
Comparative Genomics across Three Ensifer Species Using a New Complete Genome Sequence of the Medicago Symbiont Sinorhizobium (Ensifer) meliloti WSM1022

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains Used in This Study
2.2. Preparation of Biological Material for Sequencing
2.3. Genome Assembly and Annotation
2.4. Data Availability
2.5. Comparative Genomics
3. Results
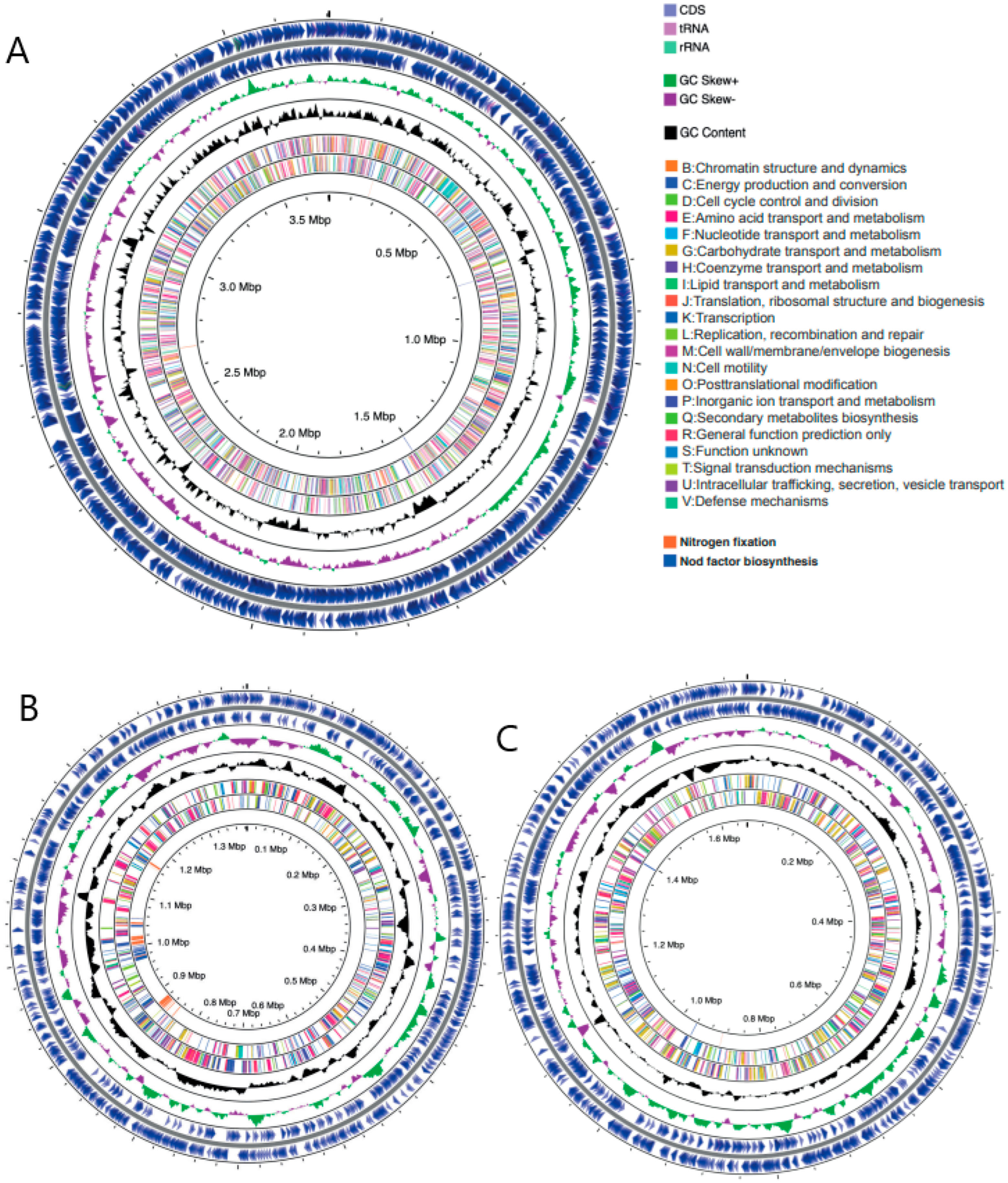
3.1. High-Quality Sinorhizobium Meliloti WSM1022 Genome Assembly and Annotation
3.2. Genomic Comparisons across the Three Rhizobial Strains
3.2.1. General Genome Feature Comparisons across the Rhizobial Strains in This Study
3.2.2. Differences in Genome Feature Arrangement
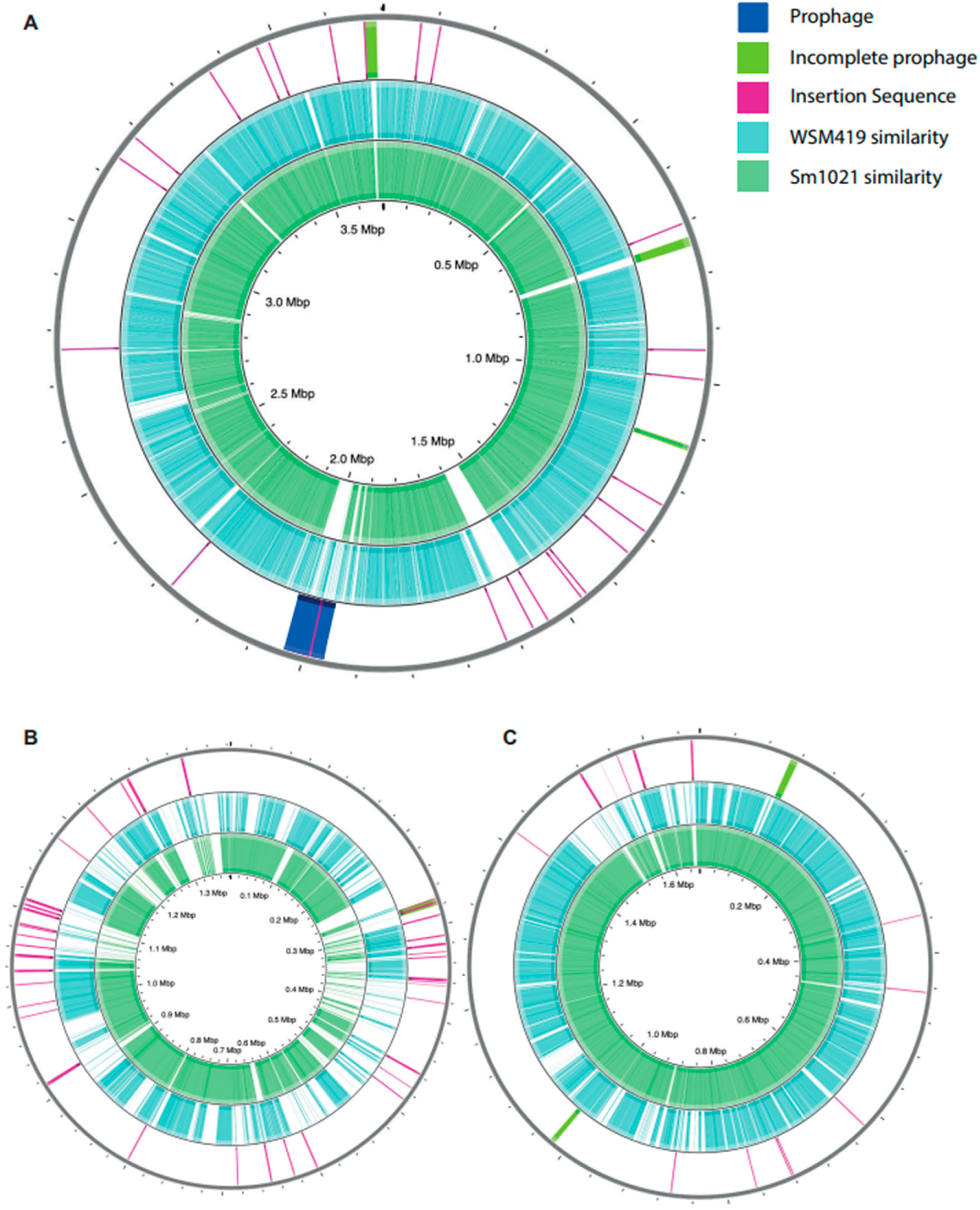
3.2.3. Insertion Sequences and Bacteriophages
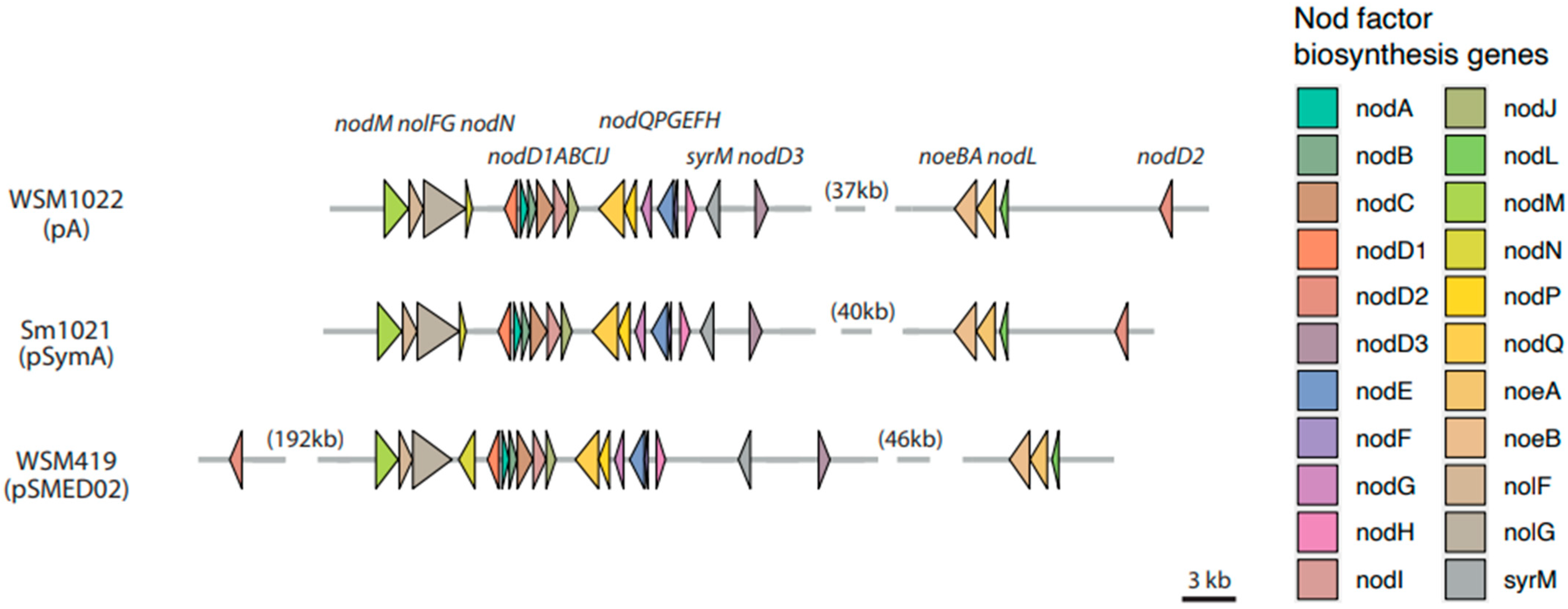
3.2.4. Nod Factor Biosynthesis and Nitrogen Fixation Genes
3.3. Orthologous Protein Predictions
3.3.1. Sinorhizobium meliloti Orthologous Protein Clusters
3.3.2. Protein Clusters Putatively Linked to Medicago truncatula High-Efficiency Compatible Nitrogen Fixer Symbionts
3.3.3. Relevant Unique Protein Clusters and Singletons
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zahran, H.H. Rhizobium-legume symbiosis and nitrogen fixation under severe conditions and in an arid climate. Microbiol. Mol. Biol. Rev. 1999, 63, 968–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foyer, C.; Nguyen, H.; Lam, H. Legumes-The art and science of environmentally sustainable agriculture. Plant Cell Environ. 2019, 42, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Galibert, F.; Finan, T.M.; Long, S.R.; Puhler, A.; Abola, P.; Ampe, F.; Barloy-Hubler, F.; Barnett, M.J.; Becker, A.; Boistard, P.; et al. The composite genome of the legume symbiont Sinorhizobium meliloti. Science 2001, 293, 668–672. [Google Scholar] [CrossRef] [Green Version]
- Young, N.D.; Udvardi, M. Translating Medicago truncatula genomics to crop legumes. Curr. Opin. Plant Biol. 2009, 2, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Arrighi, J.F.; Cartieaux, F.; Brown, S.C.; Rodier-Goud, M.; Boursot, M.; Fardoux, J.; Patrel, D.; Gully, D.; Fabre, S.; Chaintreuil, C.; et al. Aeschynomene evenia, a model plant for studying the molecular genetics of the nod-independent rhizobium-legume symbiosis. Mol. Plant Microbe Interact. 2012, 25, 851–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, D.R. Medicago truncatula—A model in the making! Curr. Opin. Plant Biol. 1999, 2, 301–304. [Google Scholar] [CrossRef]
