
Effectors Targeting the Unfolded Protein Response during Intracellular Bacterial Infection


Abstract
1. Introduction
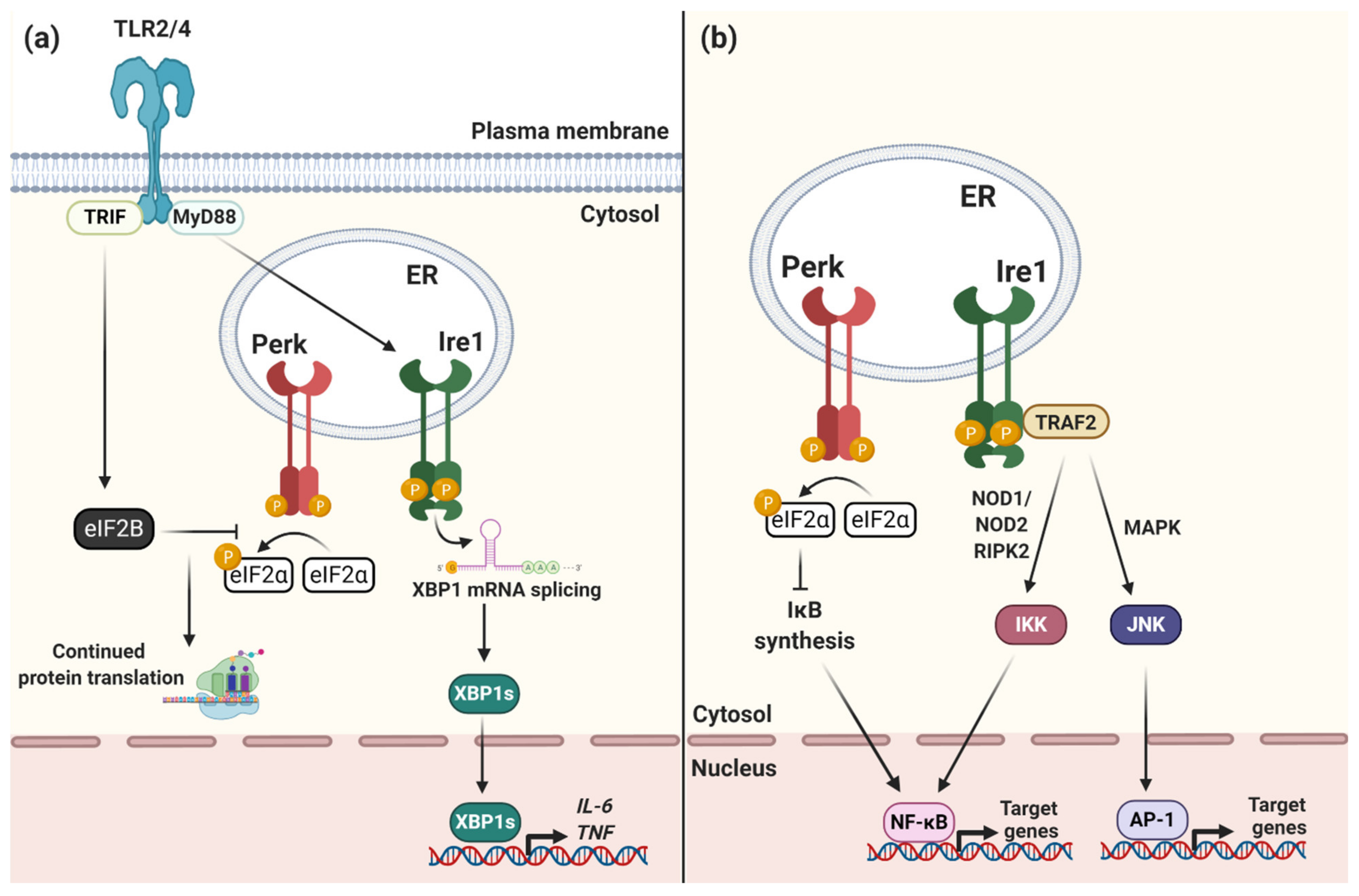
1.1. Endoplasmic Reticulum Stress and the Unfolded Protein Response
1.2. The ER Stress Sensors: Ire1, Perk and ATF6
1.3. The UPR and Impact on Immunity and Intracellular Bacterial Infection
1.4. Activation of the UPR by Brucella
1.5. Inhibition of the UPR by Legionella
1.6. Implications of Severe ER Stress for Mycobacterial Infection
1.7. Chlamydia and UPR-Mediated Inflammatory Signaling
1.8. Interactions of Salmonella with the UPR
1.9. Bacterial Toxins and ER Stress
2. Summary
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Braakman, I.; Bulleid, N.J. Protein folding and modification in the mammalian endoplasmic reticulum. Ann. Rev. Biochem. 2011, 80, 71–99. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.A.; Papa, F.R. The role of endoplasmic reticulum stress in human pathology. Ann. Rev. Pathol. Mech. Dis. 2015, 10, 173–194. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Tirasophon, W.; Welihinda, A.A.; Kaufman, R.J. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 1998, 12, 1812–1824. [Google Scholar] [CrossRef]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell. 1999, 10, 3787–3799. [Google Scholar] [CrossRef]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Snapp, E.L.; Lippincott-Schwartz, J.; Prywes, R. Stable binding of ATF6 to BiP in the endoplasmic reticulum stress response. Mol. Cell Biol. 2005, 25, 921–932. [Google Scholar] [CrossRef]
- Korennykh, A.V.; Egea, P.F.; Korostelev, A.A.; Finer-Moore, J.; Zhang, C.; Shokat, K.M.; Stroud, R.M.; Walter, P. The unfolded protein response signals through high-order assembly of Ire1. Nature 2009, 457, 687–693. [Google Scholar] [CrossRef]
- Pincus, D.; Chevalier, M.W.; Aragon, T.; van Anken, E.; Vidal, S.E.; El-Samad, H.; Walter, P. BiP binding to the ER-stress sensor Ire1 tunes the homeostatic behavior of the unfolded protein response. PLoS Biol. 2010, 8, e1000415. [Google Scholar] [CrossRef]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev. Cell 2007, 13, 365–376. [Google Scholar] [CrossRef]
- Hollien, J.; Lin, J.H.; Li, H.; Stevens, N.; Walter, P.; Weissman, J.S. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 2009, 186, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 2000, 5, 897–904. [Google Scholar] [CrossRef]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Yoshida, H.; Haze, K.; Yanagi, H.; Yura, T.; Mori, K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J. Biol. Chem. 1998, 273, 33741–33749. [Google Scholar] [CrossRef]
- Yoshida, H.; Okada, T.; Haze, K.; Yanagi, H.; Yura, T.; Negishi, M.; Mori, K. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell Biol. 2000, 20, 6755–6767. [Google Scholar] [CrossRef]
