
Culture-Independent Genotyping, Virulence and Antimicrobial Resistance Gene Identification of Staphylococcus aureus from Orthopaedic Implant-Associated Infections



Abstract
1. Introduction
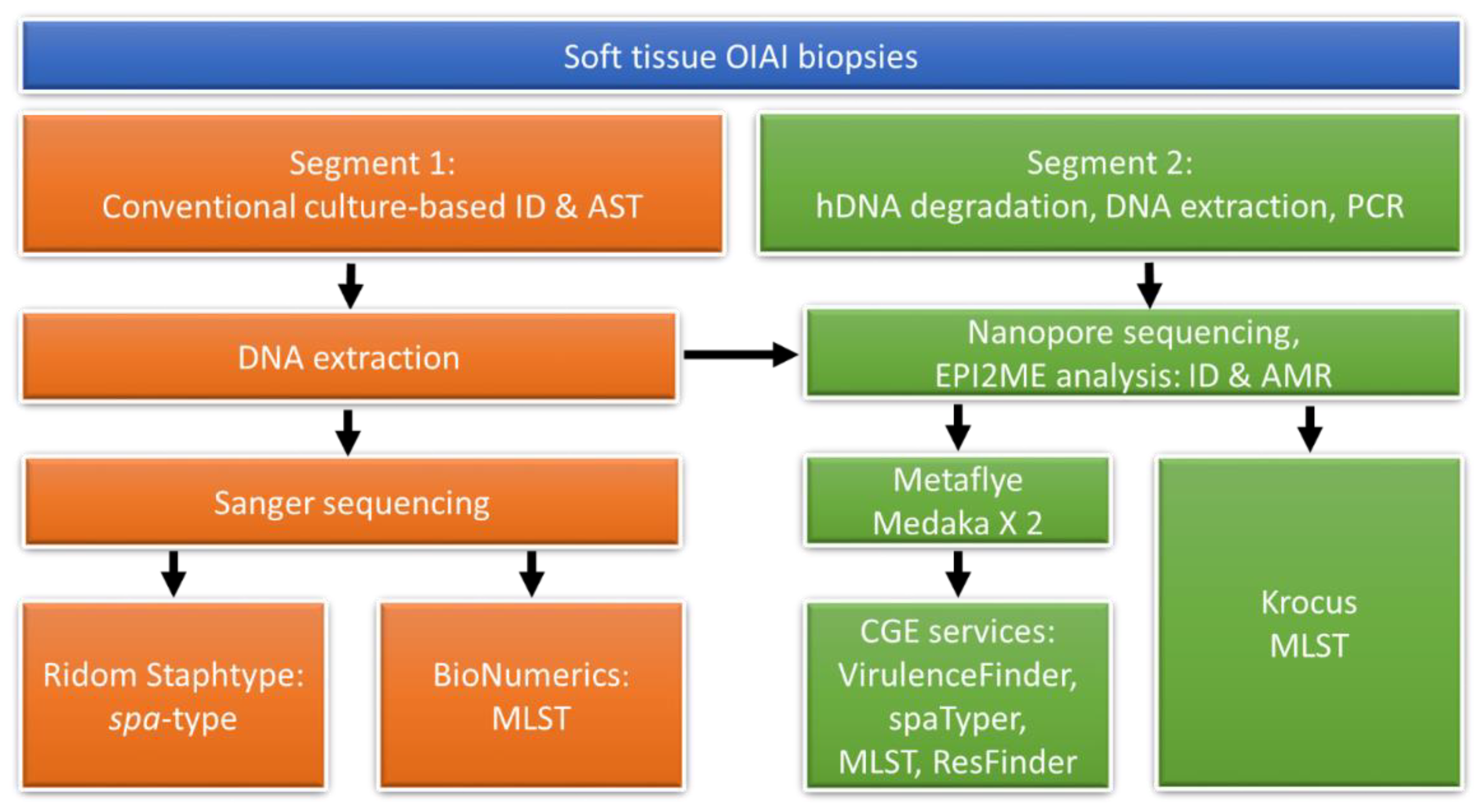
2. Materials and Methods
2.1. DNA Extraction
2.2. Spa-Typing and MLST
2.3. Nanopore Sequencing
2.4. Nanopore Sequencing Data Analysis
3. Results
3.1. Metaflye Assemblies
3.2. Spa-Type
3.3. MLST
3.4. Virulence Genes
3.5. Resistance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Karachalios, T.; Koutalos, A.; Komnos, G. Management Strategies for Infected Total Hip Arthroplasty. A Critical Appreciation of Problems and Techniques. HIP Int. 2014, 24, S44–S47. [Google Scholar] [CrossRef]
- Siljander, M.P.; Sobh, A.H.; Baker, K.C.; Baker, E.A.; Kaplan, L.M. Multidrug-Resistant Organisms in the Setting of Periprosthetic Joint Infection—Diagnosis, Prevention, and Treatment. J. Arthroplast. 2018, 33, 185–194. [Google Scholar] [CrossRef]
- Parvizi, J.; Zmistowski, B.; Berbari, E.F.; Bauer, T.W.; Springer, B.D.; Della Valle, C.J.; Garvin, K.L.; Mont, M.A.; Wongworawat, M.D.; Zalavras, C.G. New Definition for Periprosthetic Joint Infection: From the Workgroup of the Musculoskeletal Infection Society. Clin. Orthop. Relat. Res. 2011, 469, 2992–2994. [Google Scholar] [CrossRef]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the Human Intestinal Microbial Flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef]
- Noone, J.C.; Helmersen, K.; Leegaard, T.M.; Skråmm, I.; Aamot, H.V. Rapid Diagnostics of Orthopaedic Implant-Associated Infections Using Nanopore Shotgun Metagenomic Sequencing on Tissue Biopsies. Microorganisms 2021, 9, 97. [Google Scholar] [CrossRef] [PubMed]
- Arciola, C.R.; Campoccia, D.; Montanaro, L. Implant Infections: Adhesion, Biofilm Formation and Immune Evasion. Nat. Rev. Microbiol. 2018, 16, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Kolmogorov, M.; Bickhart, D.M.; Behsaz, B.; Gurevich, A.; Rayko, M.; Shin, S.B.; Kuhn, K.; Yuan, J.; Polevikov, E.; Smith, T.P.L.; et al. MetaFlye: Scalable Long-Read Metagenome Assembly Using Repeat Graphs. Nat. Methods 2020, 17, 1103–1110. [Google Scholar] [CrossRef]
- Montanaro, L.; Ravaioli, S.; Ruppitsch, W.; Campoccia, D.; Pietrocola, G.; Visai, L.; Speziale, P.; Allerberger, F.; Arciola, C.R. Molecular Characterization of a Prevalent Ribocluster of Methicillin-Sensitive Staphylococcus Aureus from Orthopedic Implant Infections. Correspondence with MLST CC30. Front. Cell. Infect. Microbiol. 2016, 6. [Google Scholar] [CrossRef]
- Skramm, I.; Moen, A.E.F.; Asbjorn, A.; Bukholm, G. Surgical Site Infections in Orthopaedic Surgery. J. Bone Jt. Surg. Am. 2014, 96, 882–888. [Google Scholar]