- Garau, G.; Reeve, W.G.; Brau, L.; Deiana, P.; Yates, R.J.; James, D.; Tiwari, R.; O’Hara, G.W.; Howieson, J.G. The symbiotic requirements of different medicago spp. suggest the evolution of sinorhizobium meliloti and s. medicae with hosts differentially adapted to soil pH. Plant Soil 2005, 276, 263–277. [Google Scholar] [CrossRef]
- O’Hara, G.W.; Terpolilli, J.; Tiwari, R.P.; Dilworth, M.J.; Howieson, J.G. The model legume Medicago truncatula A17 is poorly matched for N2-fixation with the sequenced microsymbiont Sinorhizobium meliloti 1021. New Phytol. 2008, 179, 62–66. [Google Scholar]
- Meade, H.M.; Long, S.R.; Ruvkun, G.B.; Brown, S.E.; Ausubel, M.F. Physical and genetic characterization of symbiotic and auxotrophic mutants of Rhizobium meliloti induced by transposon Tn5 mutagenesis. J. Bacteriol. 1982, 149, 114–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terpolilli, J.; Hill, Y.; Tian, R.; Howieson, J.; Bräu, L.; Goodwin, L.; Han, J.; Liolios, K.; Huntemann, M.; Pati, A.; et al. Genome sequence of Ensifer meliloti strain WSM1022; a highly effective microsymbiont of the model legume Medicago truncatula A17. Stand. Genom. Sci. 2013, 9, 315–324. [Google Scholar] [CrossRef] [Green Version]
- Reeve, W.; Chain, P.; O’Hara, G.; Ardley, J.; Nandesena, K.; Bräu, L.; Tiwari, R.; Malfatti, S.; Kiss, H.; Lapidus, A.; et al. Complete genome sequence of the Medicago microsymbiont Ensifer (Sinorhizobium) medicae strain WSM419. Stand. Genom. Sci. 2010, 2, 77–86. [Google Scholar] [CrossRef] [Green Version]
- Antipov, D.; Korobeynikov, A.; McLean, J.S.; Pevzner, P.A. HybridSPAdes: An algorithm for hybrid assembly of short and long reads. Bioinformatics 2016, 32, 1009–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Alcalde, F.; Okonechnikov, K.; Carbonell, J.; Cruz, L.M.; Götz, S.; Tarazona, S.; Dopazo, J.; Meyer, T.F.; Conesa, A. Qualimap: Evaluating next-generation sequencing alignment data. Bioinformatics 2012, 28, 2678–2679. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; O’Neill, K.R.; Haft, D.H.; DiCuccio, M.; Chetvernin, V.; Badretdin, A.; Coulouris, G.; Chitsaz, F.; Derbyshire, M.K.; Durkin, A.S.; et al. RefSeq: Expanding the Prokaryotic Genome Annotation Pipeline reach with protein family model curation. Nucleic Acids Res. 2021, 49, D1020–D1028. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef] [PubMed]
- Stothard, P.; Wishart, D.S. Circular genome visualization and exploration using CGView. Bioinformatics 2005, 21, 537–539. [Google Scholar] [CrossRef] [Green Version]
- Mewes, H.W.; Frishman, D.; Mayer, K.F.; Münsterkötter, M.; Noubibou, O.; Pagel, P.; Rattei, T.; Oesterheld, M.; Ruepp, A.; Stümpflen, V. MIPS: Analysis and annotation of proteins from whole genomes in 2005. Nucleic Acids Res. 2006, 34, D169–D172. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Tang, H. ISEScan: Automated identification of insertion sequence elements in prokaryotic genomes. Bioinformatics 2017, 33, 3340–3347. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [Green Version]
- Eichinger, V.; Nussbaumer, T.; Platzer, A.; Jehl, M.A.; Arnold, R.; Rattei, T. EffectiveDB—Updates and novel features for a better annotation of bacterial secreted proteins and Type III, IV, VI secretion systems. Nucleic Acids Res. 2016, 44, D669–D674. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Dong, Z.; Fang, L.; Luo, Y.; Wei, Z.; Guo, H.; Zhang, G.; Gu, Y.Q.; Coleman-Derr, D.; Xia, Q.; et al. OrthoVenn2: A web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2019, 47, W52–W58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pritchard, L.; Glover, R.H.; Humphris, S.; Elphinstone, J.G.; Toth, I.K. Genomics and taxonomy in diagnostics for food security: Soft-rotting enterobacterial plant pathogens. Anal. Methods 2015, 8, 12–24. [Google Scholar] [CrossRef]
- Smits, T.H.M. The importance of genome sequence quality to microbial comparative genomics. BMC Genom. 2019, 20, 662. [Google Scholar] [CrossRef] [PubMed]
- Baptista, R.P.; Kissinger, J.C. Is reliance on an inaccurate genome sequence sabotaging your experiments? PLoS Pathog. 2019, 15, e1007901. [Google Scholar] [CrossRef]
