
Evaluation of Ogataea (Hansenula) polymorpha for Hyaluronic Acid Production

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Molecular Biology Procedures
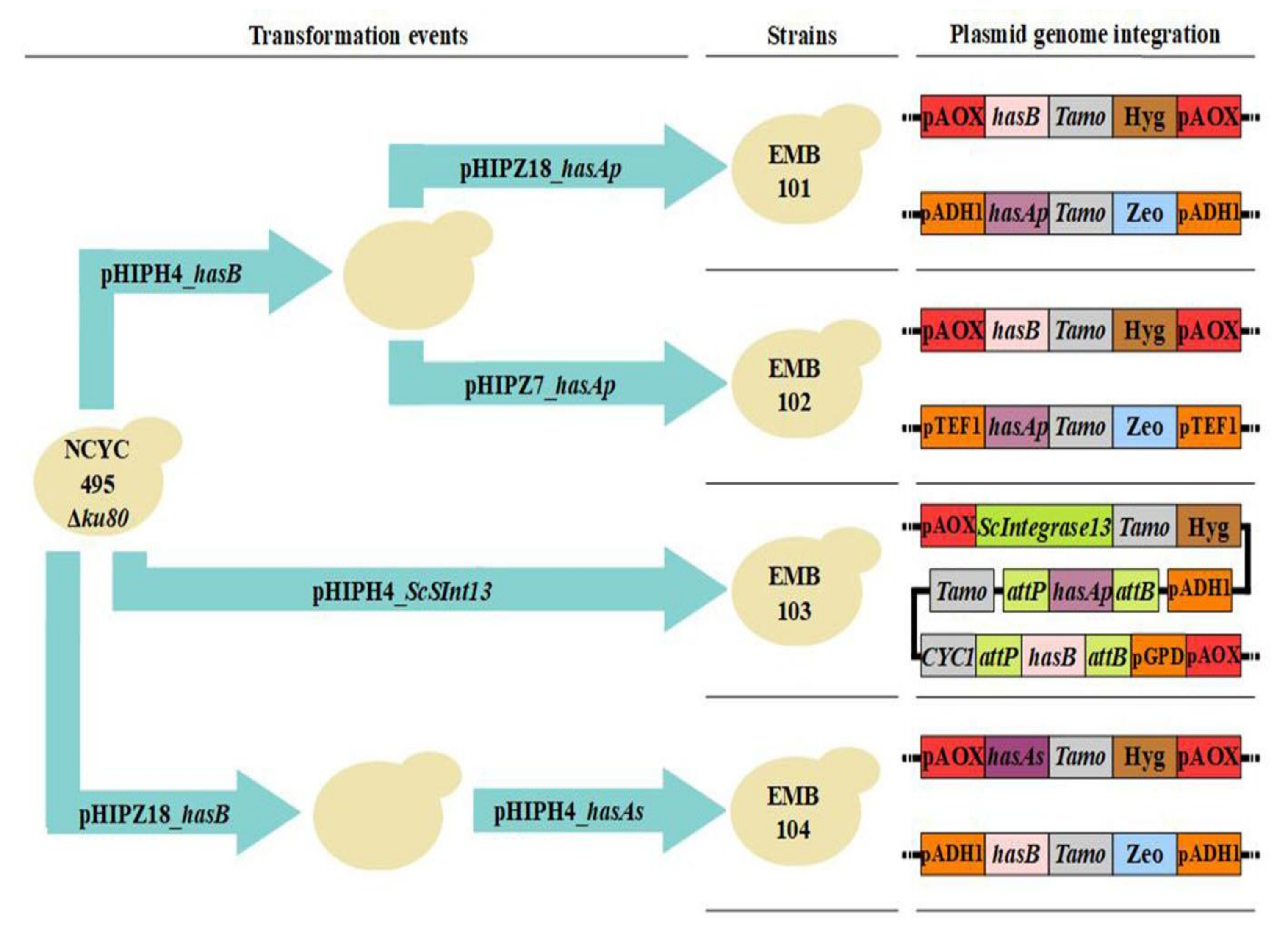
2.2. Construction of Plasmids and Strains
2.3. Media and Culture Conditions
2.4. Electrocompetent Cell Preparation and Transformation
2.5. Determination of the Growth Profile
2.6. Statistical Analysis and Data Presentation
