
Genomic Epidemiology of SARS-CoV-2 in Madrid, Spain, during the First Wave of the Pandemic: Fast Spread and Early Dominance by D614G Variants

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. RNA Isolation and RT-PCR
2.3. SARS-CoV-2 Genome Sequencing
2.4. Bioinformatic Analyses
2.5. Phylogenetic Analysis
3. Results
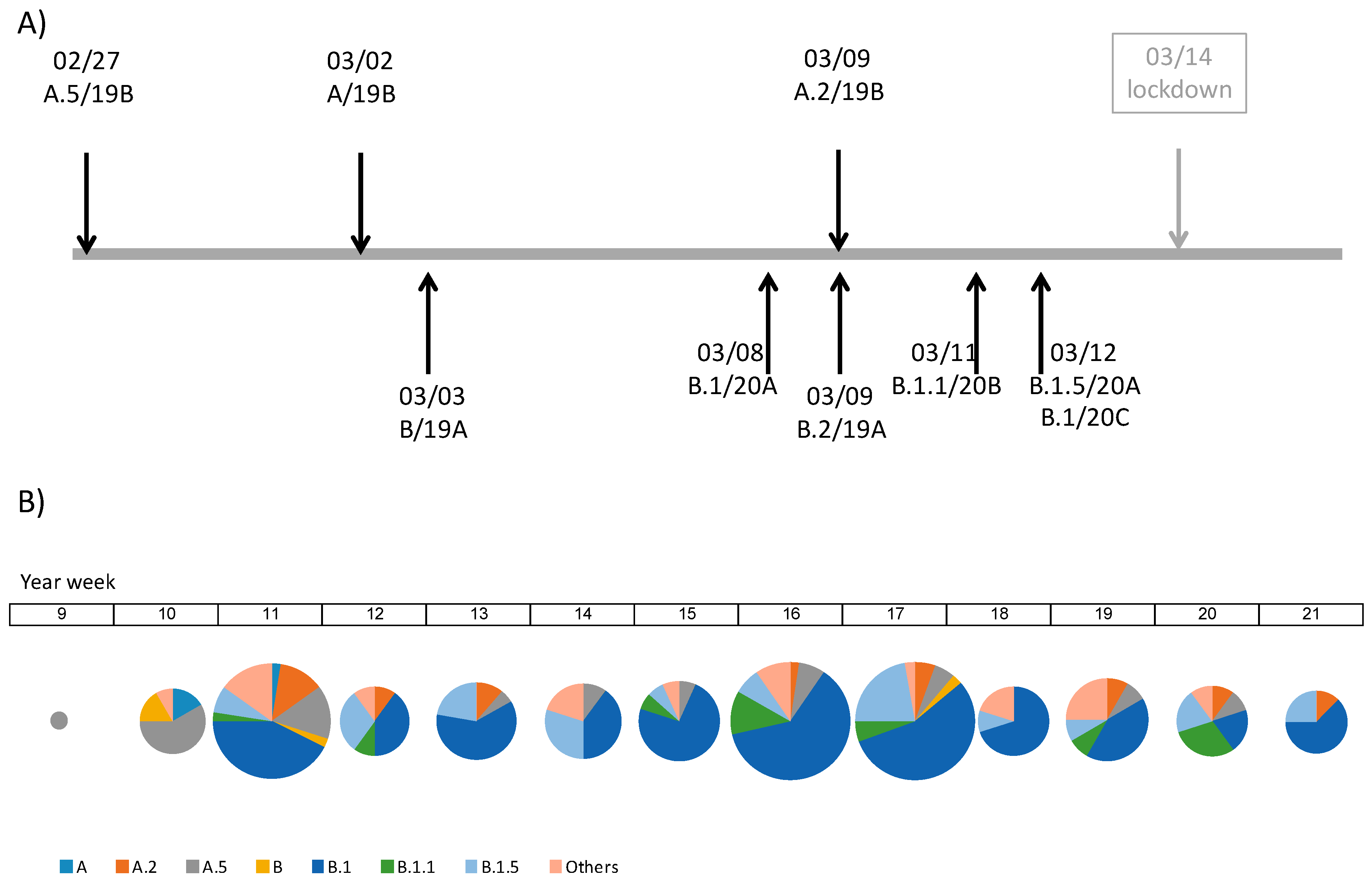
3.1. Description of SARS-CoV-2 Genomes Sampled among Patients with COVID-19 Diagnosis in Madrid during the First Pandemic Wave
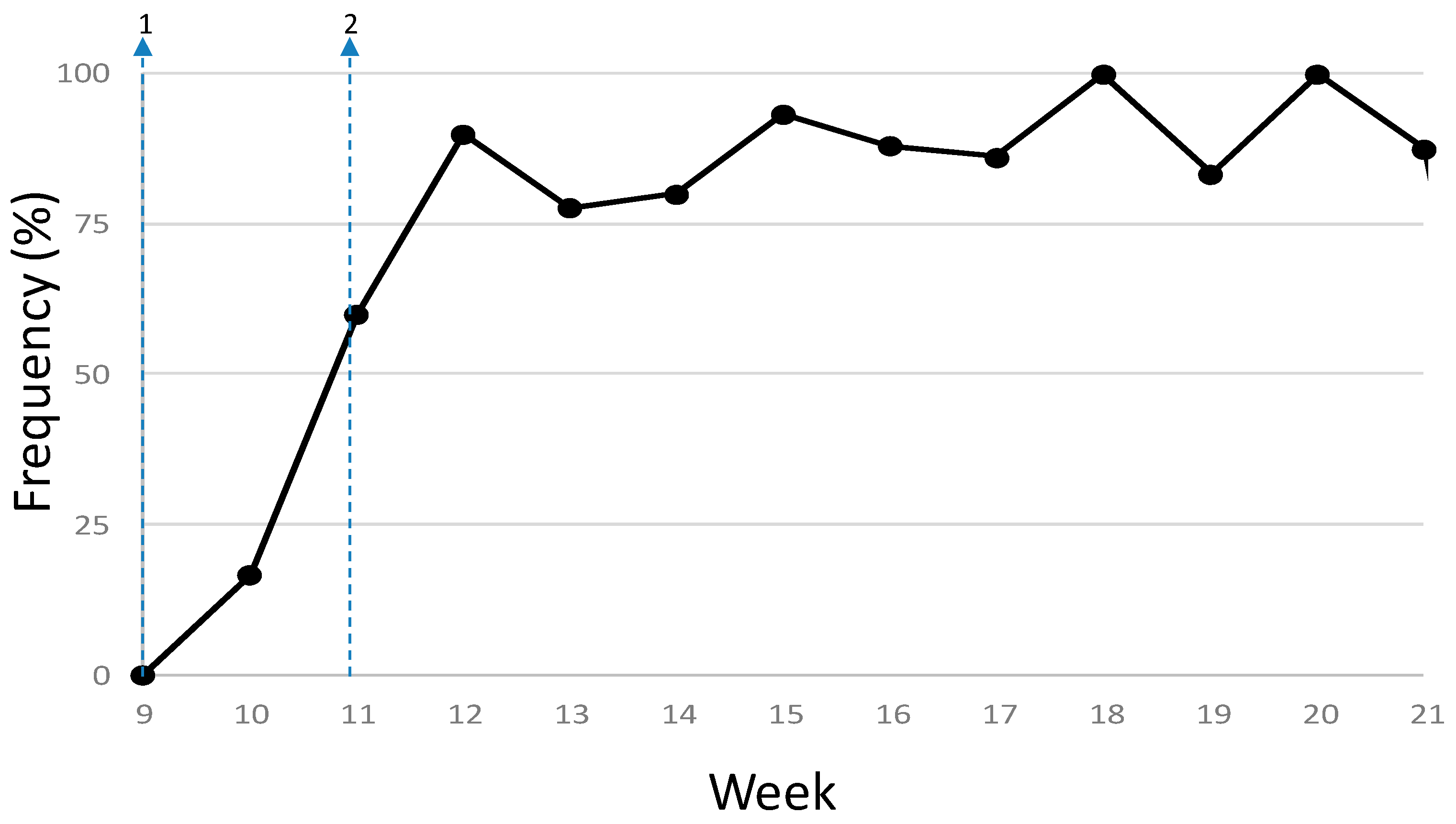
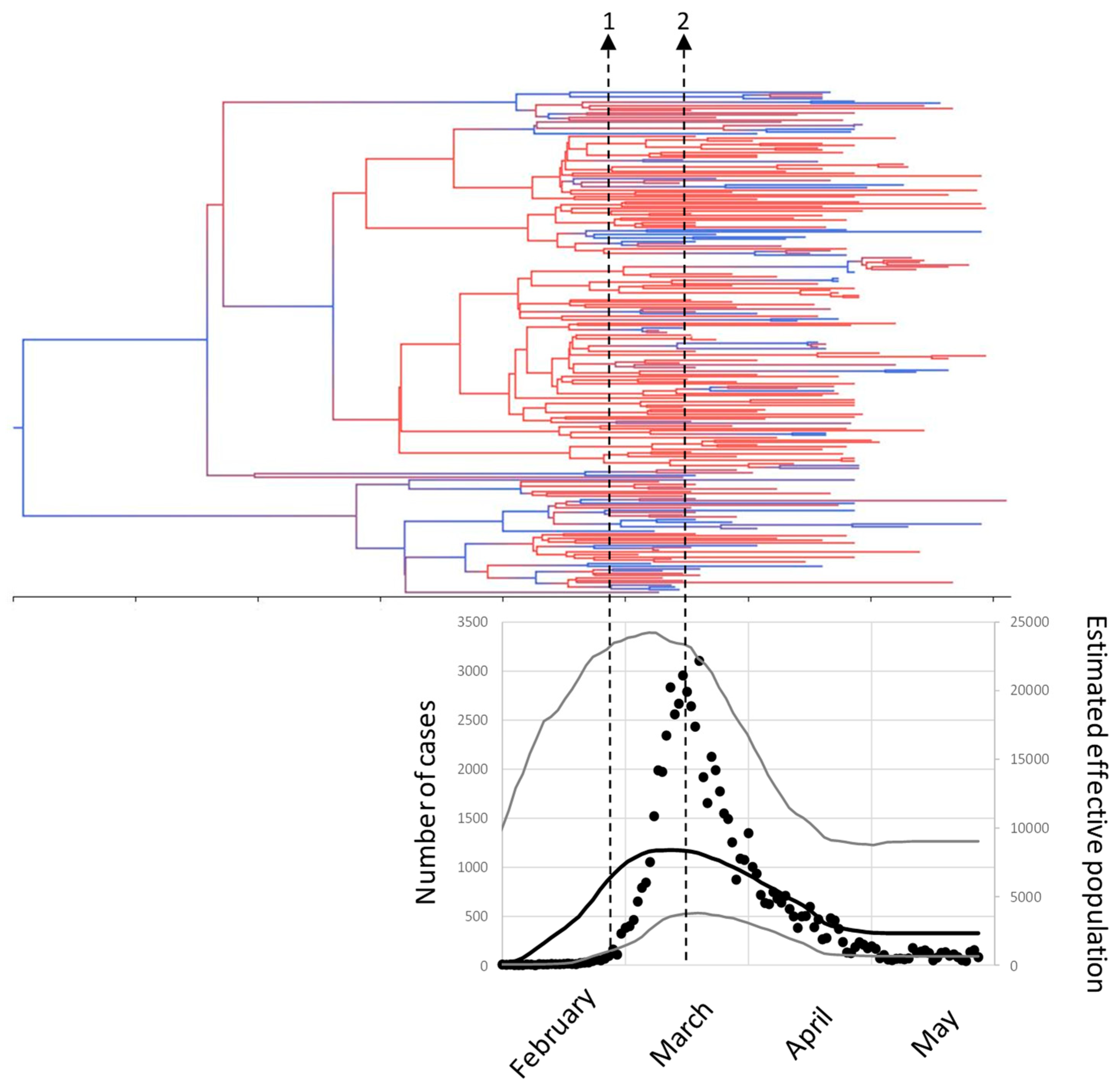
3.2. Phylodynamics of SARS-CoV-2 in Madrid
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Spiteri, G.; Fielding, J.; Diercke, M.; Campese, C.; Enouf, V.; Gaymard, A.; Bella, A.; Sognamiglio, P.; Sierra Moros, M.J.; Riutortet, A.N.; et al. First cases of coronavirus disease 2019 (COVID-19) in the WHO European Region, 24 January to 21 February 2020. Euro Surveill. 2020, 25, 2000178. [Google Scholar] [CrossRef] [PubMed]