- Sugawara, M.; Epstein, B.; Badgley, B.D.; Unno, T.; Xu, L.; Reese, J.; Gyaneshwar, P.; Denny, R.; Mudge, J.; Bharti, A.K.; et al. Comparative genomics of the core and accessory genomes of 48 Sinorhizobium strains comprising five genospecies. Genome Biol. 2013, 14, R17. [Google Scholar] [CrossRef]
- Lozano, L.; Hernández-González, I.; Bustos, P.; Santamaría, R.I.; Souza, V.; Young, J.P.; Dávila, G.; González, V. Evolutionary dynamics of insertion sequences in relation to the evolutionary histories of the chromosome and symbiotic plasmid genes of Rhizobium etli populations. Appl. Environ. Microbiol. 2010, 76, 6504–6513. [Google Scholar] [CrossRef] [Green Version]
- Dénarié, J.; Cullimore, J. Lipo-oligosaccharide nodulation factors: A minireview new class of signaling molecules mediating recognition and morphogenesis. Cell 1993, 74, 951–954. [Google Scholar] [CrossRef]
- Lindström, K.; Mousavi, S.A. Effectiveness of nitrogen fixation in rhizobia. Microb. Biotechnol. 2020, 13, 1314–1335. [Google Scholar] [CrossRef] [Green Version]
- Laranjo, M.; Alexandre, A.; Oliveira, S. Legume growth-promoting rhizobia: An overview on the Mesorhizobium genus. Microbiol. Res. 2014, 169, 2–17. [Google Scholar] [CrossRef]
- Nogales, J.; Domínguez-Ferreras, A.; Amaya-Gómez, C.V.; van Dillewijn, P.; Cuéllar, V.; Sanjuán, J.; Olivares, J.; Soto, M.J. Transcriptome profiling of a Sinorhizobium meliloti fadD mutant reveals the role of rhizobactin 1021 biosynthesis and regulation genes in the control of swarming. BMC Genom. 2010, 11, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaya-Gómez, C.V.; Hirsch, A.M.; Soto, M.J. Biofilm formation assessment in Sinorhizobium meliloti reveals interlinked control with surface motility. BMC Microbiol. 2015, 15, 58. [Google Scholar] [CrossRef] [Green Version]
- Tampakaki, A.P. Commonalities and differences of T3SSs in rhizobia and plant pathogenic bacteria. Front. Plant Sci. 2014, 5, 114. [Google Scholar] [CrossRef] [PubMed]
- Viprey, V.; del Greco, A.; Golinowski, W.; Broughton, W.J.; Perret, X. Symbiotic implications of type III protein secretion machinery in Rhizobium. Mol. Microbiol. 1998, 28, 1381–1389. [Google Scholar] [CrossRef] [PubMed]