2.7. Analysis through Scanning Electron Microscopy (SEM)
2.8. HA Quantification
3. Results and Discussion
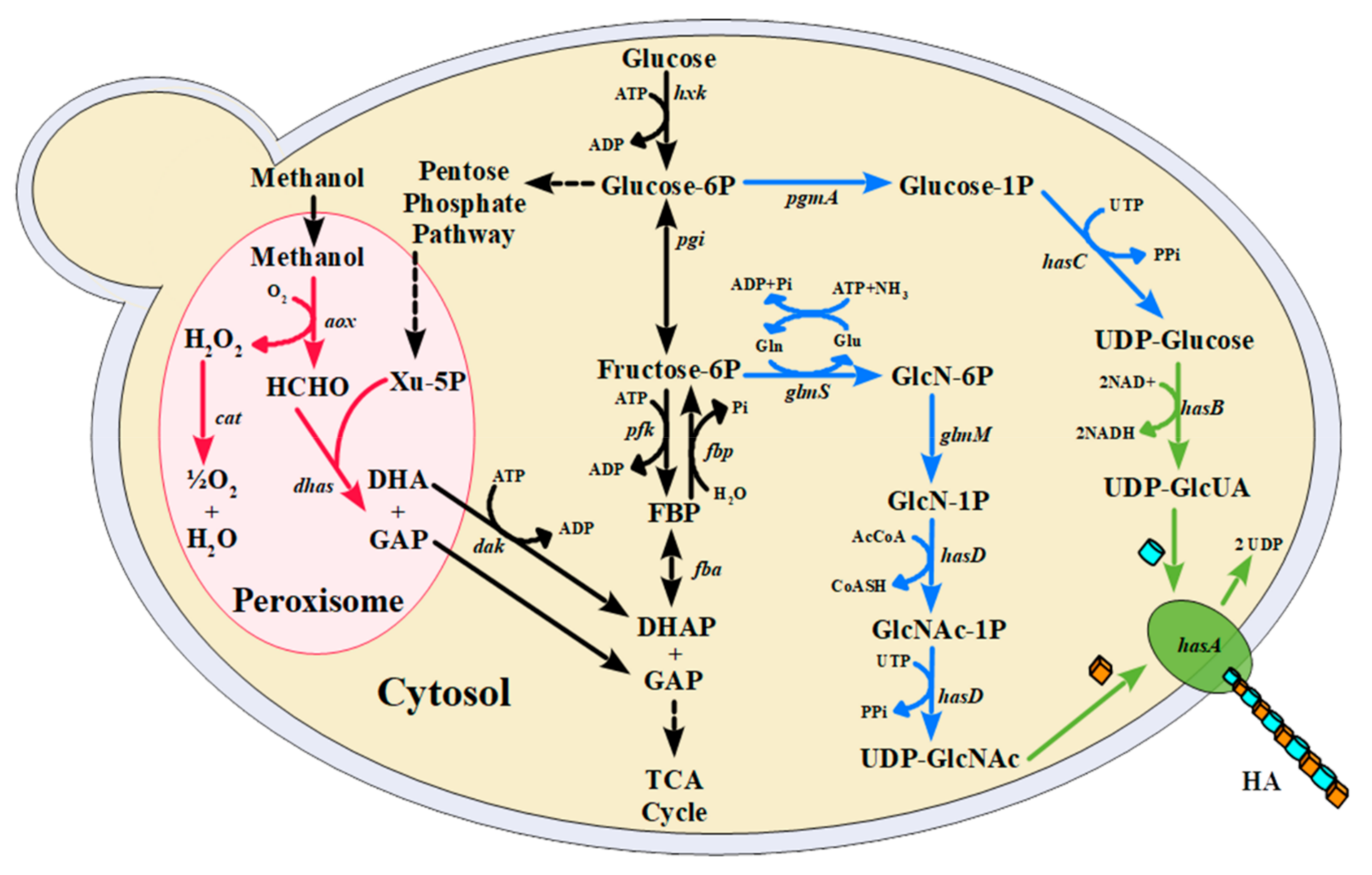
3.1. Strain Construction
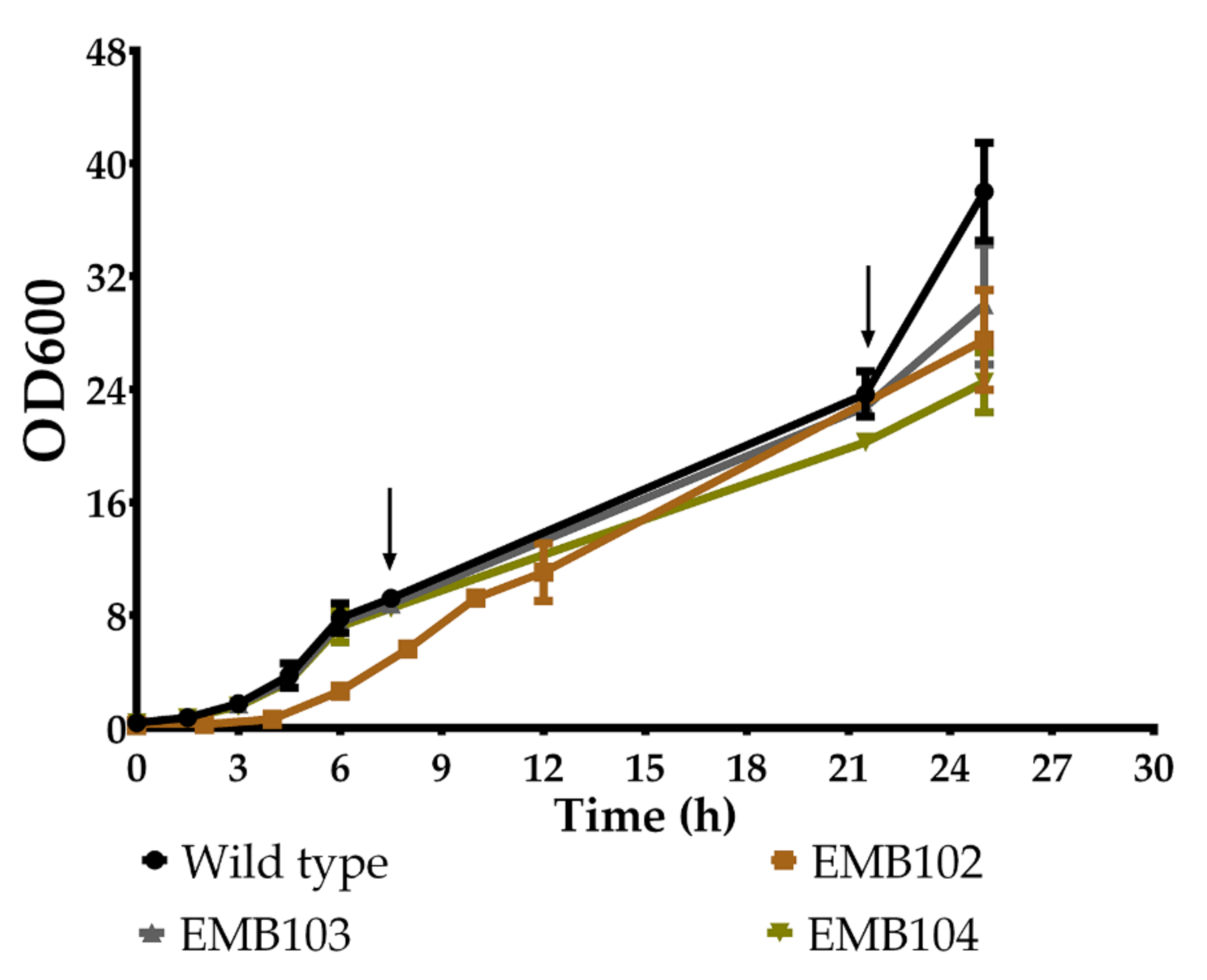
3.2. Growth Profile of Recombinant Strains
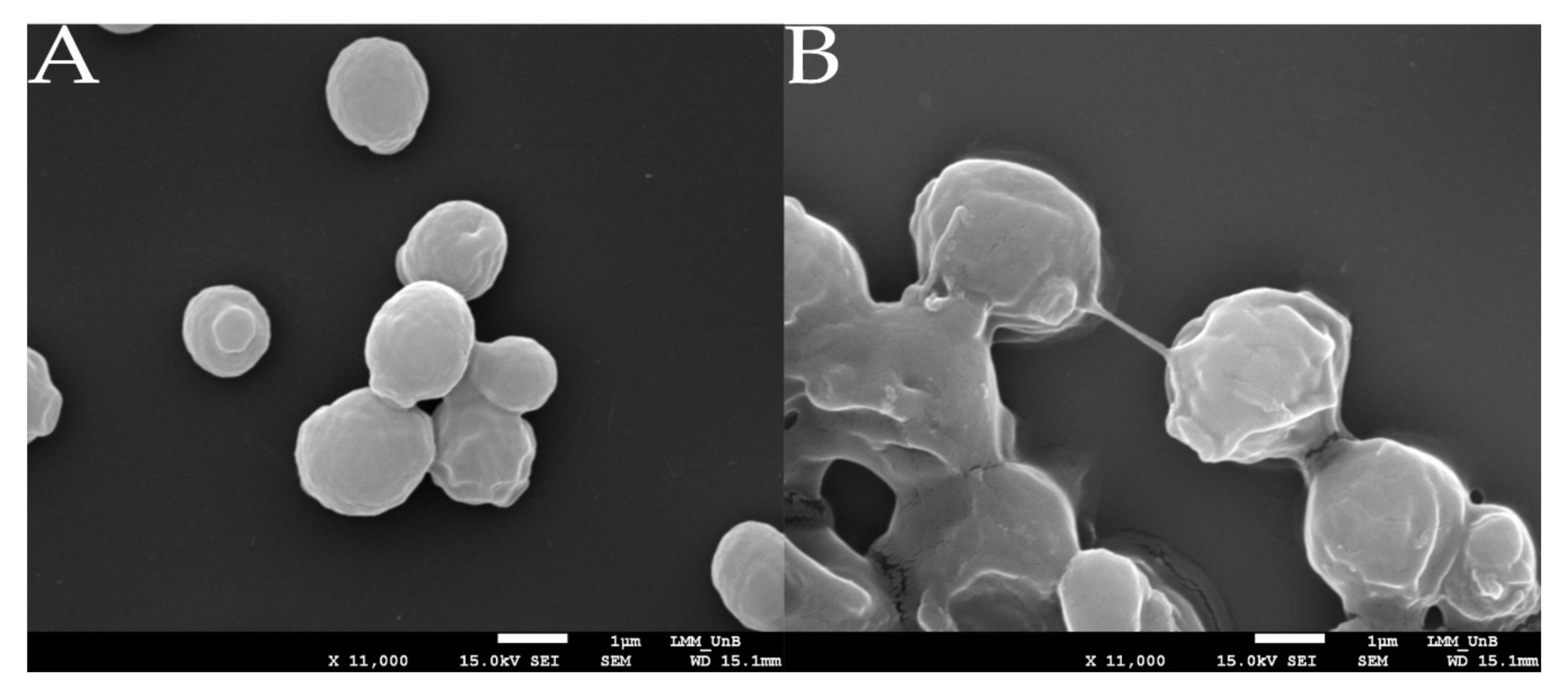
3.3. HA Production Assessed by Scanning Electron Microscopy Analysis
3.4. HA Quantification
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Carvalho, C.C.C.R. Whole cell biocatalysts: Essential workers from nature to the industry. Microb. Biotechnol. 2017, 10, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Tao, Y. Whole-cell biocatalysts by design. Microb. Cell Fact. 2017, 16, 106. [Google Scholar] [CrossRef] [PubMed]
- Fallacara, A.; Baldini, E.; Manfredini, S.; Vertuani, S. Hyaluronic acid in the third millennium. Polymers 2018, 10, 701. [Google Scholar] [CrossRef] [PubMed]
- Hyaluronic Acid Market Size, Share & Trends Analysis Report by Application (Dermal Fillers, Osteoarthritis (Single Injection, Three Injection, Five Injection), Ophthalmic, Vesicoureteral Reflux), by Region, and Segment Forecasts, 2020–2027; Grand View Research: San Francisco, CA, USA, 2020; p. 150.
