
Pangenomics of the Symbiotic Rhizobiales. Core and Accessory Functions Across a Group Endowed with High Levels of Genomic Plasticity

 ,
,  ,
, 
 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Selected Bacterial Strains and Main Genomic Features
2.2. Bioinformatics Analyses
3. Results
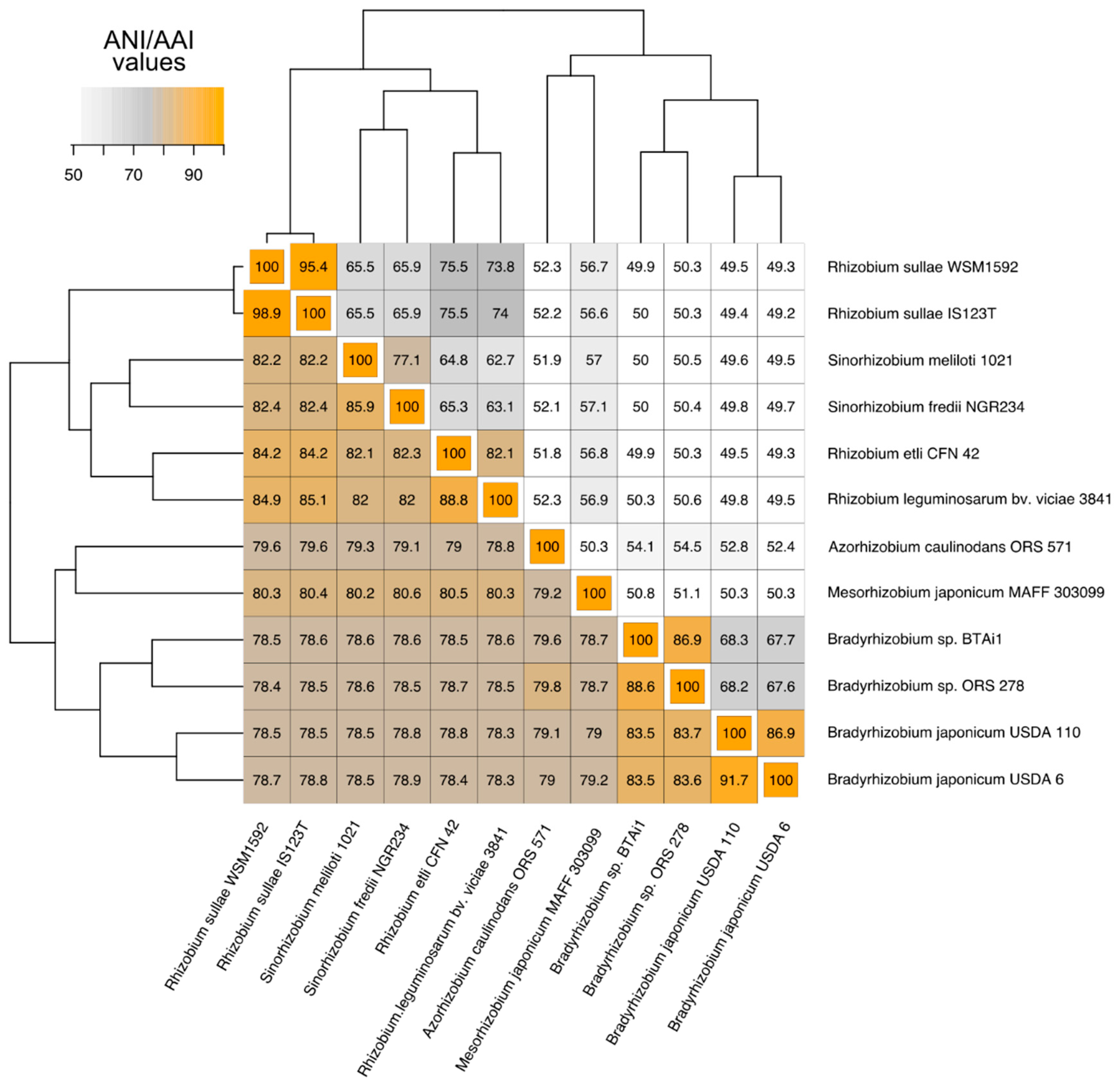
3.1. Phylogenetic Relatedness Among the 12 Rhizobiales Strains
3.2. Pangenome and Core Genome Analyses
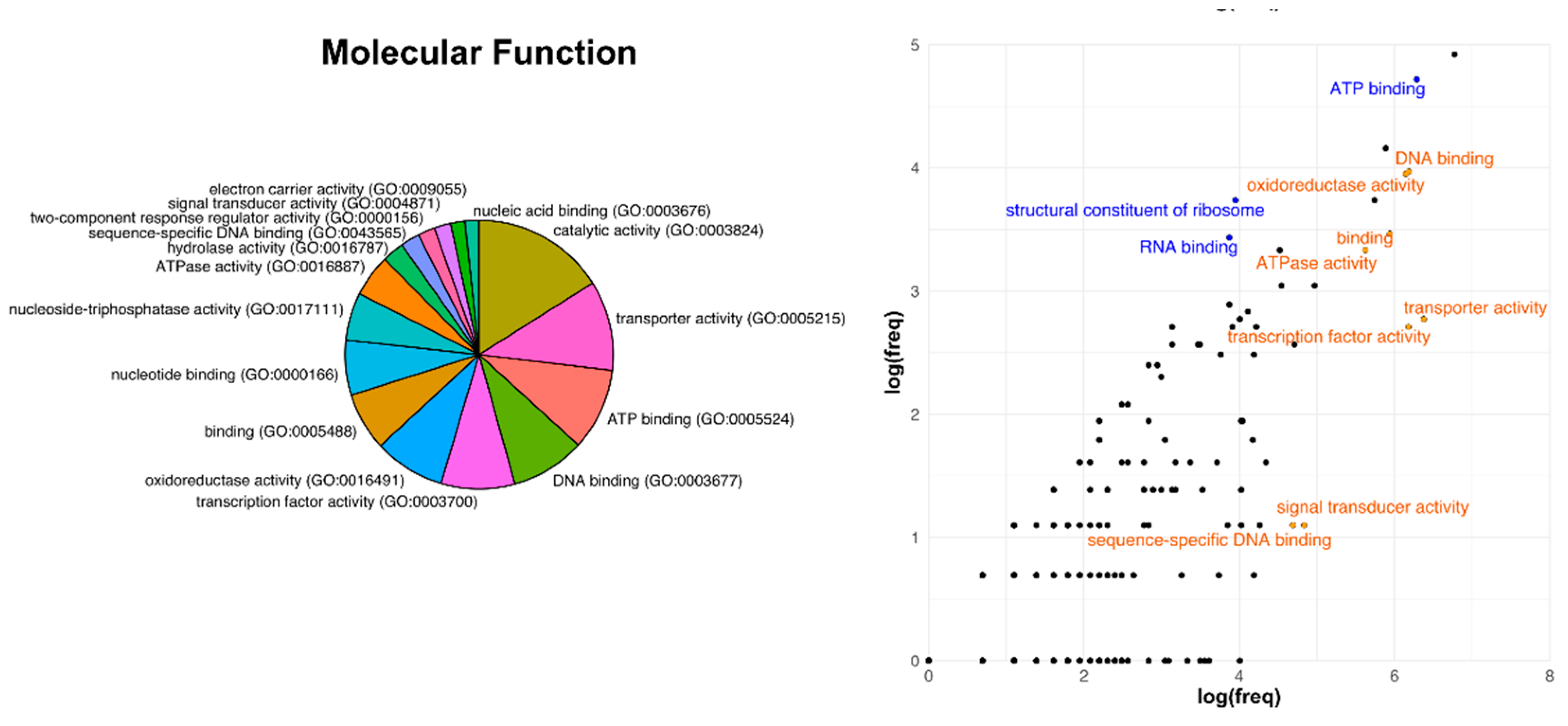
3.3. Analysis of Core Genome Function Enrichment and Depletion Using a Single Reference Genome
3.4. Functional Divergence Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Brockhurst, M.A.; Harrison, E.; Hall, J.P.J.; Richards, T.; McNally, A.; MacLean, C. The Ecology and Evolution of Pangenomes. Curr. Biol. 2019, 29, R1094–R1103. [Google Scholar] [CrossRef] [PubMed]
- McInerney, J.O.; McNally, A.; O’Connell, M.J. Why prokaryotes have pangenomes. Nat. Microbiol. 2017, 2, 17040. [Google Scholar] [CrossRef] [PubMed]
- Rasko, D.A.; Rosovitz, M.J.; Myers, G.S.A.; Mongodin, E.F.; Fricke, W.F.; Gajer, P.; Crabtree, J.; Sebaihia, M.; Thomson, N.R.; Chaudhuri, R.; et al. The pangenome structure of Escherichia coli: Comparative genomic analysis of E. coli commensal and pathogenic isolates. J. Bacteriol. 2008, 190, 6881–6893. [Google Scholar] [CrossRef] [PubMed]
- Checcucci, A.; Azzarello, E.; Bazzicalupo, M.; Galardini, M.; Lagomarsino, A.; Mancuso, S.; Marti, L.; Marzano, M.C.; Mocali, S.; Squartini, A.; et al. Mixed nodule infection in Sinorhizobium meliloti-medicago sativa symbiosis suggest the presence of cheating behavior. Front. Plant Sci. 2016, 7, 835. [Google Scholar] [CrossRef] [PubMed]
- Mosquera-Rendón, J.; Rada-Bravo, A.M.; Cárdenas-Brito, S.; Corredor, M.; Restrepo-Pineda, E.; Benítez-Páez, A. Pangenome-wide and molecular evolution analyses of the Pseudomonas aeruginosa species. BMC Genom. 2016, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Udaondo, Z.; Duque, E.; Ramos, J.L. The pangenome of the genus Clostridium. Environ. Microbiol. 2017, 19, 2588–2603. [Google Scholar] [CrossRef]
- González, V.; Santamaría, R.I.; Bustos, P.; Pérez-Carrascal, O.M.; Vinuesa, P.; Juárez, S.; Martínez-Flores, I.; Cevallos, M.Á.; Brom, S.; Martínez-Romero, E.; et al. Phylogenomic Rhizobium species are structured by a continuum of diversity and genomic clusters. Front. Microbiol. 2019, 10, 910. [Google Scholar] [CrossRef]