- Janssens, S.; Pulendran, B.; Lambrecht, B.N. Emerging functions of the unfolded protein response in immunity. Nat. Immunol. 2014, 15, 910–919. [Google Scholar] [CrossRef]
- Martinon, F.; Chen, X.; Lee, A.H.; Glimcher, L.H. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol. 2010, 11, 411–418. [Google Scholar] [CrossRef]
- Woo, C.W.; Kutzler, L.; Kimball, S.R.; Tabas, I. Toll-like receptor activation suppresses ER stress factor CHOP and translation inhibition through activation of eIF2B. Nat. Cell Biol. 2012, 14, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Weichhart, T.; Costantino, G.; Poglitsch, M.; Rosner, M.; Zeyda, M.; Stuhlmeier, K.M.; Kolbe, T.; Stulnig, T.M.; Hörl, W.H.; Hengstschläger, M. The TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity 2008, 29, 565–577. [Google Scholar] [CrossRef]
- Carpenter, S.; Ricci, E.P.; Mercier, B.C.; Moore, M.J.; Fitzgerald, K.A. Post-transcriptional regulation of gene expression in innate immunity. Nat. Rev. Immunol. 2014, 14, 361–376. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Han, Z.; Couvillon, A.D.; Kaufman, R.J.; Exton, J.H. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression. Mol. Cell Biol. 2006, 26, 3071–3084. [Google Scholar] [CrossRef] [PubMed]
- Keestra-Gounder, A.M.; Byndloss, M.X.; Seyffert, N.; Young, B.M.; Chavez-Arroyo, A.; Tsai, A.Y.; Cevallos, S.A.; Winter, M.G.; Pham, O.H.; Tiffany, C.R.; et al. NOD1 and NOD2 signalling links ER stress with inflammation. Nature 2016, 532, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Grootjans, J.; Kaser, A.; Kaufman, R.J.; Blumberg, R.S. The unfolded protein response in immunity and inflammation. Nat. Rev. Immunol. 2016, 16, 469–484. [Google Scholar] [CrossRef]
- Tam, A.B.; Mercado, E.L.; Hoffmann, A.; Niwa, M. ER stress activates NF-kappaB by integrating functions of basal IKK activity, IRE1 and PERK. PLoS ONE 2012, 7, e45078. [Google Scholar] [CrossRef]
- Fontana, M.F.; Banga, S.; Barry, K.C.; Shen, X.; Tan, Y.; Luo, Z.Q.; Vance, R.E. Secreted bacterial effectors that inhibit host protein synthesis are critical for induction of the innate immune response to virulent Legionella pneumophila. PLoS Pathog. 2011, 7, e1001289. [Google Scholar] [CrossRef] [PubMed]
- Celli, J.; Tsolis, R.M. Bacteria, the endoplasmic reticulum and the unfolded protein response: Friends or foes? Nat. Rev. Microbiol. 2015, 13, 71–82. [Google Scholar] [CrossRef] [PubMed]