- Borobia, A.M.; Carcas, A.J.; Arnalich, F.; Álvarez-Sala, R.; Monserrat-Villatoro, J.; Quintana, M.; Figueira, J.C.; Torres Santos-Olmo, R.M.; García-Rodríguez, J.; Martín-Vega, A.; et al. A cohort of patients with COVID-19 in a major teaching hospital in Europe. J. Clin. Med. 2020, 9, 1733. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Reiche, A.S.; Hernandez, M.M.; Sullivan, M.J.; Ciferri, B.; Alshammary, H.; Obla, A.; Fabre, S.; Kleiner, G.; Polanco, J.; Khanet, Z.; et al. Introductions and early spread of SARS-CoV-2 in the New York City area. Science 2020, 301, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T.; Lane, C.R.; Sherry, N.L.; Duchene, S.; Gonçalves da Silva, A.; Caly, L.; Sait, M.; Ballard, S.A.; Horan, K.; Schultz, M.B.; et al. Tracking the COVID-19 pandemic in Australia using genomics. Nat. Commun. 2020, 11l, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Alteri, C.; Cento, V.; Piralla, A.; Costabile, V.; Tallarita, M.; Colagrossi, L.; Renica, S.; Giardina, F.; Novazzi, F.; Gaiarsa, S.; et al. Genomic epidemiology of SARS-CoV-2 reveals multiple lineages and early spread of SARS-CoV-2 infections in Lombardy, Italy 2. Nat. Commun. 