- Li, N.; Wang, K.; Williams, H.N.; Sun, J.; Ding, C.; Leng, X.; Dong, K. Analysis of gene gain and loss in the evolution of predatory bacteria. Gene 2017, 598, 63–70. [Google Scholar] [CrossRef]
- Epstein, B.; Tiffin, P. Comparative genomics reveals high rates of horizontal transfer and strong purifying selection on rhizobial symbiosis genes. Proc. R. Soc. B Biol. Sci. 2021, 288, 20201804. [Google Scholar] [CrossRef]
- Levy, A.; Salas Gonzalez, I.; Mittelviefhaus, M.; Clingenpeel, S.; Herrera Paredes, S.; Miao, J.; Wang, K.; Devescovi, G.; Stillman, K.; Monteiro, F.; et al. Genomic features of bacterial adaptation to plants. Nat. Genet. 2018, 50, 138–150. [Google Scholar] [CrossRef]
- Kimura, M. The neutral theory of molecular evolution. Sci. Am. 1979, 241, 98–129. [Google Scholar] [CrossRef]
- Caffrey, B.E.; Williams, T.A.; Jiang, X.; Toft, C.; Hokamp, K.; Fares, M.A. Proteome-wide analysis of functional divergence in bacteria: Exploring a host of ecological adaptations. PLoS ONE 2012, 7, e35659. [Google Scholar] [CrossRef]
- Studer, R.A.; Robinson-Rechavi, M. How confident can we be that orthologs are similar, but paralogs differ? Trends Genet. 2009, 25, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Gharib, W.H.; Robinson-Rechavi, M. When orthologs diverge between human and mouse. Brief. Bioinform. 2011, 12, 436–441. [Google Scholar] [CrossRef]
- Nehrt, N.L.; Clark, W.T.; Radivojac, P.; Hahn, M.W. Testing the ortholog conjecture with comparative functional genomic data from mammals. PLoS Comput. Biol. 2011, 7, e1002073. [Google Scholar] [CrossRef] [PubMed]
- Stamboulian, M.; Guerrero, R.F.; Hahn, M.W.; Radivojac, P. The ortholog conjecture revisited: The value of orthologs and paralogs in function prediction. Bioinformatics 2020, 36, i219–i226. [Google Scholar] [CrossRef]
- Young, J.P.W.; Downer, H.L.; Eardly, B.D. Phylogeny of the phototrophic rhizobium strain BTAi1 by polymerase chain reaction-based sequencing of a 16S rRNA gene segment. J. Bacteriol. 1991, 173, 2271–2277. [Google Scholar] [CrossRef]
- Sprent, J.I. Evolution and diversity in the legume-rhizobium symbiosis: Chaos theory? Plant Soil 1994, 161, 1–10. [Google Scholar] [CrossRef]
- Boyd, E.S.; Peters, J.W. New insights into the evolutionary history of biological nitrogen fixation. Front. Microbiol. 2013, 4, 201. [Google Scholar] [CrossRef]