- Sze, J.H.; Brownlie, J.C.; Love, C.A. Biotechnological production of hyaluronic acid: A mini review. 3 Biotech 2016, 6, 67. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, J.D.; Carvalho, L.S.; Gomes, A.M.V.; Queiroz, L.R.; Magalhães, B.S.; Parachin, N.S. Genetic basis for hyper production of hyaluronic acid in natural and engineered microorganisms. Microb. Cell Factories 2016, 15, 119. [Google Scholar] [CrossRef]
- Jin, P.; Kang, Z.; Yuan, P.; Du, G.; Chen, J. Production of specific-molecular-weight hyaluronan by metabolically engineered Bacillus subtilis 168. Metab. Eng. 2016, 35, 21–30. [Google Scholar] [CrossRef]
- Westbrook, A.W.; Ren, X.; Oh, J.; Moo-Young, M.; Chou, C.P. Metabolic Engineering to Enhance Heterologous Production of Hyaluronic Acid in Bacillus subtilis. Metab. Eng. 2018, 47, 401–413. [Google Scholar] [CrossRef]
- Cheng, F.; Yu, H.; Stephanopoulos, G. Engineering Corynebacterium glutamicum for high-titer biosynthesis of hyaluronic acid. Metab. Eng. 2019, 55, 276–289. [Google Scholar] [CrossRef]
- Jeeva, P.; Shanmuga Doss, S.; Sundaram, V.; Jayaraman, G. Production of controlled molecular weight hyaluronic acid by glucostat strategy using recombinant Lactococcus lactis cultures. Appl. Microbiol. Biotechnol. 2019, 103, 4363–4375. [Google Scholar] [CrossRef]
- Gomes, A.M.V.; Netto, J.H.C.M.; Carvalho, L.S.; Parachin, N.S. Heterologous hyaluronic acid production in Kluyveromyces Lactis. Microorganisms 2019, 7, 294. [Google Scholar] [CrossRef] [PubMed]
- Jeong, E.; Shim, W.Y.; Kim, J.H. Metabolic engineering of Pichia Pastoris for production of hyaluronic acid with high molecular weight. J. Biotechnol. 2014, 185, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Saraya, R.; Krikken, A.M.; Kiel, J.A.K.W.; Baerends, R.J.S.; Veenhuis, M.; Klei, I.J. Novel genetic tools for Hansenula polymorpha. FEMS Yeast Res. 2012, 12, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Gellissen, G.; Kunze, G.; Gaillardin, C.; Cregg, J.; Berardi, E.; Veenhuis, M.; Vanderklei, I. New yeast expression platforms based on methylotrophic Hansenula polymorpha and Pichia pastoris and on dimorphic Arxula adenivorans and Yarrowia lipolytica–A Comparison. FEMS Yeast Res. 2005, 5, 1079–1096. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, G.; Zhao, X.; Shao, Y.; Wu, M.; Ma, T. Regulation of hyaluronic acid molecular weight and titer by temperature in engineered Bacillus subtilis. 3 Biotech 2019, 9, 225. [Google Scholar] [CrossRef]
- Gündüz Ergün, B.; Hüccetoğulları, D.; Öztürk, S.; Çelik, E.; Çalık, P. Established and upcoming yeast expression systems. In Recombinant Protein Production in Yeast; Gasser, B., Mattanovich, D., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2019; Volume 1923, pp. 1–74. ISBN 978-1-4939-9023-8. [Google Scholar]
- Avila, J.; Pérez, M.D.; Brito, N.; González, C.; Siverio, J.M. Cloning and disruption of the YNR1 gene encoding the nitrate reductase apoenzyme of the yeast Hansenula polymorpha. FEBS Lett. 1995, 366, 137–142. [Google Scholar] [CrossRef]
- Brito, N.; Avila, J.; Pérez, M.D.; González, C.; Siverio, J.M. The genes YNI1 and YNR1, encoding nitrite reductase and nitrate reductase respectively in the yeast Hansenula polymorpha, are clustered and co-ordinately regulated. Biochem. J. 1996, 317, 89–95. [Google Scholar] [CrossRef]
- Amuel, C.; Gellissen, G.; Hollenberg, C.P.; Suckow, M. Analysis of heat shock promoters in Hansenula Polymorpha: The TPS1 promoter, a novel element for heterologous gene expression. Biotechnol. Bioprocess Eng. 2000, 5, 247–252. [Google Scholar] [CrossRef]
- Stöckmann, C.; Scheidle, M.; Dittrich, B.; Merckelbach, A.; Hehmann, G.; Melmer, G.; Klee, D.; Büchs, J.; Kang, H.A.; Gellissen, G.; et al. Process development in Hansenula polymorpha and Arxula adeninivorans, a Re-Assessment. Microb. Cell Factories 2009, 8, 10. [Google Scholar] [CrossRef]