2020, 12, 434. [Google Scholar] [CrossRef] [PubMed]
- Gambaro, F.; Baidaliuk, A.; Behillil, S.; Donati, F.; Albert, M.; Alexandru, A.; Vanpeene, M.; Bizard, M.; Brisebarre, A.; Barbetet, M.; et al. Introductions and early spread of SARS-CoV-2 in France. bioRxiv 2020. [Google Scholar] [CrossRef]
- Mavian, C.; Marini, S.; Prosperi, M.; Salemi, M.A. Snapshot of SARS-CoV-2 Genome Availability up to April 2020 and its Implications: Data Analysis. JMIR Public Health Surveill. 2020, 6, e19170. [Google Scholar] [CrossRef] [PubMed]
- Díez-Fuertes, F.; Iglesias-Caballero, M.; García Pérez, J.; Monzón, S.; Jiménez, P.; Varona, S.; Cuesta, I.; Zaballos, A.; Jiménez, M.; Checa, L.; et al. A founder effect led early SARS-COV-2 transmission in Spain. J. Virol. 2021, 95, e01583-20. [Google Scholar] [CrossRef] [PubMed]
- da Silva Filipe, A.; Shepherd, J.G.; Williams, T.; Hughes, J.; Aranday-Cortes, E.; Asamaphan, P.; Ashraf, S.; Balcazar, C.; Brunker, K.; Campbell, A.; et al. Genomic epidemiology reveals multiple introductions of SARS-CoV-2 from mainland Europe into Scotland. Nat. Microbiol. 2021, 6, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Carballa, A.; Bello, X.; Pardo-Seco, J.; Pérez del Molino, M.L.; Martinón-Torres, F.; Salas, A. Phylogeography of SARS-CoV-2 pandemic in Spain: A story of multiple introductions, micro-geographic stratification, founder effects, and super-spreaders. Zool. Res. 2020, 41, 605–620. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.R.; Hoque, M.N.; Rahman, M.S.; Alam, A.S.M.R.U.; Akther, M.; Puspo, J.A.; Akter, S.; Sultana, M.; Crandall, K.A.; Hossain, M.A. Genome-wide analysis of SARS-CoV-2 virus strains circulating worldwide implicates heterogeneity. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Quick, J. nCoV-2019 Sequencing Protocol v2 (GunIt) V.2. Protocols.io. Available online: https://www.protocols.io/view/ncov-2019-sequencing-protocol-v2-bdp7i5rn?version_warning=no (accessed on 3 February 2021).