- Schmid, M.; HartMann, A. Molecular Phylogeny and Ecology of Root Associated Diazotrophic α- and β-Proteobacteria. In Associative and Endophytic Nitrogen-fixing Bacteria and Cyanobacterial Associations; Springer: Berlin/Heidelberg, Germany, 2007; Volume 5, pp. 21–40. [Google Scholar]
- Robertson, B.K.; Dreyfus, B.; Alexander, M. Ecology of stem-nodulating Rhizobium and Azorhizobium in four vegetation zones of Senegal. Microb. Ecol. 1995, 29, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Squartini, A.; Struffi, P.; Döring, H.; Selenska-Pobell, S.; Tola, E.; Giacomini, A.; Vendramin, E.; Velázquez, E.; Mateos, P.F.; Martínez-Molina, E.; et al. Rhizobium sullae sp. nov. (formerly “Rhizobium hedysari”), the root-nodule microsymbiont of Hedysarum coronarium L. Int. J. Syst. Evol. Microbiol. 2002, 52, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Muresu, R.; Porceddu, A.; Sulas, L.; Squartini, A. Nodule-associated microbiome diversity in wild populations of Sulla coronaria reveals clues on the relative importance of culturable rhizobial symbionts and co-infecting endophytes. Microbiol. Res. 2019, 221, 10–14. [Google Scholar] [CrossRef] [PubMed]
- de Diego-Diaz, B.; Treu, L.; Campanaro, S.; Duarte, V.S.; Saviane, A.; Cappellozza, S.; Squartini, A. Genome sequence of Enterococcus mundtii EM01, isolated from Bombyx mori midgut and responsible for flacherie disease in silkworms reared on an artificial diet. Genome Announc. 2018, 6. [Google Scholar] [CrossRef]
- Lee, K.B.; De Backer, P.; Aono, T.; Liu, C.T.; Suzuki, S.; Suzuki, T.; Kaneko, T.; Yamada, M.; Tabata, S.; Kupfer, D.M.; et al. The genome of the versatile nitrogen fixer Azorhizobium caulinodans ORS571. BMC Genom. 2008, 9, 271. [Google Scholar] [CrossRef] [PubMed]
- Dreyfus, B.L.; Dommergues, Y.R. Nitrogen-fixing nodules induced by Rhizobium on the stem of the tropical legume Sesbania rostrata. FEMS Microbiol. Lett. 1981, 10, 313–317. [Google Scholar] [CrossRef]
- Kaneko, T.; Nakamura, Y.; Sato, S.; Minamisawa, K.; Uchiumi, T.; Sasamoto, S.; Watanabe, A.; Idesawa, K.; Iriguchi, M.; Kawashima, K.; et al. Complete genomic sequence of nitrogen-fixing symbiotic bacterium Bradyrhizobium japonicum USDA110. DNA Res. 2002, 9, 189–197. [Google Scholar] [CrossRef]