- De Jong, M.F.; Starr, T.; Winter, M.G.; den Hartigh, A.B.; Child, R.; Knodler, L.A.; van Dijl, J.M.; Celli, J.; Tsolis, R.M. Sensing of bacterial type IV secretion via the unfolded protein response. mBio. 2013, 4, e00418-12. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Khan, M.; Magnani, D.D.; Harms, J.S.; Durward, M.; Radhakrishnan, G.K.; Liu, Y.-P.; Splitter, G.A. Brucella induces an unfolded protein response via TcpB that supports intracellular replication in macrophages. PLoS Pathog. 2013, 9, e1003785. [Google Scholar] [CrossRef] [PubMed]
- Myeni, S.; Child, R.; Ng, T.W.; Kupko, J.J.; Wehrly, T.D., 3rd; Porcella, S.F.; Knodler, L.A.; Celli, J. Brucella modulates secretory trafficking via multiple type IV secretion effector proteins. PLoS Pathog. 2013, 9, e1003556. [Google Scholar] [CrossRef] [PubMed]
- Treacy-Abarca, S.; Mukherjee, S. Legionella suppresses the host unfolded protein response via multiple mechanisms. Nat. Commun. 2015, 6, 7887. [Google Scholar] [CrossRef]
- Hempstead, A.D.; Isberg, R.R. Inhibition of host cell translation elongation by Legionella pneumophila blocks the host cell unfolded protein response. Proc. Natl. Acad Sci. USA 2015, 112, E6790–E6797. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.-H.; Shin, D.-M.; Kang, G.; Kim, K.-H.; Park, J.B.; Hur, G.M.; Lee, H.; Lim, Y.; Park, J.; Jo, E.; et al. Endoplasmic reticulum stress response is involved in Mycobacterium tuberculosis protein ESAT-6-mediated apoptosis. FEBS Lett. 2010, 584, 2445–2454. [Google Scholar] [CrossRef]
- Choi, J.; Lim, Y.; Cho, S.; Lee, J.; Jeong, J.; Kim, E.; Park, J.B.; Kim, S.H.; Park, H.S.; Kim, H.-J.; et al. Mycobacterial HBHA induces endoplasmic reticulum stress-mediated apoptosis through the generation of reactive oxygen species and cytosolic Ca 2+ in murine macrophage RAW 264.7 cells. Cell Death Dis. 2013, 4, e957. [Google Scholar] [CrossRef]
- Grover, S.; Sharma, T.; Singh, Y.; Kohli, S.; Manjunath, P.; Singh, A.; Semmler, T.; Wieler, L.H.; Tedin, K.; Ehtesham, N.Z.; et al. The PGRS Domain of Mycobacterium tuberculosis PE_PGRS Protein Rv0297 Is Involved in Endoplasmic Reticulum Stress-Mediated Apoptosis through Toll-Like Receptor 4. mBio 2018, 9. [Google Scholar] [CrossRef]
- Stamm, C.E.; Pasko, B.L.; Chaisavaneeyakorn, S.; Franco, L.H.; Nair, V.R.; Weigele, B.A.; Alto, N.M.; Shiloh, M.U. Screening Mycobacterium tuberculosis Secreted Proteins Identifies Mpt64 as a Eukaryotic Membrane-Binding Bacterial Effector. mSphere 2019, 4, e00354-19. [Google Scholar] [CrossRef] [PubMed]
- George, Z.; Omosun, Y.; Azenabor, A.A.; Goldstein, J.; Partin, J.; Joseph, K.; Ellerson, D.; He, Q.; Eko, F.; McDonald, M.A.; et al. The molecular mechanism of induction of unfolded protein response by Chlamydia. Biochem. Biophys Res. Commun. 2019, 508, 421–429. [Google Scholar] [CrossRef]