- Manfrão-Netto, J.H.C.; Gomes, A.M.V.; Parachin, N.S. Advances in using Hansenula Polymorpha as chassis for recombinant protein production. Front. Bioeng. Biotechnol. 2019, 7, 94. [Google Scholar] [CrossRef]
- Suppi, S.; Michelson, T.; Viigand, K.; Alamäe, T. Repression vs. Activation of MOX, FMD, MPP1 and MAL1 promoters by sugars in Hansenula polymorpha: The outcome depends on cell’s ability to phosphorylate sugar. FEMS Yeast Res. 2013, 13, 219–232. [Google Scholar] [CrossRef]
- Dusny, C.; Schmid, A. The MOX promoter in Hansenula polymorpha is ultrasensitive to glucose-mediated carbon catabolite repression. FEMS Yeast Res. 2016, 16, fow067. [Google Scholar] [CrossRef] [PubMed]
- Hartner, F.S.; Glieder, A. Regulation of methanol utilisation pathway genes in yeasts. Microb. Cell Fact 2006, 5, 39. [Google Scholar] [CrossRef] [PubMed]
- Vogl, T.; Fischer, J.E.; Hyden, P.; Wasmayer, R.; Sturmberger, L.; Glieder, A. Orthologous promoters from related methylotrophic yeasts surpass expression of endogenous promoters of pichia pastoris. AMB Expr. 2020, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.F.; Hellmuth, K.; Schlieker, H.; Lopez-Ulibarri, R.; Oertel, S.; Dahlems, U.; Strasser, A.W.M.; van Loon, A.P.G.M. An expression system matures: A highly efficient and cost-effective process for phytase production by recombinant strains of Hansenula polymorpha. Biotechnol. Bioeng. 1999, 63, 373–381. [Google Scholar] [CrossRef]
- Chong, B.F.; Blank, L.M.; Mclaughlin, R.; Nielsen, L.K. Microbial hyaluronic acid production. Appl. Microbiol. Biotechnol. 2005, 66, 341–351. [Google Scholar] [CrossRef]
- Liu, L.; Liu, Y.; Li, J.; Du, G.; Chen, J. Microbial production of hyaluronic acid: Current state, challenges, and perspectives. Microb. Cell Fact 2011, 10, 99. [Google Scholar] [CrossRef]
- Kurylenko, O.O.; Ruchala, J.; Vasylyshyn, R.V.; Stasyk, O.V.; Dmytruk, O.V.; Dmytruk, K.V.; Sibirny, A.A. Peroxisomes and peroxisomal transketolase and transaldolase enzymes are essential for xylose alcoholic fermentation by the methylotrophic thermotolerant yeast, Ogataea (Hansenula) polymorpha. Biotechnol. Biofuels 2018, 11, 197. [Google Scholar] [CrossRef]
- Singh, R.; Manivannan, S.; Krikken, A.M.; Boer, R.; Bordin, N.; Devos, D.P.; Klei, I.J. Hansenula polymorpha Pex37 is a peroxisomal membrane protein required for organelle fission and segregation. FEBS J. 2020, 287, 1742–1757. [Google Scholar] [CrossRef]
- Merrick, C.A.; Zhao, J.; Rosser, S.J. Serine integrases: Advancing synthetic biology. ACS Synth. Biol. 2018, 7, 299–310. [Google Scholar] [CrossRef]
- Gomide, M.S.; Sales, T.T.; Barros, L.R.C.; Limia, C.G.; de Oliveira, M.A.; Florentino, L.H.; Barros, L.M.G.; Robledo, M.L.; José, G.P.C.; Almeida, M.S.M.; et al. Genetic switches designed for eukaryotic cells and controlled by serine integrases. Commun. Biol. 2020, 3, 255. [Google Scholar] [CrossRef]
- Prasad, S.B.; Jayaraman, G.; Ramachandran, K.B. Hyaluronic acid production is enhanced by the additional co-expression of UDP-glucose pyrophosphorylase in Lactococcus lactis. Appl. Microbiol. Biotechnol. 2010, 86, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Jongsareejit, B.; Bhumiratana, A.; Morikawa, M.; Kanaya, S. Cloning of hyaluronan synthase (Sz-Has) gene from Streptococcus zooepidemicus in Escherichia coli. ScienceAsia 2007, 33, 389. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D.W. Molecular Cloning-A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press, Cold Spring Harbor: New York, NY, USA, 2001. [Google Scholar]
- Green, M.R.; Sambrook, J. The hanahan method for preparation and transformation of competent, E. Coli: High-efficiency transformation. Cold Spring Harbor Protoc. 2018, 2018. pdb.prot101188. [Google Scholar] [CrossRef] [PubMed]
- Van der Klei, I.J. The Hansenula polymorpha Expression System. Available online: https://www.rug.nl/research/molecular-cell-biology/research/the-hansenula-polymorpha-expression-system (accessed on 19 May 2020).