- Kaneko, T.; Maita, H.; Hirakawa, H.; Uchiike, N.; Minamisawa, K.; Watanabe, A.; Sato, S. Complete genome sequence of the soybean symbiont bradyrhizobium japonicum strain USDA6 T. Genes 2011, 2, 763–787. [Google Scholar] [CrossRef]
- Giraud, E.; Moulin, L.; Vallenet, D.; Barbe, V.; Cytryn, E.; Avarre, J.C.; Jaubert, M.; Simon, D.; Cartieaux, F.; Prin, Y.; et al. Legumes symbioses: Absence of Nod genes in photosynthetic bradyrhizobia. Science 2007, 316, 1307–1312. [Google Scholar] [CrossRef]
- Kaneko, T.; Nakamura, Y.; Sato, S.; Asamizu, E.; Kato, T.; Sasamoto, S.; Watanabe, A.; Idesawa, K.; Ishikawa, A.; Kawashima, K.; et al. Complete genome structure of the nitrogen-fixing symbiotic bacterium Mesorhizobium loti. DNA Res. 2000, 7, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Young, J.P.W.; Crossman, L.C.; Johnston, A.W.B.; Thomson, N.R.; Ghazoui, Z.F.; Hull, K.H.; Wexler, M.; Curson, A.R.J.; Todd, J.D.; Poole, P.S.; et al. The genome of Rhizobium leguminosarum has recognizable core and accessory components. Genome Biol. 2006, 7, R34. [Google Scholar] [CrossRef] [PubMed]
- González, V.; Santamaría, R.I.; Bustos, P.; Hernández-González, I.; Medrano-Soto, A.; Moreno-Hagelsieb, G.; Janga, S.C.; Ramírez, M.A.; Jiménez-Jacinto, V.; Collado-Vides, J.; et al. The partitioned Rhizobium etli genome: Genetic and metabolic redundancy in seven interacting replicons. Proc. Natl. Acad. Sci. USA 2006, 103, 3834–3839. [Google Scholar] [CrossRef]
- Sablok, G.; Rosselli, R.; Seeman, T.; van Velzen, R.; Polone, E.; Giacomini, A.; La Porta, N.; Geurts, R.; Muresu, R.; Squartini, A. Draft genome sequence of the nitrogen-fixing Rhizobium sullae type strain IS123T focusing on the key genes for symbiosis with its host Hedysarum coronarium L. Front. Microbiol. 2017, 8, 1348. [Google Scholar] [CrossRef]
- Yates, R.; Howieson, J.; De Meyer, S.E.; Tian, R.; Seshadri, R.; Pati, A.; Woyke, T.; Markowitz, V.; Ivanova, N.; Kyrpides, N.; et al. High-quality permanent draft genome sequence of Rhizobium sullae strain WSM1592; a Hedysarum coronarium microsymbiont from Sassari, Italy. Stand. Genom. Sci. 2015, 10, 44. [Google Scholar] [CrossRef]
- Schmeisser, C.; Liesegang, H.; Krysciak, D.; Bakkou, N.; Le Quéré, A.; Wollherr, A.; Heinemeyer, I.; Morgenstern, B.; Pommerening-Röser, A.; Flores, M.; et al. Rhizobium sp. strain NGR234 possesses a remarkable number of secretion systems. Appl. Environ. Microbiol. 2009, 75, 4035–4045. [Google Scholar] [CrossRef] [PubMed]