- Antoniou, A.N.; Lenart, I.; Kriston-Vizi, J.; Iwawaki, T.; Turmaine, M.; McHugh, K.; Sadfer, A.; Blake, N.; Bowness, P.; Bajaj-Elliott, M.; et al. Salmonella exploits HLA-B27 and host unfolded protein responses to promote intracellular replication. Ann. Rheum. Dis. 2019, 78, 74–82. [Google Scholar] [CrossRef]
- Bernal-Bayard, J.; Cardenal-Muñoz, E.; Ramos-Morales, F. The Salmonella type III secretion effector, Salmonella leucine-rich repeat protein (SlrP), targets the human chaperone ERdj3. J. Biol. Chem. 2010, 285, 16360–16368. [Google Scholar] [CrossRef]
- Pillich, H.; Loose, M.; Zimmer, K.P.; Chakraborty, T. Activation of the unfolded protein response by Listeria monocytogenes. Cell Microbiol. 2012, 14, 949–964. [Google Scholar] [CrossRef] [PubMed]
- Akazawa, Y.; Isomoto, H.; Matsushima, K.; Kanda, T.; Minami, H.; Yamaghchi, N.; Taura, N.; Shiozawa, K.; Ohnita, K.; Takeshima, F.; et al. Endoplasmic reticulum stress contributes to Helicobacter pylori VacA-induced apoptosis. PLoS ONE 2013, 8, e82322. [Google Scholar] [CrossRef]
- Massey, S.; Burress, H.; Taylor, M.; Nemec, K.N.; Ray, S.; Haslam, D.B.; Teter, K. Structural and functional interactions between the cholera toxin A1 subunit and ERdj3/HEDJ.; a chaperone of the endoplasmic reticulum. Infect. Immun. 2011, 79, 4739–4747. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, T.; Grabon, A.; Taylor, M.; Teter, K. cAMP-Independent Activation of the Unfolded Protein Response by Cholera Toxin. Infect. Immun. 2020. [Google Scholar] [CrossRef]
- Paton, A.W.; Beddoe, T.; Thorpe, C.M.; Whisstock, J.C.; Wilce, M.C.; Rossjohn, J.; Talbot, U.M.; Paton, J.C. AB5 subtilase cytotoxin inactivates the endoplasmic reticulum chaperone BiP. Nature 2006, 443, 548–552. [Google Scholar] [CrossRef]
- Wolfson, J.J.; May, K.L.; Thorpe, C.M.; Jandhyala, D.M.; Paton, J.C.; Paton, A.W. Subtilase cytotoxin activates PERK.; IRE1 and ATF6 endoplasmic reticulum stress-signalling pathways. Cell Microbiol. 2008, 10, 1775–1786. [Google Scholar] [CrossRef] [PubMed]
- Morinaga, N.; Yahiro, K.; Matsuura, G.; Moss, J.; Noda, M. Subtilase cytotoxin, produced by Shiga-toxigenic Escherichia coli, transiently inhibits protein synthesis of Vero cells via degradation of BiP and induces cell cycle arrest at G1 by downregulation of cyclin D1. Cell Microbiol. 2008, 10, 921–929. [Google Scholar] [CrossRef]
- Yu, M.; Haslam, D.B. Shiga toxin is transported from the endoplasmic reticulum following interaction with the luminal chaperone HEDJ/ERdj3. Infect. Immun. 2005, 73, 2524–2532. [Google Scholar] [CrossRef]
- Falguieres, T.; Johannes, L. Shiga toxin B-subunit binds to the chaperone BiP and the nucleolar protein B23. Biol. Cell 2006, 98, 125–134. [Google Scholar] [CrossRef]
