Version]
- Mazzola, M.; Hewavitharana, S.; Strauss, S.L. Brassica seed meal soil amendments transform the rhizosphere microbiome and improve apple production through resistance to pathogen reinfestation. Phytopathology 2015, 105, 460–467. [Google Scholar] [CrossRef]
- Ventorino, V.; Parillo, R.; Testa, A.; Viscardi, S.; Espresso, F.; Pepe, O. Chestnut green waste composting for sustainable forest management: Microbiota dynamics and impact on plant disease control. J. Environ. Manag. 2015, 166, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, R.L.; Pieterse, C.M.J.; Bakker, P.A.H.M. The rhizosphere microbiome and plant health. Trends Plant Sci. 2012, 17, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Hol, W.H.G.; De Boer, W.; Termorshuizen, A.J.; Meyer, K.M.; Schneider, J.H.M.; Van Dam, N.M.; van Veen, J.A.; van der Putten, W.H. Reduction of rare soil microbes modifies plant herbivore interactions. Ecol. Lett. 2010, 13, 292–301. [Google Scholar] [CrossRef]
- Martinuz, A.; Schouten, A.; Menjivar, R.D.; Sikora, R.A. Effectiveness of systemic resistance toward Aphis gossypii (Hom., Aphididae) as induced by combined applications of the endophytes Fusarium oxysporum Fo162 and Rhizobiumetli G12. Biol. Control 2012, 62, 206–212. [Google Scholar] [CrossRef]
- Mendes, R.; Kruijt, M.; de Bruijin, I.; Dekkers, E.; van der Voort, M.; Schneider, J.H.M.; Piceno, Y.M.; DeSantis, T.Z.; Andersen, G.L.; Bakker, P.A.H.M.; et al. Deciphering the rhizosphere microbiome for disease suppressive bacteria. Science 2011, 332, 1097–1100. [Google Scholar] [CrossRef]
- Partida-Martinez, L.P.; Heil, M. The microbe-free plant: Fact or artifact? Front. Plant Sci. 2011, 2, 100. [Google Scholar] [CrossRef]
- Wang, L.; Mazzola, M. Field evaluation of reduced rate Brassicaceae seed meal amendment and rootstock genotype on the microbiome and control of apple replant disease. Phytopathology 2019, 109, 1378–1391. [Google Scholar] [CrossRef]
- Shin, S.; Zheng, P.; Fazio, G.; Mazzola, M.; Main, D.; Zhu, Y. Transcriptome changes specially associated with apple (Malus domestica) root defense response during Pythium ultimum infection. Physiol. Mol. Plant Pathol. 2016, 94, 16–26. [Google Scholar] [CrossRef]
- Ruzicka, D.R.; Barrios-Masias, F.H.; Hausmann, N.T.; Jackson, L.E.; Schachtman, D.P. Tomato root transcriptome response to a nitrogen-enriched soil patch. BMC Plant Biol. 2010, 10, 75. [Google Scholar] [CrossRef]
- Viger, M.; Hancock, R.D.; Miglietta, F.; Taylor, G. More plant growth but less plant defence? First global gene expression data for plants grown in soil amendment with biochar. GCB Bioenergy 2015, 7, 658–672. [Google Scholar] [CrossRef]
- Zhu, Y.; Shin, S.; Mazzola, M. Genotype responses of two apple rootstocks to infection by Pythium ultimum causing apple replant disease. Can. J. Plant Pathol. 2016, 38, 483–491. [Google Scholar] [CrossRef]
- Zhu, Y.; Shao, J.; Zhou, Z.; Davis, R.E. Comparative transcriptome analysis reveals a performed defense system in apple root of a resistant genotype of G.935 in the absence of pathogen. Int. J. Plant Genom. 2017, 8950746. [Google Scholar] [CrossRef]
- Emmett, B.; Nelson, E.B.; Kessler, A.; Bauerle, T. Fine-root system development and susceptibility to pathogen colonization. Planta 2014, 239, 325–340. [Google Scholar] [CrossRef]
- Atucha, A.; Emmett, B.; Bauerle, T.L. Growth rate of fine root systems influences rootstock tolerance to replant disease. Plant Soil 2014, 376, 337–346. [Google Scholar] [CrossRef]
- Mazzola, M. Elucidation of the microbial complex having a causal role in the development of apple replant disease in Washington. Phytopathology 1998, 88, 930–938. [Google Scholar] [CrossRef]