- Pueppke, S.G.; Broughton, W.J. Rhizobium sp. strain NGR234 and R. fredii USDA257 share exceptionally broad, nested host ranges. Mol. Plant Microbe Interact. 1999, 12, 293–318. [Google Scholar] [CrossRef] [PubMed]
- Galibert, F.; Finan, T.M.; Long, S.R.; Pühler, A.; Abola, P.; Ampe, F.; Barloy-Hubler, F.; Barnet, M.J.; Becker, A.; Boistard, P.; et al. The composite genome of the legume symbiont Sinorhizobium meliloti. Science 2001, 293, 668–672. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, K.T.; Tiedje, J.M. Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 2567–2572. [Google Scholar] [CrossRef] [PubMed]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Moreira, B.; Vinuesa, P. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 2013, 79, 7696–7701. [Google Scholar] [CrossRef]
- Kaas, R.S.; Friis, C.; Ussery, D.W.; Aarestrup, F.M. Estimating variation within the genes and inferring the phylogeny of 186 sequenced diverse Escherichia coli genomes. BMC Genom. 2012, 13, 577. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Börnigen, D.; Morgan, X.C.; Huttenhower, C. PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat. Commun. 2013, 4, 1–11. [Google Scholar] [CrossRef]
- Ward, N.; Moreno-Hagelsieb, G. Quickly finding orthologs as reciprocal best hits with BLAT, LAST, and UBLAST: How much do we miss? PLoS ONE 2014, 9, e101850. [Google Scholar] [CrossRef]
- Ankenbrand, M.J.; Keller, A. BcgTree: Automatized phylogenetic tree building from bacterial core genomes. Genome 2016, 59, 783–791. [Google Scholar] [CrossRef]
- Arai, W.; Taniguchi, T.; Goto, S.; Moriya, Y.; Uehara, H.; Takemoto, K.; Ogata, H.; Takami, H. Maple 2.3.0: An improved system for evaluating the functionomes of genomes and metagenomes. Biosci. Biotechnol. Biochem. 2018, 82, 1515–1517. [Google Scholar] [CrossRef] [PubMed]
- Takami, H.; Taniguchi, T.; Arai, W.; Takemoto, K.; Moriya, Y.; Goto, S. An automated systemfor evaluation of the potential functionome: MAPLE version 2.1.0. DNA Res. 2016, 23, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [PubMed]
- Gu, X. A simple statistical method for estimating type-II (cluster-specific) functional divergence of protein sequences. Mol. Biol. Evol. 2006, 23, 1937–1945. [Google Scholar] [CrossRef]