- Lee, S.Y.; Lee, M.S.; Cherla, R.P.; Tesh, V.L. Shiga toxin 1 induces apoptosis through the endoplasmic reticulum stress response in human monocytic cells. Cell Microbiol. 2008, 10, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Celli, J.; de Chastellier, C.; Franchini, D.-M.; Pizarro-Cerda, J.; Moreno, E.; Gorvel, J.-P. Brucella evades macrophage killing via VirB-dependent sustained interactions with the endoplasmic reticulum. J. Exp. Med. 2003, 198, 545–556. [Google Scholar] [CrossRef]
- Celli, J. The changing nature of the Brucella-containing vacuole. Cell Microbiol. 2015, 17, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, G.K.; Harms, J.S.; Splitter, G.A. Modulation of microtubule dynamics by a TIR domain protein from the intracellular pathogen Brucella melitensis. Biochem. J. 2011, 439, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.-M.; Pei, J.; Ancona, V.; Shaw, B.D.; Ficht, T.A.; de Figueiredo, P. RNAi screen of endoplasmic reticulum–associated host factors reveals a role for IRE1α in supporting Brucella replication. PLoS Pathog. 2008, 4, e1000110. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Lin, F.; Cabello, A.L.; da Costa, L.F.; Feng, X.; Feng, H.Q.; Zhang, M.; Iwawaki, T.; Rice-Ficht, A.; Ficht, T.A.; et al. Activation of Host IRE1alpha-Dependent Signaling Axis Contributes the Intracellular Parasitism of Brucella melitensis. Front. Cell Infect. Microbiol. 2018, 8, 103. [Google Scholar] [CrossRef] [PubMed]
- Byndloss, M.X.; Tsai, A.Y.; Walker, G.T.; Miller, C.N.; Young, B.M.; English, B.C.; Seyffert, N.; Kerrinnes, T.; de Jong, M.F.; Atluri, L.V.; et al. Brucella abortus Infection of Placental Trophoblasts Triggers Endoplasmic Reticulum Stress-Mediated Cell Death and Fetal Loss via Type IV Secretion System-Dependent Activation of CHOP. mBio 2019, 10, e01538-19. [Google Scholar] [CrossRef] [PubMed]
- Mondino, S.; Schmidt, S.; Rolando, M.; Escoll, P.; Gomez-Valero, L.; Buchrieser, C. Legionnaires’ Disease: State of the Art Knowledge of Pathogenesis Mechanisms of Legionella. Annu. Rev. Pathol. 2020, 15, 439–466. [Google Scholar] [CrossRef]
- Isberg, R.R.; O’Connor, T.J.; Heidtman, M. The Legionella pneumophila replication vacuole: Making a cosy niche inside host cells. Nat. Rev. Microbiol. 2009, 7, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Belyi, Y.; Niggeweg, R.; Opitz, B.; Vogelsgesang, M.; Hippenstiel, S.; Wilm, M.; Aktories, K. Legionella pneumophila glucosyltransferase inhibits host elongation factor 1A. Proc. Natl. Acad Sci. USA 2006, 103, 16953–16958. [Google Scholar] [CrossRef]
- Van der Wel, N.; Hava, D.; Houben, D.; Fluitsma, D.; van Zon, M.; Pierson, J. M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell 2007, 129, 1287–1298. [Google Scholar] [CrossRef]