- Hewavitharana, S.S.; Rudell, D.; Mazzola, M. Carbon source-dependent antifungal and nematicidal volatiles derived during anaerobic soil disinfestation. Eur. J. Plant Pathol. 2014, 140, 39–52. [Google Scholar] [CrossRef]
- Brown, J.; Davis, J.B.; Erickson, D.A.; Gosselin, T.K. Registration of ‘Pacific Gold’ oriental condiment mustard. Crop. Sci. 2004, 44, 2271–2272. [Google Scholar] [CrossRef]
- Brown, J.; Davis, J.B.; Erickson, D.A.; Brown, A.P.; Seip, L. Registration of ‘IdaGold’ mustard. Crop. Sci. 1997, 38, 541. [Google Scholar]
- Yepes, L.M.; Aldwinckle, H.S. Micropropagation of thirteen Malus cultivars and rootstocks, and effect of antibiotics on proliferation. Plant Growth Regul. 1994, 15, 55–67. [Google Scholar] [CrossRef]
- Gasic, K.; Hernandez, A.; Korban, S. RNA extraction from different apple tissues rich in polyphenols and polysaccharides for cDNA library construction. Plant Mol. Biol. Rep. 2004, 22, 437–438. [Google Scholar] [CrossRef]
- Velasco, R.; Zharkikh, A.; Affourtit, J.; Dhingra, A.; Cestaro, A.; Kalyanaraman, A.; Fontana, P.; Bhatnagar, S.K.; Troggio, M.; Pruss, D.; et al. The genome of the domesticated apple (Malus × domestica Borkh). Nature Genet. 2010, 42, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Lee, T.; Cheng, C.H.; Buble, K.; Zheng, P.; Yu, J.; Humann, J.; Ficklin, S.P.; Gasic, K.; Scott, K.; et al. 15 years of GDR: New data and functionality in the Genome Database for Rosaceae. Nucleic Acids Res. 2019, 47, D1137–D1145. [Google Scholar] [CrossRef]
- Andrews, S.; Fast, Q.C. A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 3 March 2021).
- Robinson, M.D.; Smyth, G.K. Small-sample estimation of negative binomial dispersion, with applications to sage data. Biostatistics 2008, 9, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Madden, T.; Coulouris, G.; Avagyan, V.; Ma, N.; Tao, T.; Agarwala, R. BLAST Command Line Applications User Manual. Bethesda (MD): National Center for Biotechnology Information (US). 2008. Available online: http://www.ncbi.nlm.nih.gov/books/NBK279674/ (accessed on 24 February 2021).
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Wojciech Szcześniak, M.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Alexa, A.; Rahnenfuhrer, J. Gene set enrichment analysis with topGO. In R Package Version 3.5.3; R Foundation for Statistical Computing: Vienna, Austria, 2007. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Martin, K.; Rygiewicz, P.T. Fungal-specific PCR primers developed for analysis of the ITS region of environmental DNA extracts. BMC Microbiol. 2005, 5, 28. [Google Scholar] [CrossRef]
- Vilgalys, R.; Hester, M. Rapid genetic identification and mapping of enzymatically amplified ribosomal DNA from several Cryptococcus species. J. Bacteriol. 1990, 172, 4238–4246. [Google Scholar] [CrossRef]
- Øvreas, L.; Torsvik, V. Microbial diversity and community structure in two different agricultural soil communities. Microb. Ecol. 1998, 36, 303–315. [Google Scholar] [CrossRef]
- Robertson, C.E.; Harris, J.K.; Wagner, B.D.; Granger, D.; Browne, K.; Tatem, B.; Feazel, L.M.; Park, K.; Pace, N.R.; Frank, D.N. Explicet: Graphical user interface software for metadata-driven management, analysis and visualization of microbiome data. Bioinformatics 2013, 29, 3100–3101. [Google Scholar] [CrossRef] [PubMed]
- Wagner, B.D.; Roberson, C.E.; Harris, J.K. Application of two-part statistic for comparison of sequence variant counts. PLoS ONE 2011, 6, e20296. [Google Scholar] [CrossRef] [PubMed]
- Kieber, J.J.; Rothenberg, M.; Roman, G.; Feldmann, K.A.; Ecker, J.R. CTR1, a negative regulator of the ethylene response pathway in Arabidopsis, encodes a member of the Raf family of protein kinases. Cell 1993, 72, 427–441. [Google Scholar] [CrossRef]