- Shepard, S.S.; Meno, S.; Bahl, J.; Wilson, M.M.; Barnes, J.; Neuhaus, E. Viral deep sequencing needs an adaptive approach: IRMA, the iterative refinement meta-assembler. BMC Genom. 2016, 17, 1–18. [Google Scholar] [CrossRef]
- Seemann, T. Snippy n.d. Available online: https://github.com/tseemann/snippy (accessed on 3 February 2021).
- De Maio, N.; Walker, C.; Borges, R.; Weilguny, L.; Slodkowicz, G.; Goldman, N. Issues with SARS-CoV-2 Sequencing Data. Available online: https://virological.org/t/issues-with-sars-cov-2-sequencing-data/473 (accessed on 3 February 2021).
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Hodcroft, E.B.; Hadfield, J.; Neher, R.A.; Bedford, T. Year-Letter Genetic Clade Naming for SARS-CoV-2 on Nextstrain.org. Available online: https://virological.org/t/year-letter-genetic-clade-naming-for-sars-cov-2-on-nextstrain-org/498 (accessed on 3 February 2021).
- Callaway, E. Making sense of coronavirus mutations. Nature 2020, 585, 174–177. [Google Scholar] [CrossRef] [PubMed]
- Di Giallonardo, F.; Duchene, S.; Puglia, I.; Curini, V.; Profeta, F.; Cammà, C.; Marcacci, M.; Calistri, P.; Holmes, E.C.; Lorusso, A. Genomic Epidemiology of the First Wave of SARS-CoV-2 in Italy. Viruses 2020, 12, 1438. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef] [PubMed]
- Pollán, M.; Pérez-Gómez, B.; Pastor-Barriuso, R.; Oteo, J.; Hernán, M.A.; Pérez-Olmeda, M.; Sanmartín, J.L.; Fernández-García, A.; Cruz, I.; Fernández de Larrea, N.; et al. Prevalence of SARS-CoV-2 in Spain (ENE-COVID): A nationwide, population-based seroepidemiological study. Lancet 2020, 396, 535–544. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Clade | H12O | HLP | HRyC | Total |
|---|---|---|---|---|
| 19A | 6 (5.8) | 5 (5.6) | 0 (0) | 11 (4.9) |
| 19B | 14 (13.5) | 13 (14.4) | 14 (46.7) | 41 (18.3) |
| 20A | 76 (73.1) | 68 (75.6) | 11 (36.7) | 155 (69.2) |
| 20B | 5 (4.8) | 4 (4.4) | 5 (16.7) | 14 (6.3) |
| 20C | 3 (2.9) | 0 (0) | 0 (0) | 3 (1.3) |
| Lineage | ||||
| A.2 | 7 (6.7) | 5 (5.6) | 2 (6.7) | 14 (6.3) |
| A.5 | 7 (6.7) | 7 (7.8) | 10 (33.3) | 24 (10.7) |
| B.1 | 60 (57.7) | 49 (54.4) | 3 (10) | 112 (50.0) |
| B.1.1 | 5 (4.8) | 4 (4.4) | 5 (16.7) | 14 (6.3) |
| B.1.5 | 15 (14.4) | 11 (12.2) | 5 (16.7) | 31 (13.8) |
| Others | 10 (9.6) | 14 (15.6) | 5 (16.7) | 29 (12.9) |
| Transmission Node | Diversification Date | 95% HPD Interval | First Sequenced Sample | Detection Lag (Days) | |
|---|---|---|---|---|---|
| H12_57/LP55/LP88/H12_2208/LP57/LP68 | 25/02/2020 | 29/01/2020 | 20/03/2020 | 27/03/2020 | 32 |
| LP19/LP22 | 29/02/2020 | 17/01/2020 | 07/03/2020 | 07/03/2020 | 7 |
| R64/H12_45/H12_59 | 02/03/2020 | 10/02/2020 | 17/03/2020 | 17/03/2020 | 15 |