- Deakin, W.J.; Broughton, W.J. Symbiotic use of pathogenic strategies: Rhizobial protein secretion systems. Nat. Rev. Microbiol. 2009, 7, 312–320. [Google Scholar] [CrossRef]
- Teulet, A.; Busset, N.; Fardoux, J.; Gully, D.; Chaintreuil, C.; Cartieaux, F.; Jauneau, A.; Comorge, V.; Okazaki, S.; Kaneko, T.; et al. The rhizobial type III effector ErnA confers the ability to form nodules in legumes. Proc. Natl. Acad. Sci. USA 2019, 116, 21758–21768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudrappa, T.; Biedrzycki, M.L.; Kunjeti, S.G.; Donofrio, N.M.; Czymmek, K.J.; Paré, P.W.; Bais, H.P. The rhizobacterial elicitor acetoin induces systemic resistance in Arabidopsis thaliana. Commun. Integr. Biol. 2010, 3, 130–138. [Google Scholar] [CrossRef] [Green Version]
- Patil, A.; Kale, A.; Ajane, G.; Sheikh, R.; Patil, S. Soil Biology Book Series; Springer: Cham, Switzerland, 2017; Volume 50. [Google Scholar]
- Inoue, T.; Shingaki, R.; Hirose, S.; Waki, K.; Mori, H.; Fukui, K. Genome-wide screening of genes required for swarming motility in Escherichia coli K-12. J. Bacteriol. 2007, 189, 950–957. [Google Scholar] [CrossRef] [Green Version]
- Matilla, M.A.; Ramos, J.L.; Duque, E.; Alché, J.d.; Espinosa-Urgel, M.; Ramos-González, M.I. Temperature and pyoverdine-mediated iron acquisition control surface motility of Pseudomonas putida. Environ. Microbiol. 2007, 9, 1842–1850. [Google Scholar] [CrossRef]
- Storey, E.P.; Boghozian, R.; Little, J.L.; Lowman, D.W.; Chakraborty, R. Characterization of ‘Schizokinen’; a dihydroxamate-type siderophore produced by Rhizobium leguminosarum IARI 917. Biometals 2006, 19, 637–649. [Google Scholar] [CrossRef]
- Viguier, C.; Cuív, P.O.; Clarke, P.; O’Connell, M. RirA is the iron response regulator of the rhizobactin 1021 biosynthesis and transport genes in Sinorhizobium meliloti 2011. FEMS Microbiol. Lett. 2005, 246, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carson, K.C.; Holliday, S.; Glenn, A.R.; Dilworth, M.J. Siderophore and organic acid production in root nodule bacteria. Arch. Microbiol. 