- Shinya, T.; Yamaguchi, K.; Desaki, Y.; Yamada, K.; Narisawa, T.; Kobayashi, Y.; Maeda, K.; Suzuki, M.; Tanimoto, T.; Takeda, J.; et al. Selective regulation of the chitin-induced defense response by the Arabidopsis receptor-like cytoplasmic kinase PBL27. Plant J. 2014, 79, 56–66. [Google Scholar] [CrossRef]
- Turner, J.G.; Ellis, C.; Devoto, A. The jasmonate signal pathway. Plant Cell 2002, 14, 153–164. [Google Scholar] [CrossRef]
- Wang, K.L.-C.; Li, H.; Ecker, J.R. Ethylene biosynthesis and signaling networks. Plant Cell 2002, 14, 131–151. [Google Scholar] [CrossRef]
- Devoto, A.; Turner, J.G. Regulation of jasmonate-mediated plant responses in Arabidopsis. Ann. Bot. 2003, 92, 329–337. [Google Scholar] [CrossRef]
- Okubara, P.; Paulitz, T. Root defense responses to fungal pathogens: A molecular perspective. Plant Soil 2005, 274, 215–226. [Google Scholar] [CrossRef]
- Pauwels, L.; Barbero, G.F.; Geerinck, J.; Tilleman, S.; Grunewald, W.; Pérez, A.C.; Chico, J.M.; Bossche, R.V.; Sewell, J.; Gil, E.; et al. NINJA connects the corepressor TOPLESS to jasmonate signaling. Nature 2010, 464, 788–791. [Google Scholar] [CrossRef]
- Zeilmaker, T.; Ludwig, N.R.; Elberse, J.; Seidl, M.F.; Berke, L.; Van Doorn, A.; Schuurink, R.C.; Snel, B.; Van den Ackerveken, G. Downy mildew resistant 6 and DMR6-like oxygenase 1 are partially redundant but distinct suppressors of immunity in Arabidopsis. Plant J. 2015, 81, 210–222. [Google Scholar] [CrossRef]
- De Geyter, N.; Gholami, A.; Goormachtig, S.; Goossens, A. Transcriptional machineries in jasmonate-elicited plant secondary metabolism. Trends Plant Sci. 2012, 17, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, U.A.; Maxwell, C.A.; Joseph, C.M.; Phillips, D.A. Chrysoeriol and luteolin released from alfalfa seeds induce nod genes in Rhizobium meliloti. Plant Physiol. 1990, 92, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Peer, W.A.; Murphy, A.S. Flavonoids and auxin transport: Modulators or regulators? Trends Plant Sci. 2007, 12, 556–563. [Google Scholar] [CrossRef]
- Mierziak, J.; Kostyn, K.; Kulma, A. Flavonoids as important molecules of plant interactions with the environment. Molecules 2014, 19, 16240–16265. [Google Scholar] [CrossRef]
- Goritschnig, S.; Weihmann, T.; Zhang, Y.; Fobert, P.; McCourt, P.; Li, X. A novel role for protein farnesylation in plant innate immunity. Plant Physiol. 2008, 148, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Mazzola, M.; Brown, J.; Zhao, X.; Izzo, A.D.; Fazio, G. Interaction of Brassicaceous seed meal and apple rootstock on recovery of Pythium spp. and Pratylenchus penetrans from roots grown in replant soils. Plant Dis. 2009, 93, 51–57. [Google Scholar] [CrossRef]
- Göhre, V.; Robatzek, S. Breaking the barriers: Microbial effector molecules subvert plant immunity. Annu. Rev. Phytopathol. 2008, 46, 189–215. [Google Scholar] [CrossRef]
- Schwessinger, B.; Zipfel, C. News from the frontline: Recent insights into PAMP-triggered immunity in plants. Curr. Opin. Plant Biol. 2008, 11, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, K.; Sato, M.; Glazebrook, J.; Cohen, J.D.; Katagiri, F. Interplay between MAMP-triggered and SA-mediated defense responses. Plant J. 2008, 53, 763–775. [Google Scholar] [CrossRef]
- Nürnberger, T.; Kemmerling, B. Pathogen-associated molecular patterns (PAMP) and PAMP-triggered immunity. Annu. Plant Rev. 2009, 34, 16–47. [Google Scholar]