- Rouli, L.; Merhej, V.; Fournier, P.E.; Raoult, D. The bacterial pangenome as a new tool for analysing pathogenic bacteria. New Microbes New Infect. 2015, 7, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Galardini, M.; Mengoni, A.; Brilli, M.; Pini, F.; Fioravanti, A.; Lucas, S.; Lapidus, A.; Cheng, J.F.; Goodwin, L.; Pitluck, S.; et al. Exploring the symbiotic pangenome of the nitrogen-fixing bacterium Sinorhizobium meliloti. BMC Genom. 2011, 12, 235. [Google Scholar] [CrossRef]
- Kaluza, K.; Hahn, M.; Hennecke, H. Repeated sequences similar to insertion elements clustered around the nif region of the Rhizobium japonicum genome. J. Bacteriol. 1985, 162, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Minamisawa, K.; Isawa, T.; Nakatsuka, Y.; Ichikawa, N. New Bradyrhizobium japonicum strains that possess high copy numbers of the repeated sequence RSα. Appl. Environ. Microbiol. 1998, 64, 1845–1851. [Google Scholar] [CrossRef] [PubMed]
- Perret, X.; Viprey, V.; Freiberg, C.; Broughton, W.J. Structure and evolution of NGRRS-1, a complex, repeated element in the genome of Rhizobium sp. strain NGR234. J. Bacteriol. 1997, 179, 7488–7496. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Individual Genomes | Core Genome (700) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Total Genes | On
Chromosome | On
Plasmids | N. Plasmids | % On Plasmids | N. Unique Genes | % of Unique Genes in Genome | On
Plasmids | % On Plasmids | |
| A. caul. ORS571 | 4780 | 4780 | 0 | 0 | 0 | 2239 | 46.84 | 0 | 0 |
| B. jap. USDA 110 | 8374 | 8374 | 0 | 0 | 0 | 1908 | 22.78 | 0 | 0 |
| B. jap. USDA 6 | 8888 | 8888 | 0 | 0 | 0 | 2424 | 27.27 | 0 | 0 |
| B. jap. BtAi1 | 7694 | 7466 | 228 | 1 | 2.96 | 1935 | 25.15 | 0 | 0 |
| Br. sp. ORS 278 | 6783 | 6783 | 0 | 0 | 0 | 1354 | 19.96 | 0 | 0 |
| M. loti MAFF3030. | 7343 | 6814 | 529 | 2 | 7.20 | 3804 | 51.80 | 0 | 0 |
| R. sullae IS123 | 7780 | nd | nd | nd | nd | 1455 | 18.70 | nd | nd |
| R. etli CFN42 | 6022 | 4094 | 1928 | 6 | 32.02 | 1047 | 17.39 | 36 | 5.14 |
| R. leg. viciae 3841 | 7342 | 4797 | 2545 | 6 | 34.66 | 1796 | 24.46 | 37 | 5.29 |
| R.sullae WSM1592 | 6995 | nd | nd | nd | nd | 547 | 8.10 | nd | nd |
| R. fredii NGR234 | 6437 | 3691 | 2746 | 2 | 42.66 | 1775 | 27.57 | 44 | 6.29 |
| S. meliloti 1021 | 6287 | 3425 | 2862 | 2 | 45.52 | 1686 | 26.82 | 50 | 7.14 |
| N. of Paralog Copies | N. of Paralogs | % Paralogs in Genome | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 3 | 4 | 5 | 6 | 7 | 8 | 11 | 13 | |||