1992, 157, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.T.; Trzebiatowski, J.R.; Cruickshank, R.W.; Gouzy, J.; Brown, S.D.; Elliot, R.M.; Fleetwood, D.J.; McCallum, N.G.; Rossbach, U.; Stuart, G.S.; et al. Comparative sequence analysis of the symbiosis island of Mesorhizobium loti strain R7A. J. Bacteriol. 2002, 184, 3086–3095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christie, P.J.; Whitaker, N.; González-Rivera, C. Mechanism and structure of the bacterial type IV secretion systems. Biochim. Biophys. Acta 2014, 1843, 1578–1591. [Google Scholar] [CrossRef] [Green Version]
- Zehner, S.; Schober, G.; Wenzel, M.; Lang, K.; Göttfert, M. Expression of the Bradyrhizobium japonicum type III secretion system in legume nodules and analysis of the associated tts box promoter. Mol. Plant Microbe Interact. 2008, 21, 1087–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WSM1022 | Sm1021 | WSM419 | |
|---|---|---|---|
| Species | S. meliloti | S. meliloti | S. medicae |
| Genome size (bp, total) | 6,751,834 | 6,691,694 | 6,817,576 |
| No. of contigs | 3 | 3 | 4 |
| No. of chromosomes | 1 | 1 | 1 |
| No. of plasmids | 2 | 2 | 3 |
| GC content (%) | 62.22 | 62.17 | 61.15 |
| RefSeq/GenBank assembly accession | GCF_0013315775.1 | GCF_00006965.1 | GCF_000017145.1 |
| Genes (total) | 6328 | 6293 | 6464 |
| CDS (total) | 6260 | 6225 | 6396 |
| CDS (with protein) | 6058 | 5981 | 6068 |
| Genes (RNA) | 68 | 68 | 68 |
| rRNAs (5S, 15S, 23S) | 3, 3, 3 | 3, 3, 3 | 3, 3, 3 |
| tRNAs | 55 | 55 | 55 |
| ncRNAs | 4 | 4 | 4 |
| Pseudo Genes (total, without protein) | 202 | 244 | 328 |
| Pseudo Genes (frameshifted) | 134 of 202 | 173 of 244 | 218 of 328 |
| Pseudo Genes (incomplete) | 97 of 202 | 97 of 244 | 170 of 328 |
| Pseudo Genes (internal stop) | 18 of 202 | 28 of 244 | 41 of 326 |
| Pseudo Genes (multiple problems) | 45 of 202 | 49 of 244 | 94 of 326 |
| COG ID | COG Symbol | WSM1022 Protein | Sm1021 Protein | WSM419 Protein | |
|---|---|---|---|---|---|
| Type III | COG4669 | EscJ | QKN17985.1 | - | - |
| COG4790 | EscR/YscR | QKN17990.1 | - | - | |
| COG4794 | EscS/YscS | QKN17991.1 | - | - | |
| COG4791 | EscT/YscT | QKN17992.1 | - | - | |
| COG4792 | EscU/YscU | QKN17973.1 | - | - | |
| COG4789 | EscV | QKN17977.1 | - | - | |