- Conrad, W.H.; Osman, M.M.; Shanahan, J.K.; Chu, F.; Takaki, K.K.; Cameron, J.; Brenner, M.; Peters, P.J. Mycobacterial ESX-1 secretion system mediates host cell lysis through bacterium contact-dependent gross membrane disruptions. Proc. Natl. Acad Sci. USA 2017, 114, 1371–1376. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Zhao, D.; Barrow, P.A.; Zhou, X. The endoplasmic reticulum stress response: A link with tuberculosis? Tuberculosis 2016, 97, 52–56. [Google Scholar] [CrossRef]
- Seimon, T.A.; Kim, M.-J.; Blumenthal, A.; Koo, J.; Ehrt, S.; Wainwright, H.; Bekker, L.; Kaplan, G.; Nathan, C.; Tabas, I.; et al. Induction of ER stress in macrophages of tuberculosis granulomas. PLoS ONE 2010, 5, e12772. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.-J.; Choi, J.-A.; Choi, H.-H.; Cho, S.-N.; Kim, H.-J.; Jo, E.-K.; Park, J.-K.; Song, C.-H. Endoplasmic reticulum stress pathway-mediated apoptosis in macrophages contributes to the survival of Mycobacterium tuberculosis. PLoS ONE 2011, 6, e28531. [Google Scholar] [CrossRef] [PubMed]
- Motaung, B.; Walzl, G.; Loxton, A.G. The level of the endoplasmic reticulum stress chaperone protein, binding immunoglobulin protein (BiP), decreases following successful tuberculosis treatment. Int. J. Infect. Dis. 2019, 81, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.H.; Choi, J.A.; Lim, Y.J.; Lee, J.; Cho, S.N.; Oh, S.M.; Go, D.; Kim, S.-H.; Song, C.-H. Calreticulin modulates the intracellular survival of mycobacteria by regulating ER-stress-mediated apoptosis. Oncotarget 2017, 8, 58686–58698. [Google Scholar] [CrossRef][Green Version]
- Dumoux, M.; Clare, D.K.; Saibil, H.R.; Hayward, R.D. Chlamydiae assemble a pathogen synapse to hijack the host endoplasmic reticulum. Traffic 2012, 13, 1612–1627. [Google Scholar] [CrossRef]
- Dickinson, M.S.; Anderson, L.N.; Webb-Robertson, B.M.; Hansen, J.R.; Smith, R.D.; Wright, A.T.; Hybiske, K. Proximity-dependent proteomics of the Chlamydia trachomatis inclusion membrane reveals functional interactions with endoplasmic reticulum exit sites. PLoS Pathog. 2019, 15, e1007698. [Google Scholar] [CrossRef]
- Pham, O.H.; Lee, B.; Labuda, J.; Keestra-Gounder, A.M.; Byndloss, M.X.; Tsolis, R.M.; McSorley, S.J. NOD1/NOD2 and RIP2 Regulate Endoplasmic Reticulum Stress-Induced Inflammation during Chlamydia Infection. mBio 2020, 11. [Google Scholar] [CrossRef]
- George, Z.; Omosun, Y.; Azenabor, A.A.; Partin, J.; Joseph, K.; Ellerson, D.; He, Q.; Eko, F.; Bandea, C.; Svoboda, P.; et al. The roles of unfolded protein response pathways in Chlamydia pathogenesis. J. Infect. Dis. 2017, 215, 456–465. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Webster, S.J.; Ellis, L.; O’Brien, L.M.; Tyrrell, B.; Fitzmaurice, T.J.; Elder, M.J.; Clare, S.; Chee, R.; Gaston, J.S.H.; Goodall, J.C. IRE1alpha mediates PKR activation in response to Chlamydia trachomatis infection. Microbes Infect. 2016, 18, 472–483. [Google Scholar] [CrossRef]