- Pieterse, C.M.J.; Leon-Reyes, A.; Van der Ent, S.; Van Wees, S.C.M. Networking by small-molecules hormones in plant immunity. Nat. Chem. Biol. 2009, 5, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Spoel, S.H.; Dong, X. Making sense of hormone crosstalk during plant immune responses. Cell Host Microbe 2008, 3, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Pajerowska-Mukhtar, K.; Hendrickson Culler, A.; Dong, X. Salicylic acid inhibits pathogen growth in plants through repression of the auxin signaling pathway. Curr. Biol. 2007, 17, 1784–1790. [Google Scholar] [CrossRef] [PubMed]
- Navarro, L.; Bari, R.; Achard, P.; Lison, P.; Nemri, A.; Harberd, N.P.; Jones, J.D.G. DELLAs control plant immune responses by modulating the balance of jasmonic acid and salicylic acid signaling. Curr. Biol. 2008, 18, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, M.; Ishikawa, A.; Jikumaru, Y.; Seki, M.; Umezawa, T.; Asami, T.; Maruyama-Nakashita, A.; Kudo, T.; Shinozaki, K.; Yoshida, S.; et al. Antagonistic interaction between systemic acquired resistance and the abscisic acid-mediated abiotic stress response in Arabidopsis. Plant Cell 2008, 20, 1678–1692. [Google Scholar] [CrossRef]
- Campos, M.L.; de Almeida, M.; Rossi, M.L.; Martinelli, A.P.; Litholdo, C.G.; Figueira, A.; Rampelotti-Ferreira, F.T.; Vendramim, J.D.; Benedito, V.A.; Peres, L.E.P. Brassinosteroids interact negatively with jasmonates in the formation of anti-herbivory traits in tomato. J. Exp. Bot. 2009, 60, 4346–4360. [Google Scholar] [CrossRef] [PubMed]
- de Torres-Zabala, M.; Bennett, M.H.; Truman, W.H.; Grant, M.R. Antagonism between salicylic and abscisic acid reflects early host-pathogen conflict and moulds plant defence responses. Plant J. 2009, 59, 375–386. [Google Scholar] [CrossRef]
- De Vleesschauwer, D.; Yang, Y.; Cruz, C.V.; Höfte, M. Abscisic acid-induced resistance against the brown spot pathogen Cochliobolus miyabeanus in rice involves MAP kinase-mediated repression of ethylene signaling. Plant Physiol. 2010, 152, 2036–2052. [Google Scholar] [CrossRef]
- Jiang, C.J.; Shimono, M.; Sugano, S.; Kojima, M.; Yazawa, K.; Yoshida, R.; Inoue, H.; Hayashi, N.; Sakakibara, H.; Takatsuji, H. Abscisic acid interacts antagonistically with salicylic acid signaling pathway in rice-Magnaporthe grisea interaction. Mol. Plant Microbe Interact. 2010, 23, 791–798. [Google Scholar] [CrossRef]
- Tian, D.; Traw, M.B.; Chen, J.Q.; Kreitman, M.; Bergelson, J. Fitness costs of R-gene-mediated resistance in Arabidopsis thaliana. Nature 2003, 423, 74–77. [Google Scholar] [CrossRef]
- Van Hulten, M.; Pelser, M.; van Loon, L.C.; Pieterse, C.M.J.; Ton, J. Costs and benefits of priming for defense in Arabidopsis. Proc. Natl. Acad. Sci. USA 2006, 103, 5602–5607. [Google Scholar] [CrossRef]
- Sani, E.; Herzyk, P.; Perrella, G.; Colot, V.; Amtmann, A. Hyperosmotic priming of Arabidopsis seedlings establishes a long-term somatic memory accompanied by specific changes of the epigenome. Genome Biol. 2013, 14, R59. [Google Scholar] [CrossRef]
- Mazzola, M.; Brown, J. Efficacy of Brassicaceous seed meal formulations for the control of apple replant disease in organic and conventional orchard production systems. Plant Dis. 2010, 94, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Mazzola, M.; Zhao, X. Brassica juncea seed meal particle size influences chemistry but not soil biology-based suppression of individual agents inciting apple replant disease. Plant Soil 2010, 337, 313–324. [Google Scholar] [CrossRef]
- Mazzola, M.; Manici, L.M. Apple replant disease: Role of microbial ecology in cause and control. Annu. Rev. Phytopathol. 2012, 50, 45–65. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Hwang, B.K. The pepper MLO gene, CaMLO2, is involved in the susceptibility cell-death response and bacterial and oomycete proliferation. Plant J. 2012, 72, 843–855. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.A.; von Roepenack-Lahaye, E.; Parr, A.; Daniels, M.J.; Dow, J.M. Prior exposure to lipopolysaccharide potentiates expression of plant defenses in response to bacteria. Plant J. 2002, 29, 487–495. [Google Scholar] [CrossRef]