- Thammavongsa, V.; Kim, H.K.; Missiakas, D.; Schneewind, O. Staphylococcal Manipulation of Host Immune Responses. Nat. Rev. Microbiol. 2015, 13, 529–543. [Google Scholar] [CrossRef] [PubMed]
- Tuchscherr, L.; Pöllath, C.; Siegmund, A.; Deinhardt-Emmer, S.; Hoerr, V.; Svensson, C.M.; Figge, M.T.; Monecke, S.; Löffler, B. Clinical S. Aureus Isolates Vary in Their Virulence to Promote Adaptation to the Host. Toxins 2019, 11, 135. [Google Scholar] [CrossRef] [PubMed]
- Aamot, H.V.; Noone, J.C.; Skråmm, I.; Truls, M. Are Conventional Microbiological Diagnostics Sufficiently Expedient in the Era of Rapid Diagnostics? Evaluation of Conventional Microbiological Diagnostics of Orthopedic Implant- Associated Infections (OIAI) Are Conventional Microbiological Diagnostics. Acta Orthop. 2020. [Google Scholar] [CrossRef]
- Pedersen, S.K.; Wagenaar, J.A.; Vigre, H.; Roer, L.; Mikoleit, M.; Aidara-Kane, A.; Cawthorne, A.L.; Aarestrup, F.M.; Hendriksen, R.S. Proficiency of WHO Global Foodborne Infections Network External Quality Assurance System Participants in the Identification and Susceptibility Testing of Thermo-Tolerant Campylobacter Spp. from 2003–2012. bioRxiv 2018, 56, 1–9. [Google Scholar] [CrossRef]
- Hendriksen, R.S.; Seyfarth, A.M.; Jensen, A.B.; Whichard, J.; Karlsmose, S.; Joyce, K.; Mikoleit, M.; Delong, S.M.; Weill, F.X.; Aidara-Kane, A.; et al. Results of Use of Who Global Salm-Surv External Quality Assurance System for Antimicrobial Susceptibility Testing of Salmonella Isolates from 2000 to 2007. J. Clin. Microbiol. 2009, 47, 79–85. [Google Scholar] [CrossRef]
- Diekema, D.J.; Lee, K.; Raney, P.; Herwaldt, L.A.; Doern, G.V.; Tenover, F.C. Accuracy and Appropriateness of Antimicrobial Susceptibility Test Reporting for Bacteria Isolated from Blood Cultures. J. Clin. Microbiol. 2004, 42, 2258–2260. [Google Scholar] [CrossRef] [PubMed]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for Predictions of Phenotypes from Genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef]
- Aamot, H.V.; Johnsen, B.O.; Skråmm, I. Rapid Diagnostics of Orthopedic Implant-Associated Infections Using Unyvero ITI Implant and Tissue Infection Application Is Not Optimal for Staphylococcus Species Identification. BMC Res. Notes 2019, 12, 725. [Google Scholar] [CrossRef] [PubMed]
- Helmersen, K.; Aamot, H.V. DNA Extraction of Microbial DNA Directly from Infected Tissue: An Optimized Protocol for Use in Nanopore Sequencing. Sci. Rep. 2020, 10, 2985. [Google Scholar] [CrossRef] [PubMed]
- Blomfeldt, A.; Jørgensen, S.B.; Helmersen, K.; Eskonsipo, P.K.J.; Aamot, H.V. Is Increased Staphylococcus Aureus Colonisation during Military Service Caused by Specific Genotypes? Molecular Examination of Long-Term Carriage in a Prospective Cohort Study. APMIS 2020, 7, 170–177. [Google Scholar] [CrossRef]