- Bischof, L.J.; Kao, C.Y.; Los, F.C.; Gonzalez, M.R.; Shen, Z.; Briggs, S.P.; van der Goot, F.G.; Aroian, R.V. Activation of the unfolded protein response is required for defenses against bacterial pore-forming toxin in vivo. PLoS Pathog. 2008, 4, e1000176. [Google Scholar] [CrossRef] [PubMed]
- Baird, M.; Woon Ang, P.; Clark, I.; Bishop, D.; Oshima, M.; Cook, M.C.; Hemmings, C.; Takeishi, S.; Worthley, D.; Boussioutas, A.; et al. The unfolded protein response is activated in Helicobacter-induced gastric carcinogenesis in a non-cell autonomous manner. Lab. Invest. 2013, 93, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Halder, P.; Datta, C.; Kumar, R.; Sharma, A.K.; Basu, J.; Kundu, M. The secreted antigen, HP0175, of Helicobacter pylori links the unfolded protein response (UPR) to autophagy in gastric epithelial cells. Cell Microbiol. 2015, 17, 714–729. [Google Scholar] [CrossRef]
- Dixit, G.; Mikoryak, C.; Hayslett, T.; Bhat, A.; Draper, R.K. Cholera toxin up-regulates endoplasmic reticulum proteins that correlate with sensitivity to the toxin. Exp. Biol. Med. 2008, 233, 163–175. [Google Scholar] [CrossRef]
- Gallagher, C.M.; Garri, C.; Cain, E.L.; Ang, K.K.; Wilson, C.G.; Chen, S.; Hearn, B.R.; Jaishankar, P.; Aranda-Diaz, A.; Arkin, M.R.; et al. Ceapins are a new class of unfolded protein response inhibitors, selectively targeting the ATF6alpha branch. Elife 2016, 5, e11878. [Google Scholar] [CrossRef]
- Harding, H.P.; Zyryanova, A.F.; Ron, D. Uncoupling proteostasis and development in vitro with a small molecule inhibitor of the pancreatic endoplasmic reticulum kinase, PERK. J. Biol. Chem. 2012, 287, 44338–44344. [Google Scholar] [CrossRef] [PubMed]
- Paxman, R.; Plate, L.; Blackwood, E.A.; Glembotski, C.; Powers, E.T.; Wiseman, R.L.; Kelly, J.W. Pharmacologic ATF6 activating compounds are metabolically activated to selectively modify endoplasmic reticulum proteins. Elife 2018, 7, e37168. [Google Scholar] [CrossRef]
- Plate, L.; Cooley, C.B.; Chen, J.J.; Paxman, R.J.; Gallagher, C.M.; Madoux, F.; Genereux, J.C.; Dobbs, W.; Garza, D.; Spicer, T.P.; et al. Small molecule proteostasis regulators that reprogram the ER to reduce extracellular protein aggregation. Elife 2016, 5, e15550. [Google Scholar] [CrossRef] [PubMed]
- Kudo, T.; Kanemoto, S.; Hara, H.; Morimoto, N.; Morihara, T.; Kimura, R.; Tabira, T.; Imaizumi, K.; Takeda, M. A molecular chaperone inducer protects neurons from ER stress. Cell Death Differ. 2008, 15, 364–375. [Google Scholar] [CrossRef]