- Conrath, U.; Becker, G.J.; Langenbach, C.J.; Jaskiewicz, M.R. Priming for enhanced defense. Annu. Rev. Phytopathol. 2015, 53, 97–119. [Google Scholar] [CrossRef]
- Hilker, M.; Schwachtje, J.; Baier, M.; Balazadeh, S.; Bäurle, I.; Geiselhardt, S.; Hincha, D.K.; Kunze, R.; Mueller-Roeber, B.; Rillig, M.C.; et al. Priming and memory of stress responses in organisms lacking a nervous system. Biol. Rev. 2015, 4, 1118–1133. [Google Scholar] [CrossRef]
- Oostendorp, M.; Kunz, W.; Dietrich, B.; Staub, T. Induced disease resistance in plants by chemicals. Eur. J. Plant Pathol. 2001, 107, 19–28. [Google Scholar] [CrossRef]
- Pastor, V.; Luna, E.; Mauch-Mani, B.; Ton, J.; Flors, V. Primed plants do not forget. Environ. Exp. Bot. 2012, 94, 46–56. [Google Scholar] [CrossRef]
- Balmer, A.; Pastor, V.; Gamir, J.; Flors, V.; Mauch-Mani, B. The ‘prime-ome’: Towards a holistic approach to priming. Trends Plant Sci. 2015, 20, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Pieterse, C.M.J.; Zamioudis, C.; Berendsen, R.J.; Weller, D.M.; Van Wees, S.C.M.; Bakker, P.A.H.M. Induced systemic resistance by beneficial microbes. Annu. Rev. Phytopathol. 2014, 52, 347–375. [Google Scholar] [CrossRef]
- Galletti, S.; Sala, E.; Leoni, O.; Burzi, P.L.; Cerato, C. Trichoderma spp. tolerance to Brassica carinata seed meal for a combined use in biofumigation. Biol. Control 2008, 45, 319–327. [Google Scholar] [CrossRef]
- Kobayashi, D.Y.; Reedy, R.M.; Palumbo, J.D.; Zhou, J.M.; Yuen, G.Y. A clp gene homologue belonging to the crp gene family globally regulates lytic enzyme production, antimicrobial activity, and biological control activity expressed by Lysobacter enzymogenes strain C3. Appl. Environ. Microbiol. 2005, 71, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, D.Y.; Yuen, G.Y. The role of clp-regulated factors in antagonism against Magnaporthe poae and biological control of summer patch disease of Kentucky bluegrass by Lysobacter enzymogenes C3. Can. J. Microbiol. 2005, 51, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Sun, Q.; Zhao, C.; Wen, D.; Tang, X. Aerobic degradation of pyridine by a new bacterial strain, Shinella zoogloeoides BC026. J. Ind. Microbiol. Biotechnol. 2009, 36, 1391–1400. [Google Scholar] [CrossRef] [PubMed]
- Camargo, F.A.O.; Bento, F.M.; Okeke, B.C.; Frankenberger, W.T. Hexavalent chromium reduction by an actinomycete, Arthrobacter crystallopoietes ES 32. Biol. Trace Elem. Res. 2003, 97, 183–194. [Google Scholar] [CrossRef]
- Van Horn, C.; Somera, T.; Mazzola, M. Comparative analysis of the rhizosphere and endophytic microbiomes across apple rootstock genotypes in replant orchard soils. Phytobiomes J. 2021. [Google Scholar] [CrossRef]
- Lanfranco, L.; Bonfante, P.; Genre, A. The mutualistic interaction between plants and arbuscular mycorrhizal fungi. Microbiol. Spectrum 2016, 4. [Google Scholar] [CrossRef]
- Fung, R.W.M.; Gonzalo, M.; Fekete, C.; Kovacs, L.G.; He, Y.; Marsh, E.; McIntyre, L.M.; Schachtman, D.P.; Qiu, W. Powdery Mildew Induces Defense-Oriented Reprogramming of the Transcriptome in a Susceptible but not in a Resistant Grapevine. Plant Physiol. 2008, 146, 236–249. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sampling Timepoint | Apple Rootstock Genotype | Comparisons z | No. of DEG | No. of Upregulated y | No. of Downregulated |
|---|---|---|---|---|---|
| 48 h | M.26 | M.26-48 h-NTC vs. M.26 48 h-SM | 7 | 6 | 1 |
| G.210 | G.210-48 h-NTC vs. G.210-48 h-SM | 6 | 2 | 4 | |