- Nash, H.A. Integration and excision of Bacteriophage λ: The Mechanism of conservative site specific recombination. Annu. Rev. Genet. 1981, 15, 143–167. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, K.; Yuan, P.; Perry, K.; Sharp, R.; Van Duyne, G.D. Attachment site recognition and regulation of directionality by the serine integrases. Nucleic Acids Res. 2013, 41, 8341–8356. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Nielsen, A.A.K.; Fernandez-Rodriguez, J.; McClune, C.J.; Laub, M.T.; Lu, T.K.; Voigt, C.A. Permanent genetic memory with >1-byte capacity. Nat. Methods 2014, 9, 1261–1266. [Google Scholar] [CrossRef]
- Saraya, R.; Gidijala, L.; Veenhuis, M.; van der Klei, I.J. Tools for genetic engineering of the yeast Hansenula polymorpha. In Yeast Metabolic Engineering; Mapelli, V., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2014; Volume 1152, pp. 43–62. ISBN 978-1-4939-0562-1. [Google Scholar]
- Murtey, M.D.; Ramasamy, P. Sample preparations for scanning electron microscopy–life sciences. In Modern Electron Microscopy in Physical and Life Sciences; Janecek, M., Kral, R., Eds.; InTech: London, UK, 2016; ISBN 978-953-51-2252-4. [Google Scholar]
- Im, J.-H.; Song, J.-M.; Kang, J.-H.; Kang, D.-J. Optimization of medium components for high-molecular-weight hyaluronic acid production by Streptococcus Sp. ID9102 via a statistical approach. J. Ind. Microbiol. Biotechnol. 2009, 8, 1337. [Google Scholar] [CrossRef]
- Bitter, T.; Muir, H.M. A modified uronic acid carbazole reaction. Analytical Biochem. 1962, 4, 330–334. [Google Scholar] [CrossRef]
- Hmar, R.V.; Prasad, S.B.; Jayaraman, G.; Ramachandran, K.B. Chromosomal integration of hyaluronic acid synthesis (Has) genes enhances the molecular weight of hyaluronan produced in Lactococcus lactis. Biotechnol. J. 2014, 9, 1554–1564. [Google Scholar] [CrossRef]
- Wang, Y.; Hu, L.; Huang, H.; Wang, H.; Zhang, T.; Chen, J.; Du, G.; Kang, Z. Eliminating the capsule-like layer to promote glucose uptake for hyaluronan production by engineered Corynebacterium glutamicum. Nat. Commun. 2020, 11, 3120. [Google Scholar] [CrossRef]
- Yang, Z.; Blenner, M. Genome editing systems across yeast species. Curr. Opin. Biotechnol. 2020, 66, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Taylor, G.; Evans, S.K.; Fogg, P.C.M.; Smith, M.C.M. Application of serine integrases for secondary metabolite pathway assembly in Streptomyces. Synth. Syst. Biotechnol. 2020, 5, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Brown, W.R.A. Comparison and optimization of ten phage encoded serine integrases for genome engineering in Saccharomyces Cerevisiae. BMC Biotechnol. 2016, 16, 13. [Google Scholar] [CrossRef] [PubMed]
- Snoeck, N.; De Mol, M.L.; Van Herpe, D.; Goormans, A.; Maryns, I.; Coussement, P.; Peters, G.; Beauprez, J.; De Maeseneire, S.L.; Soetaert, W. Serine Integrase Recombinational Engineering (SIRE): A versatile toolbox for genome editing. Biotechnol. Bioeng. 2019, 116, 364–374. [Google Scholar] [CrossRef]
- Chung, J.Y.; Zhang, Y.; Adler, B. The capsule biosynthetic locus of Pasteurella multocida A:1. FEMS Microbiol. Lett. 1998, 166, 289–296. [Google Scholar] [CrossRef]
- Zhang, L.; Toscano Selão, T.; Nixon, P.J.; Norling, B. Photosynthetic conversion of CO2 to hyaluronic acid by engineered strains of the cyanobacterium Synechococcus sp. PCC 7002. Algal Res. 2019, 44, 101702. [Google Scholar] [CrossRef]
- Badle, S.S.; Jayaraman, G.; Ramachandran, K.B. Ratio of intracellular precursors concentration and their flux influences hyaluronic acid molecular weight in Streptococcus zooepidemicus and recombinant Lactococcus lactis. Bioresour. Technol. 2014, 163, 222–227. [Google Scholar] [CrossRef]