- Enright, M.C.; Day, N.P.J.; Davies, C.E.; Peacock, S.J.; Spratt, B.G. Multilocus Sequence Typing for Characterization of Methicillin-Resistant and Methicillin-Susceptible Clones of Staphylococcus Aureus. J. Clin. Microbiol. 2000, 38, 1008–1015. [Google Scholar] [CrossRef]
- Page, A.J.; Keane, J.A. Rapid Multi-Locus Sequence Typing Direct from Uncorrected Long Reads Using Krocus. PeerJ 2018, 2018, e5233. [Google Scholar] [CrossRef] [PubMed]
- Stefani, S.; Chung, D.R.; Lindsay, J.A.; Friedrich, A.W.; Kearns, A.M.; Westh, H.; MacKenzie, F.M. Meticillin-Resistant Staphylococcus Aureus (MRSA): Global Epidemiology and Harmonisation of Typing Methods. Int. J. Antimicrob. Agents 2012, 39, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Kono, N.; Arakawa, K. Nanopore Sequencing: Review of Potential Applications in Functional Genomics. Dev. Growth Differ. 2019, 61, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Kurlenda, J.; Grinholc, M. Alternative Therapies in Staphylococcus Aureus Diseases. Acta Biochim. Pol. 2012, 59, 171–184. [Google Scholar] [CrossRef]
- Kane, T.L.; Carothers, K.E.; Lee, S.W. Virulence Factor Targeting of the Bacterial Pathogen Staphylococcus Aureus for Vaccine and Therapeutics. Curr Drug Targets 2018, 19, 111–127. [Google Scholar] [CrossRef]
- Truong-Bolduc, Q.C.; Dunman, P.M.; Strahilevitz, J.; Projan, S.J.; Hooper, D.C. MgrA Is a Multiple Regulator of Two New Efflux Pumps in Staphylococcus Aureus. J. Bacteriol. 2005, 187, 2395–2405. [Google Scholar] [CrossRef]
- McAleese, F.; Petersen, P.; Ruzin, A.; Dunman, P.M.; Murphy, E.; Projan, S.J.; Bradford, P.A. A Novel MATE Family Efflux Pump Contributes to the Reduced Susceptibility of Laboratory-Derived Staphylococcus Aureus Mutants to Tigecycline. Antimicrob. Agents Chemother. 2005, 49, 1865–1871. [Google Scholar] [CrossRef] [PubMed]
- Dawson, R.J.P.; Locher, K.P. Structure of the Multidrug ABC Transporter Sav1866 from Staphylococcus Aureus in Complex with AMP-PNP. FEBS Lett. 2007, 581, 935–938. [Google Scholar] [CrossRef]
- Costa, S.S.; Viveiros, M.; Amaral, L.; Couto, I. Multidrug Efflux Pumps in Staphylococcus Aureus: An Update. Open Microbiol. J. 2013, 7, 59–71. [Google Scholar] [CrossRef]
- Tessé, S.; Trueba, F.; Berthet, N.; Hot, C.; Chesneau, O. Resistance Genes Underlying the LSA Phenotype of Staphylococcal Isolates from France. Antimicrob. Agents Chemother. 2013, 57, 4543–4546. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, F.A.; Helmersen, K.; Visnovska, T.; Jørgensen, S.B.; Aamot, H.V. Rapid Nanopore-Based DNA Sequencing Protocol of Antibiotic-Resistant Bacteria for Use in Surveillance and Outbreak Investigation. Microb. Genom. 2020. Accepted, pending publication. [Google Scholar]