| 72 h | M.26 | M.26-72 h-NTC vs. M.26 72 h-SM | 122 | 64 | 58 |
| G.210 | G.210-72 h-NTC vs. G.210-72 h-SM | 177 | 106 | 71 | |
| 7 d | M.26 | M.26-7 d-NTC vs. M.26-7 d-SM | 10 | 6 | 4 |
| G.210 | G.210-7 d-NTC vs. G.210-7 d-SM | 232 | 33 | 199 |
| Gene ID in GDR z | Protein Function Annotation y | Log2(FC) x | Timepoint |
|---|---|---|---|
| MDP0000196909 | TMV resistance protein N | 2.03 | 48 h |
| MDP0000711750 | CBL-interacting serine/threonine-protein kinase 12 | −2.61 | 48 h |
| MDP0000306089 | Calcium-binding EF-hand family protein | 2.33 | 72 h |
| MDP0000283789 | Disease resistance protein RGA3 | 1.94 | 72 h |
| MDP0000162375 | Disease resistance protein | 2.31 | 72 h |
| MDP0000683814 | Ethylene-responsive transcription factor RAP2-4 | 2.49 | 72 h |
| MDP0000278380 | Pathogenesis-related protein 5 | 2.88 | 72 h |
| MDP0000198976 | Serine/threonine-protein kinase CTR1 | 5.56 | 72 h |
| MDP0000273596 | Serine/threonine-protein kinase PBL27 | 2.17 | 72 h |
| MDP0000268026 | TMV resistance protein N | 3.33 | 72 h |
| MDP0000191786 | 4-coumarate-CoA ligase-like 5 | −5.46 | 72 h |
| MDP0000185169 | Benzyl alcohol O-benzoyltransferase | −5.45 | 72 h |
| MDP0000189033 | Ethylene-responsive transcription factor 2 | −4.13 | 72 h |
| MDP0000234530 | MLO-like protein 6 | −10.06 | 72 h |
| MDP0000266840 | NADP-dependent alkenal double bond reductase P2 | −2.66 | 72 h |
| MDP0000231619 | Probable pectinesterase/pectinesterase inhibitor 7 | −5.13 | 72 h |
| MDP0000318604 | Probable S-adenosylmethionine-dependent methyltransferase | −2.85 | 72 h |
| MDP0000675952 | Probable S-adenosylmethionine-dependent methyltransferase | −4.2 | 72 h |
| MDP0000931970 | Receptor-like protein kinase THESEUS 1 | −2.38 | 72 h |
| MDP0000755567 | Indole-3-acetic acid-induced protein ARG2 | 2.29 | 7 d |
| Gene ID in GDR z | Protein Function Annotation y | Log2(FC) x | Timepoint |
|---|---|---|---|
| MDP0000319359 | Calcium-dependent lipid-binding (CaLB domain) family protein | 6.41 | 48 h |
| MDP0000296303 | Abscisic acid-insensitive 5-like protein | 2.17 | 72 h |
| MDP0000306089 | Calcium-binding EF-hand family protein | 2.51 | 72 h |
| MDP0000216765 | CBL-interacting serine/threonine-protein kinase 9 | 2.19 | 72 h |
| MDP0000127009 | Disease resistance protein RGA2 | 1.82 | 72 h |
| MDP0000137225 | Disease resistance protein | 3.02 | 72 h |
| MDP0000640906 | Disease resistance protein | 2.95 | 72 h |
| MDP0000162375 | Disease resistance protein | 2.22 | 72 h |
| MDP0000228070 | 9-cis-epoxycarotenoid dioxygenase | 8.60 | 72 h |
| MDP0000929213 | 9-cis-epoxycarotenoid dioxygenase | 3.68 | 72 h |
| MDP0000484601 | GTP-binding protein brassinazole insensitive pale green 2 | 1.89 | 72 h |
| MDP0000290028 | LRR receptor-like serine/threonine-protein kinase GSO2 | 4.90 | 72 h |
| MDP0000119875 | Ninja-family protein AFP2 | 2.37 | 72 h |
| MDP0000875341 | Ninja-family protein AFP3 | 4.79 | 72 h |
| MDP0000588503 | Pathogenesis-related protein 10 | 2.18 | 72 h |
| MDP0000234689 | S-type anion channel SLAH3 | 3.23 | 72 h |
| MDP0000268026 | TMV resistance protein N | 2.59 | 72 h |
| MDP0000657441 | WRKY transcription factor 68 | 2.47 | 72 h |
| MDP0000166337 | Abscisic acid 8′-hydroxylase 4 | −9.91 | 72 h |
| MDP0000197409 | Carotenoid cleavage dioxygenase 7 | −3.39 | 72 h |
| MDP0000139334 | Carotenoid cleavage dioxygenase 7 | −3.47 | 72 h |
| MDP0000652331 | Cytochrome b561 and DOMON | −2.24 | 72 h |
| MDP0000138669 | Ethylene-responsive transcription factor RAP2-11 | −6.41 | 72 h |
| MDP0000295857 | Fusaric acid resistance protein | −4.77 | 72 h |
| MDP0000239643 | MLO-like protein 6 | −3.88 | 72 h |