| COG1157 | FliI | QKN17988.1 | WP_010968730.1 | WP_011974463.1 | |
| COG1317 | FliH | - | - | - | |
| Type IV | COG3838 | VirB2 | QKN18893.1 | WP_010967695.1 | WP_011971086.1 |
| COG3702 | VirB3 | QKN18440.1 | WP_010967694.1 | WP_011971088.1 | |
| COG3701 | TrbF | - | - | WP_011970261.1 | |
| COG3504 | VirB9 | QKN18446.1 | WP_010967688.1 | WP_011971094.1 | |
| COG3704 | VirB6 | QKN18443.1 | WP_013845459.1 | WP_011971092.1 | |
| COG3736 | VirB8 | QKN18445.1 | WP_010967689.1 | WP_011971093.1 | |
| COG2948 | VirB10 | QKN18447.1 | WP_010967687.1 | WP_011971095.1 | |
| COG3451 | VirB4 | - | - | - | |
| COG0630 | VirB11 | QKN18448.1 | WP_010967686.1 | - | |
| COG3505 | VirD4 | QKN18557.1 | WP_010967483.1 | WP_024325706.1 | |
| COG3157 | Hcp | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baxter, L.; Roy, P.; Picot, E.; Watts, J.; Jones, A.; Wilkinson, H.; Schäfer, P.; Gifford, M.; Lagunas, B. Comparative Genomics across Three Ensifer Species Using a New Complete Genome Sequence of the Medicago Symbiont Sinorhizobium (Ensifer) meliloti WSM1022. Microorganisms 2021, 9, 2428. https://doi.org/10.3390/microorganisms9122428
Baxter L, Roy P, Picot E, Watts J, Jones A, Wilkinson H, Schäfer P, Gifford M, Lagunas B. Comparative Genomics across Three Ensifer Species Using a New Complete Genome Sequence of the Medicago Symbiont Sinorhizobium (Ensifer) meliloti WSM1022. Microorganisms. 2021; 9(12):2428. https://doi.org/10.3390/microorganisms9122428
Chicago/Turabian StyleBaxter, Laura, Proyash Roy, Emma Picot, Jess Watts, Alex Jones, Helen Wilkinson, Patrick Schäfer, Miriam Gifford, and Beatriz Lagunas. 2021. "Comparative Genomics across Three Ensifer Species Using a New Complete Genome Sequence of the Medicago Symbiont Sinorhizobium (Ensifer) meliloti WSM1022" Microorganisms 9, no. 12: 2428. https://doi.org/10.3390/microorganisms9122428
APA StyleBaxter, L., Roy, P., Picot, E., Watts, J., Jones, A., Wilkinson, H., Schäfer, P., Gifford, M., & Lagunas, B. (2021). Comparative Genomics across Three Ensifer Species Using a New Complete Genome Sequence of the Medicago Symbiont Sinorhizobium (Ensifer) meliloti WSM1022. Microorganisms, 9(12), 2428. https://doi.org/10.3390/microorganisms9122428