- Cross, B.C.; Bond, P.J.; Sadowski, P.G.; Jha, B.K.; Zak, J.; Goodman, J.M.; Silverman, R.H.; Neubert, T.A.; Baxendale, I.R.; Ron, D.; et al. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc. Natl. Acad Sci. USA 2012, 109, E869–E878. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Wang, L.; Wang, E.S.; Perera, B.G.; Igbaria, A.; Morita, S.; Prado, K.; Thamsen, M.; Caswell, D.; Macias, H.; et al. Allosteric inhibition of the IRE1alpha RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 2014, 158, 534–548. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Bacterium | Toxin or Effector | Effect | Mode of Action | Reference |
|---|---|---|---|---|
| Brucella | VceC | Induces the UPR | Binds BiP | [31] |
| TcpB (BtpA/Btp1] | Induces the UPR | Restructures ER tubules | [32] | |
| BspC, BspG, BspH, BspI, BspK | Induces the UPR | Unknown | [33] | |
| Legionella | Lgt1, Lgt2, Lgt3 | Inhibits XBP1 splicing | Inhibits translation elongation | [34,35] |
| Mycobacteria | ESAT-6, Heparin-Binding Haemagglutinin (HBHA) | Induces the UPR | Increases intracellular Ca2+ and Reactive Oxygen Species (ROS) | [36,37] |
| Rv027 | Induces the UPR | Increases intracellular Ca2+ and ROS | [38] | |
| Mpt64 | Inhibits CHOP expression | Unknown, binds PIPs on ER membrane | [39] | |
| Chlamydia | CT288, Tarp | Activates Ire1 | Drives Ire1 oligomerisation | [40] |
| Salmonella | SlrP | Induces the UPR | Binds ERdj3, BiP cochaperone | [41,42] |
| Listeria | Listeriolysin O (LLO) | Induces the UPR | Increases intracellular Ca2+ | [43] |
| Helicobacter | VacA | Activates Perk | Unknown | [44] |
| Vibrio cholerae | Cholera toxin (CT) | Induces the UPR | Binds BiP and ERdj3 | [45,46] |
| E. coli | Subtilase cytotoxin | Induces the UPR | Cleaves BiP | [47,48,49] |
| Shiga-like toxins (SLT) | Induces the UPR | Binds BiP and ERdj3 | [50,51,52] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alshareef, M.H.; Hartland, E.L.; McCaffrey, K. Effectors Targeting the Unfolded Protein Response during Intracellular Bacterial Infection. Microorganisms 2021, 9, 705. https://doi.org/10.3390/microorganisms9040705
Alshareef MH, Hartland EL, McCaffrey K. Effectors Targeting the Unfolded Protein Response during Intracellular Bacterial Infection. Microorganisms. 2021; 9(4):705. https://doi.org/10.3390/microorganisms9040705
Chicago/Turabian StyleAlshareef, Manal H., Elizabeth L. Hartland, and Kathleen McCaffrey. 2021. "Effectors Targeting the Unfolded Protein Response during Intracellular Bacterial Infection" Microorganisms 9, no. 4: 705. https://doi.org/10.3390/microorganisms9040705
APA StyleAlshareef, M. H., Hartland, E. L., & McCaffrey, K. (2021). Effectors Targeting the Unfolded Protein Response during Intracellular Bacterial Infection. Microorganisms, 9(4), 705. https://doi.org/10.3390/microorganisms9040705