| MDP0000266930 | NADPH-dependent oxidoreductase 2-alkenal reductase | −3.17 | 72 h |
| MDP0000872370 | NADPH-dependent oxidoreductase 2-alkenal reductase | −3.00 | 72 h |
| MDP0000675952 | Probable S-adenosylmethionine-dependent methyltransferase | −5.92 | 72 h |
| MDP0000261194 | Probable S-adenosylmethionine-dependent methyltransferase | −6.22 | 72 h |
| MDP0000729108 | Pathogenesis-related protein 10 | −11.18 | 72 h |
| MDP0000296675 | Protein LURP-one-related 4 | −∞ | 72 h |
| MDP0000930978 | Protein SAR deficient 1 | −10.33 | 72 h |
| MDP0000139821 | Disease resistance protein | −2.48/−2.32 | 72 h/7 d |
| MDP0000309171 | Disease resistance protein | −2.82/−3.34 | 72 h/7 d |
| MDP0000792101 | Wound-induced protein | −5.45/−5.61 | 72 h/7 d |
| MDP0000275716 | Cellulose synthase-like protein B4 | −3.35 | 7 d |
| MDP0000411073 | Cellulose synthase-like protein E2 | −3.91 | 7 d |
| MDP0000221346 | Cellulose synthase-like protein G2 | −3.33 | 7 d |
| MDP0000314000 | Cysteine-rich receptor-like protein kinase 29 | −3.43 | 7 d |
| MDP0000685403 | Cysteine-rich receptor-like protein kinase 29 | −10.06 | 7 d |
| MDP0000927688 | Cysteine-rich repeat secretory protein 38 | −7.61 | 7 d |
| MDP0000416561 | Cysteine-rich repeat secretory protein 38 | −8.13 | 7 d |
| MDP0000842997 | ABC transporter C family member 3 | −2.79 | 7 d |
| MDP0000865714 | Disease resistance protein | −16.43 | 7 d |
| MDP0000706371 | Disease resistance protein | −2.73 | 7 d |
| MDP0000126026 | Disease resistance protein | −3.37 | 7 d |
| MDP0000224187 | Disease resistance RPP13-like protein 4 | −3.27 | 7 d |
| MDP0000193383 | Endochitinase EP3 | −10.32 | 7 d |
| MDP0000143462 | Glycine-rich cell wall structural protein | −13.37 | 7 d |
| MDP0000260110 | Major allergen Mal d 1 | −3.06 | 7 d |
| MDP0000831518 | Major allergen Mal d 1 | −4.14 | 7 d |
| MDP0000831519 | Major allergen Mal d 1 | −4.46 | 7 d |
| MDP0000295542 | Major allergen Mal d 1 | −5.61 | 7 d |
| MDP0000864747 | Major allergen Mal d 1 | −7.23 | 7 d |
| MDP0000313454 | Major allergen Pru ar 1 | −9.78 | 7 d |
| MDP0000124524 | Pathogenesis-related protein STH-2 | −6.88 | 7 d |
| MDP0000119517 | Pathogenesis-related protein STH-2 | −7.37 | 7 d |
| MDP0000221319 | Pectin acetylesterase 8 | −7.66 | 7 d |
| MDP0000249386 | Polygalacturonase | 3.07 | 7 d |
| MDP0000755567 | Indole-3-acetic acid-induced protein ARG2 | 2.39 | 7 d |
| MDP0000683814 | Ethylene-responsive transcription factor RAP2-4 | 2.17 | 7 d |
| MDP0000282421 | U-box domain-containing protein 9 | 1.94 | 7 d |
| MDP0000897962 | Probable LRR receptor-like serine/threonine-protein kinase | −2.68 | 7 d |
| MDP0000229843 | Protein DMR6-like oxygenase | −5.85 | 7 d |
| MDP0000576922 | Protein DMR6-like oxygenase | −3.28 | 7 d |
| MDP0000218810 | Protein DMR6-like oxygenase | −3.34 | 7 d |
| MDP0000593536 | Protein DMR6-like oxygenase | −3.67 | 7 d |
| MDP0000566057 | Protein DMR6-like oxygenase | −4.53 | 7 d |
| MDP0000147913 | Protein DMR6-like oxygenase | −5.70 | 7 d |
| MDP0000694318 | Rust resistance kinase Lr10 | −5.93 | 7 d |
| MDP0000156351 | U-box domain-containing protein 21 | −4.99 | 7 d |
| MDP0000206106 | Wall-associated receptor kinase | −7.21 | 7 d |
| Gene ID in GDR z | Log2(FC) y in | Biological Process x | Protein Function Annotation w | |
|---|---|---|---|---|
| M.26 | G.210 | |||
| MDP0000306089 | 2.33 | 2.51 | Plant-pathogen interaction | Calcium-binding EF-hand family protein |
| MDP0000162375 | 2.31 | 2.22 | Disease resistance | Disease resistance protein |
| MDP0000268026 | 3.33 | 2.59 | Protein binding | TMV resistance protein N |
| MDP0000302115 | 4.12 | 3.92 | N/A v | Disease resistance protein |
| MDP0000570102 | 2.09 | 2.51 | Flavonoid biosynthetic process | N/A |