- Kaur, M.; Jayaraman, G. Hyaluronan production and molecular weight is enhanced in pathway-engineered strains of lactate dehydrogenase-deficient Lactococcus lactis. Metab. Eng. Commun. 2016, 3, 15–23. [Google Scholar] [CrossRef]
- Park, J.-N.; Choo, J.; Kang, H.A. Functional analysis of a Hansenula polymorpha MNN2-2 homologue encoding a putative UDP-N-acetylglucosamine transporter localized in the endoplasmic reticulum. J. Microbiol. 2011, 49, 1012–1017. [Google Scholar] [CrossRef]
- Woo, J.E.; Seong, H.J.; Lee, S.Y.; Jang, Y.-S. Metabolic engineering of Escherichia coli for the production of hyaluronic acid from glucose and galactose. Front. Bioeng. Biotechnol. 2019, 7, 351. [Google Scholar] [CrossRef]
- Lehnen, M.; Ebert, B.E.; Blank, L.M. Elevated temperatures do not trigger a conserved metabolic network response among thermotolerant yeasts. BMC Microbiol. 2019, 19, 100. [Google Scholar] [CrossRef] [PubMed]
- Ravin, N.V.; Eldarov, M.A.; Kadnikov, V.V.; Beletsky, A.V.; Schneider, J.; Mardanova, E.S.; Smekalova, E.M.; Zvereva, M.I.; Dontsova, O.A.; Mardanov, A.V.; et al. Genome sequence and analysis of methylotrophic yeast Hansenula polymorpha DL1. BMC Genom. 2013, 14, 837. [Google Scholar] [CrossRef] [PubMed]
- Rußmayer, H.; Buchetics, M.; Gruber, C.; Valli, M.; Grillitsch, K.; Modarres, G.; Guerrasio, R.; Klavins, K.; Neubauer, S.; Drexler, H.; et al. Systems-level organization of yeast methylotrophic lifestyle. BMC Biol. 2015, 13, 80. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EMB Strains | Promoter Driving hasA | Source of hasA | Promoter Driving hasB | Source of hasB |
|---|---|---|---|---|
| EMB102 | pTEF1 | P. multocida | pAOX | Xenopus laevis |
| EMB103 | pADH1 | P. multocida | pGPD (S. cerevisiae) | X. laevis |
| EMB104 | pAOX | S. zooepidemicus | pADH1 | X. laevis |
| Strains | μMAX (h−1) * | Final OD * | Doubling Time (h) * | Hyaluronic Acid (HA) Titer (μg/mL) ** | Final OD ** |
|---|---|---|---|---|---|
| WT | 0.50 ± 0.02 a | 38.00 ± 3.46 a | 1.35 ± 0.06 a | NA | 21.00 ± 1.41 a |
| EMB102 | 0.45 ± 0.04 a | 27.50 ± 3.54 a,b | 1.55 ± 0.15 a | 151.20 ± 13.04 a,b | 27.00 ± 0.00 a |
| EMB103 | 0.52 ± 0.01 a | 30.00 ± 4.24 a,b | 1.34 ± 0.01 a | 197.76 ± 5.66 b | 27.50 ± 0.71 a |
| EMB104 | 0.49 ± 0.02 a | 24.50 ± 2.12 b | 1.41 ± 0.07 a | 123.20 ± 26.56 a | 21.50 ± 3.54 a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manfrão-Netto, J.H.C.; Queiroz, E.B.; Rodrigues, K.A.; Coelho, C.M.; Paes, H.C.; Rech, E.L.; Parachin, N.S. Evaluation of Ogataea (Hansenula) polymorpha for Hyaluronic Acid Production. Microorganisms 2021, 9, 312. https://doi.org/10.3390/microorganisms9020312
Manfrão-Netto JHC, Queiroz EB, Rodrigues KA, Coelho CM, Paes HC, Rech EL, Parachin NS. Evaluation of Ogataea (Hansenula) polymorpha for Hyaluronic Acid Production. Microorganisms. 2021; 9(2):312. https://doi.org/10.3390/microorganisms9020312
Chicago/Turabian StyleManfrão-Netto, João Heitor Colombelli, Enzo Bento Queiroz, Kelly Assis Rodrigues, Cintia M. Coelho, Hugo Costa Paes, Elibio Leopoldo Rech, and Nádia Skorupa Parachin. 2021. "Evaluation of Ogataea (Hansenula) polymorpha for Hyaluronic Acid Production" Microorganisms 9, no. 2: 312. https://doi.org/10.3390/microorganisms9020312
APA StyleManfrão-Netto, J. H. C., Queiroz, E. B., Rodrigues, K. A., Coelho, C. M., Paes, H. C., Rech, E. L., & Parachin, N. S. (2021). Evaluation of Ogataea (Hansenula) polymorpha for Hyaluronic Acid Production. Microorganisms, 9(2), 312. https://doi.org/10.3390/microorganisms9020312