| A. caulinodans ORS571 | 16 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 36 | 0.75 |
| B. japonicum USDA110 | 57 | 5 | 1 | 3 | 0 | 0 | 1 | 1 | 1 | 180 | 2.15 |
| B. japonicum USDA 6 | 50 | 4 | 1 | 0 | 0 | 2 | 1 | 0 | 0 | 138 | 1.55 |
| Br. sp. BTAi1 | 55 | 9 | 0 | 2 | 0 | 1 | 0 | 0 | 0 | 154 | 2.00 |
| Br. sp. ORS278 | 28 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 65 | 0.96 |
| M. loti MAFF303099 | 50 | 6 | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 134 | 1.82 |
| R. sullae IS123T | 56 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 115 | 1.48 |
| R. etli CFN42 | 41 | 2 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 106 | 1.76 |
| R. leg. bv. viciae 3841 | 58 | 8 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 148 | 2.02 |
| R. sullae WSM1592 | 29 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 61 | 0.90 |
| S. fredii NGR234 | 65 | 13 | 0 | 5 | 2 | 0 | 2 | 0 | 0 | 207 | 3.22 |
| S. meliloti 1021 | 49 | 6 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 128 | 2.04 |
| Category | Whole Genome | Core Genome | ||
|---|---|---|---|---|
| number | % | number | % | |
| Cell Processes | 1478 | 20.13 | 100 | 14.29 |
| Extrachromosomal | 130 | 1.77 | 0 | 0 |
| Macromolecule Metabolism | 414 | 5.64 | 144 | 20.57 |
| Membrane/Exported/Lipoproteins | 664 | 9.04 | 28 | 4.00 |
| Metabolism of Small Molecules | 1552 | 21.14 | 279 | 39.86 |
| Regulation | 708 | 9.64 | 33 | 4.71 |
| Ribosome Constituents | 55 | 0.75 | 43 | 6.14 |
| RNA | 61 | 0.83 | 0 | 0 |
| Unknown Function | 2280 | 31.05 | 73 | 10.43 |
| Total | 7342 | 700 | ||
| Gene code (from B.j.USDA6) | Diverged in the Following Rhizobia | Definition | Category | Biological
Process | Molecular Function |
|---|---|---|---|---|---|
| BJ6T_01910 | B. j. USDA110, B. j. USDA 6, B. sp. BTAi1, B.. sp. ORS278, M. l. MAFF303099 | Cobyrinic acid synthase | Coenzyme transport and metabolism | undefined | protein binding (GO:0005515) |
| BJ6T_03330 | A. c_ORS571, B. j. USDA110, B. j. USDA 6, B. sp. BTAi1 | protease II | Amino acid transport and metabolism | proteolysis (GO:0006508) | serine-type endopeptidase activity (GO:0004252) |
| BJ6T_04060 | B. j. USDA 6, B.sp. BTAi1, B.. sp. ORS278 | ATP synthase Gamma chain | Energy production and conversion | ATP synthesis coupled proton transport (GO:0015986) | proton-transporting ATPase, rotational mechanism (GO:0046961) |