| MDP0000755567 | 2.29 | 2.39 | Response to stress | Late embryogenesis abundant like-5 (ATLEA5) |
| MDP0000385497 | 2.14 | 2.12 | Response to stress | Late embryogenesis abundant like-5 (ATLEA5) |
| MDP0000234689 | 2.62 | 3.23 | Response to salt stress and water deprivation | SLAC1 homologue 3 (SLAH3) |
| MDP0000190029 | 2.06 | 2.06 | DNA binding | Nuclear factor Y, Subunit C2 |
| MDP0000581293 | 2.76 | 2.12 | DNA binding | REVEILLE 1-like |
| MDP0000153123 | 1.97 | 2.40 | Metal ion binding | Heme-binding protein |
| MDP0000821892 | 1.91 | 2.34 | Metal ion binding | Metallothionein |
| MDP0000212510 | 2.83 | 2.23 | Sugar transport | Monosaccharid transporter 2 |
| MDP0000281884 | 2.70 | 2.56 | Sugar transport | Monosaccharid transporter 2 |
| MDP0000403033 | 2.38 | 2.61 | Ion channel | Chloride channel C |
| MDP0000307717 | 3.06 | 3.60 | Lipid transfer | Lipid-transfer protein |
| MDP0000270246 | 2.45 | 2.13 | Protein insertion into membrane | Membrane protein |
| MDP0000322201 | 2.69 | 2.44 | N-acetyltransferase | Acyl-CoA N-acyltransferases (NAT) superfamily protein |
| MDP0000210084 | 2.77 | 2.94 | Involved in leaf vasculature patterning | Glycine-rich protein DOT1-like |
| MDP0000827125 | 2.59 | 3.60 | N/A | N/A |
| MDP0000286637 | 2.62 | 3.24 | Transport | Integral membrane HPP family protein |
| MDP0000239643 | −3.99 | −3.88 | Cell death | MLO family protein |
| MDP0000171573 | −3.09 | −2.63 | Fructose and mannose metabolism | GroES-like zinc-binding alcohol dehydrogenase family protein |
| MDP0000145305 | −2.81 | −3.75 | DNA replication initiation | Dienelactone hydrolase |
| MDP0000675952 | −4.20 | −5.92 | Methyltransfer | Methyltransferase |
| MDP0000258061 | −6.21 | −6.46 | Oxidation-reduction process | 2-oxoglutarate (2OG) and Fe(II)-dependent oxygenase superfamily protein |
| MDP0000139947 | −6.11 | −5.058 | Protein phosphorylation | Protein kinase THE1 |
| MDP0000870778 | −2.14 | −2.02 | Protein phosphorylation | Receptor-like protein kinase FERONIA |
| MDP0000252274 | -∞ | −2.61 | Isoprenoid biosynthetic process | Terpenoid synthase |
| MDP0000295857 | −5.02 | −4.77 | Phosphate ion transmembrane transport | P-hydroxybenzoic acid efflux pump subunit |
| MDP0000464148 | −3.01 | −3.18 | Integral component of membrane | Transmembrane protein |
| MDP0000816071 | −7.76 | −6.15 | N/A | LOB domain-containing protein |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Somera, T.S.; Hargarten, H.; Honaas, L.; Mazzola, M. Comparative Analysis of the Apple Root Transcriptome as Affected by Rootstock Genotype and Brassicaceae Seed Meal Soil Amendment: Implications for Plant Health. Microorganisms 2021, 9, 763. https://doi.org/10.3390/microorganisms9040763
Wang L, Somera TS, Hargarten H, Honaas L, Mazzola M. Comparative Analysis of the Apple Root Transcriptome as Affected by Rootstock Genotype and Brassicaceae Seed Meal Soil Amendment: Implications for Plant Health. Microorganisms. 2021; 9(4):763. https://doi.org/10.3390/microorganisms9040763
Chicago/Turabian StyleWang, Likun, Tracey S. Somera, Heidi Hargarten, Loren Honaas, and Mark Mazzola. 2021. "Comparative Analysis of the Apple Root Transcriptome as Affected by Rootstock Genotype and Brassicaceae Seed Meal Soil Amendment: Implications for Plant Health" Microorganisms 9, no. 4: 763. https://doi.org/10.3390/microorganisms9040763
APA StyleWang, L., Somera, T. S., Hargarten, H., Honaas, L., & Mazzola, M. (2021). Comparative Analysis of the Apple Root Transcriptome as Affected by Rootstock Genotype and Brassicaceae Seed Meal Soil Amendment: Implications for Plant Health. Microorganisms, 9(4), 763. https://doi.org/10.3390/microorganisms9040763