- Durand, G.; Javerliat, F.; Bes, M.; Veyrieras, J.B.; Guigon, G.; Mugnier, N.; Schicklin, S.; Kaneko, G.; Santiago-Allexant, E.; Bouchiat, C.; et al. Routine Whole-Genome Sequencing for Outbreak Investigations of Staphylococcus Aureus in a National Reference Center. Front. Microbiol. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Liou, C.H.; Wu, H.C.; Liao, Y.C.; Lauderdale, T.L.Y.; Huang, I.W.; Chen, F.J. Nanomlst: Accurate Multilocus Sequence Typing Using Oxford Nanopore Technologies Minion with a Dual-Barcode Approach to Multiplex Large Numbers of Samples. Microb. Genom. 2020, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- NORM NORM-VET. 2019. Available online: https://www.vetinst.no/en/surveillance-programmes/norm-norm-vet-report (accessed on 27 March 2021).



{kind=link}
{kind=link}
| Patient | Sanger BioNumerics Isolate | Nanopore CGE MLST Isolate | Nanopore CGE MLST Metagenomic | Nanopore Krocus Isolate | Nanopore Krocus Metagenomic |
| ID 111 | 15 | 5510? * yqil 99.8% | 5510? * yqil 99.8% | 15 | 15 |
| ID 114 | 22 | 22 | 957 | 22 | 22 |
| ID 128 | 6325 | 6325? * arcC 99.8% | 6325? * arcC 99.8% | 6325? * arcC 99.8% | Not found glpF 99.9% |
| ID 139 | 30 | 4618 | 4618 | 30 | 30 |
| ID 140 | 6326 | 6326? * arcC, gmk, pta, tpi 99.8% | 6326 | 6326? * arcC, gmk, pta, tpi 99.3% | 6326 or 582 |
| ID 141 | 30 | 4618 | 4618 | 30 * aroE 99.9% | 30 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noone, J.C.; Ferreira, F.A.; Aamot, H.V. Culture-Independent Genotyping, Virulence and Antimicrobial Resistance Gene Identification of Staphylococcus aureus from Orthopaedic Implant-Associated Infections. Microorganisms 2021, 9, 707. https://doi.org/10.3390/microorganisms9040707
Noone JC, Ferreira FA, Aamot HV. Culture-Independent Genotyping, Virulence and Antimicrobial Resistance Gene Identification of Staphylococcus aureus from Orthopaedic Implant-Associated Infections. Microorganisms. 2021; 9(4):707. https://doi.org/10.3390/microorganisms9040707
Chicago/Turabian StyleNoone, J. Christopher, Fabienne Antunes Ferreira, and Hege Vangstein Aamot. 2021. "Culture-Independent Genotyping, Virulence and Antimicrobial Resistance Gene Identification of Staphylococcus aureus from Orthopaedic Implant-Associated Infections" Microorganisms 9, no. 4: 707. https://doi.org/10.3390/microorganisms9040707
APA StyleNoone, J. C., Ferreira, F. A., & Aamot, H. V. (2021). Culture-Independent Genotyping, Virulence and Antimicrobial Resistance Gene Identification of Staphylococcus aureus from Orthopaedic Implant-Associated Infections. Microorganisms, 9(4), 707. https://doi.org/10.3390/microorganisms9040707