| BJ6T_11840 | A.c._ORS571, B. j. USDA110, B. j. USDA 6, B. sp. BTAi1, B. sp. ORS278 | DNA polymerase I | Replication, recombination and repair | DNA repair (GO:0006281), DNA replication (GO:0006260) | undefined |
| BJ6T_16480 | B. j. USDA110, B. j. USDA 6, B.. sp. BTAi1, B.. sp. ORS278 | inositol monophosphatase family protein | Carbohydrate transport and metabolism | phosphatidylinositol phosphorylation (GO:0046854) | histidinol-phosphatase activity (GO:0004401) |
| BJ6T_87180 | A. c._ORS571 B. j. USDA110, B. j. USDA 6, B. sp. BTAi1, B.. sp. ORS278, M. l. MAFF303099 | putative glutathione synthetase | Coenzyme transport and metabolism | glutathione biosynthetic process (GO:0006750) | glutamate-cysteine ligase activity (GO:0004357) |
| BJ6T_59850 | B. j.USDA110, B. j. USDA 6, B. sp. BTAi1, B. sp. ORS278 | hypothetical protein | undefined | Cell wall/membrane/envelope biogenesis | undefined |
| BJ6T_67020 | A. c._ORS571 B. j. USDA110, B. j. USDA 6, B. sp. BTAi1, B.. sp. ORS278, M. l. MAFF303099 | hypothetical protein | undefined | undefined | undefined |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosselli, R.; La Porta, N.; Muresu, R.; Stevanato, P.; Concheri, G.; Squartini, A. Pangenomics of the Symbiotic Rhizobiales. Core and Accessory Functions Across a Group Endowed with High Levels of Genomic Plasticity. Microorganisms 2021, 9, 407. https://doi.org/10.3390/microorganisms9020407
Rosselli R, La Porta N, Muresu R, Stevanato P, Concheri G, Squartini A. Pangenomics of the Symbiotic Rhizobiales. Core and Accessory Functions Across a Group Endowed with High Levels of Genomic Plasticity. Microorganisms. 2021; 9(2):407. https://doi.org/10.3390/microorganisms9020407
Chicago/Turabian StyleRosselli, Riccardo, Nicola La Porta, Rosella Muresu, Piergiorgio Stevanato, Giuseppe Concheri, and Andrea Squartini. 2021. "Pangenomics of the Symbiotic Rhizobiales. Core and Accessory Functions Across a Group Endowed with High Levels of Genomic Plasticity" Microorganisms 9, no. 2: 407. https://doi.org/10.3390/microorganisms9020407
APA StyleRosselli, R., La Porta, N., Muresu, R., Stevanato, P., Concheri, G., & Squartini, A. (2021). Pangenomics of the Symbiotic Rhizobiales. Core and Accessory Functions Across a Group Endowed with High Levels of Genomic Plasticity. Microorganisms, 9(2), 407. https://doi.org/10.3390/microorganisms9020407