| H12_28/H12_29 | 05/03/2020 | 19/02/2020 | 12/03/2020 | 12/03/2020 | 7 |
| H12_54/H12_70/ H12_71/H12_66 | 05/03/2020 | 09/03/2020 | 11/04/2020 | 01/04/2020 | 26 |
| H12_43/H12_56/ H12_85 | 10/03/2020 | 11/02/2020 | 05/04/2020 | 06/04/2020 | 26 |
| R62/LP105 | 14/03/2020 | 09/02/2020 | 15/04/2020 | 21/04/2020 | 37 |
| LP90/LP100 | 16/03/2020 | 12/02/2020 | 15/04/2020 | 26/04/2020 | 40 |
| H12_47/H12_88/ H12_55 | 20/03/2020 | 19/02/2020 | 09/04/2020 | 09/04/2020 | 19 |
| H12_42/H12_81 | 26/03/2020 | 03/03/2020 | 05/04/2020 | 06/04/2020 | 10 |
| H12_63/H12_64/ H12_62 | 29/03/2020 | 09/03/2020 | 12/04/2020 | 12/04/2020 | 13 |
| H12_82/H12_83 | 30/03/2020 | 05/03/2020 | 16/04/2020 | 16/04/2020 | 16 |
| H12_72/H12_73 | 03/04/2020 | 12/04/2020 | 21/05/2020 | 13/04/2020 | 10 |
| * R82/LP77/LP78/LP82/LP84/LP51 | 03/04/2020 | 09/03/2020 | 20/04/2020 | 05/04/2020 | 2 |
| R56/R84 | 05/04/2020 | 13/03/2020 | 20/04/2020 | 20/04/2020 | 15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viedma, E.; Dahdouh, E.; González-Alba, J.M.; González-Bodi, S.; Martínez-García, L.; Lázaro-Perona, F.; Recio, R.; Rodríguez-Tejedor, M.; Folgueira, M.D.; Cantón, R.; et al. Genomic Epidemiology of SARS-CoV-2 in Madrid, Spain, during the First Wave of the Pandemic: Fast Spread and Early Dominance by D614G Variants. Microorganisms 2021, 9, 454. https://doi.org/10.3390/microorganisms9020454
Viedma E, Dahdouh E, González-Alba JM, González-Bodi S, Martínez-García L, Lázaro-Perona F, Recio R, Rodríguez-Tejedor M, Folgueira MD, Cantón R, et al. Genomic Epidemiology of SARS-CoV-2 in Madrid, Spain, during the First Wave of the Pandemic: Fast Spread and Early Dominance by D614G Variants. Microorganisms. 2021; 9(2):454. https://doi.org/10.3390/microorganisms9020454
Chicago/Turabian StyleViedma, Esther, Elias Dahdouh, José María González-Alba, Sara González-Bodi, Laura Martínez-García, Fernando Lázaro-Perona, Raúl Recio, María Rodríguez-Tejedor, María Dolores Folgueira, Rafael Cantón, and et al. 2021. "Genomic Epidemiology of SARS-CoV-2 in Madrid, Spain, during the First Wave of the Pandemic: Fast Spread and Early Dominance by D614G Variants" Microorganisms 9, no. 2: 454. https://doi.org/10.3390/microorganisms9020454
APA StyleViedma, E., Dahdouh, E., González-Alba, J. M., González-Bodi, S., Martínez-García, L., Lázaro-Perona, F., Recio, R., Rodríguez-Tejedor, M., Folgueira, M. D., Cantón, R., Delgado, R., García-Rodríguez, J., Galán, J. C., Mingorance, J., & on behalf of the SARS-CoV-2 Working Groups. (2021). Genomic Epidemiology of SARS-CoV-2 in Madrid, Spain, during the First Wave of the Pandemic: Fast Spread and Early Dominance by D614G Variants. Microorganisms, 9(2), 454. https://doi.org/10.3390/microorganisms9020454