
Functional Characterisation of Bile Metagenome: Study of Metagenomic Dark Matter

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bile Samples and Participants
2.2. Fosmid Libraries Preparation
2.2.1. Extraction of Total DNA for Library Construction
2.2.2. High-Molecular-Weight DNA Purification and Fractionation
2.2.3. Library Construction and Isolation
2.3. Functional Analysis of Bile Metagenome: Antibiotic and Multidrug Resistance Genes (ARGs and MDRs)
2.4. Functional Analysis of Bile Metagenome: Metagenomic Dark Matter
3. Results and Discussion
3.1. Functional Analysis of Bile Metagenome: Antibiotic and Multidrug Resistance Genes
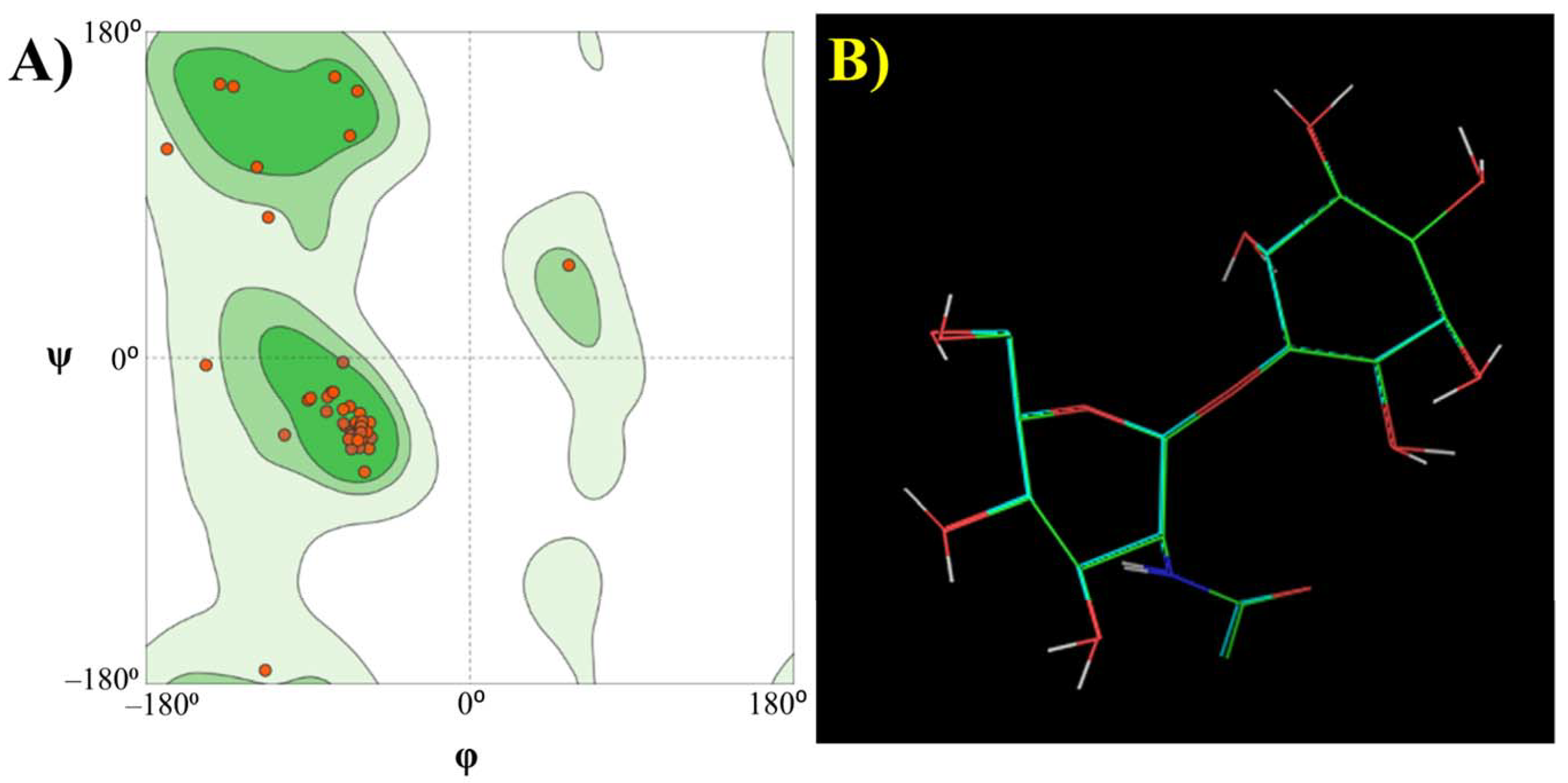
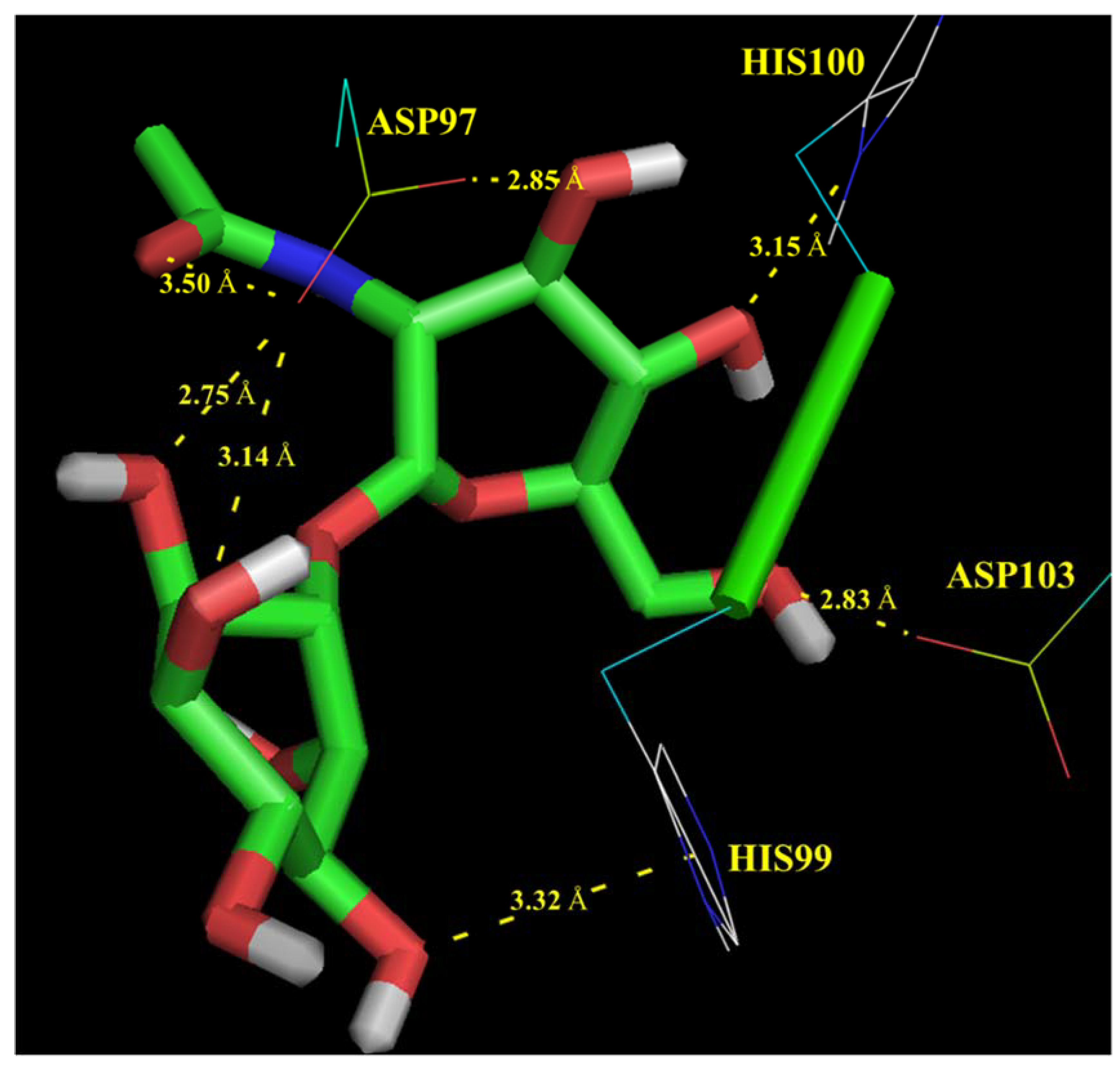
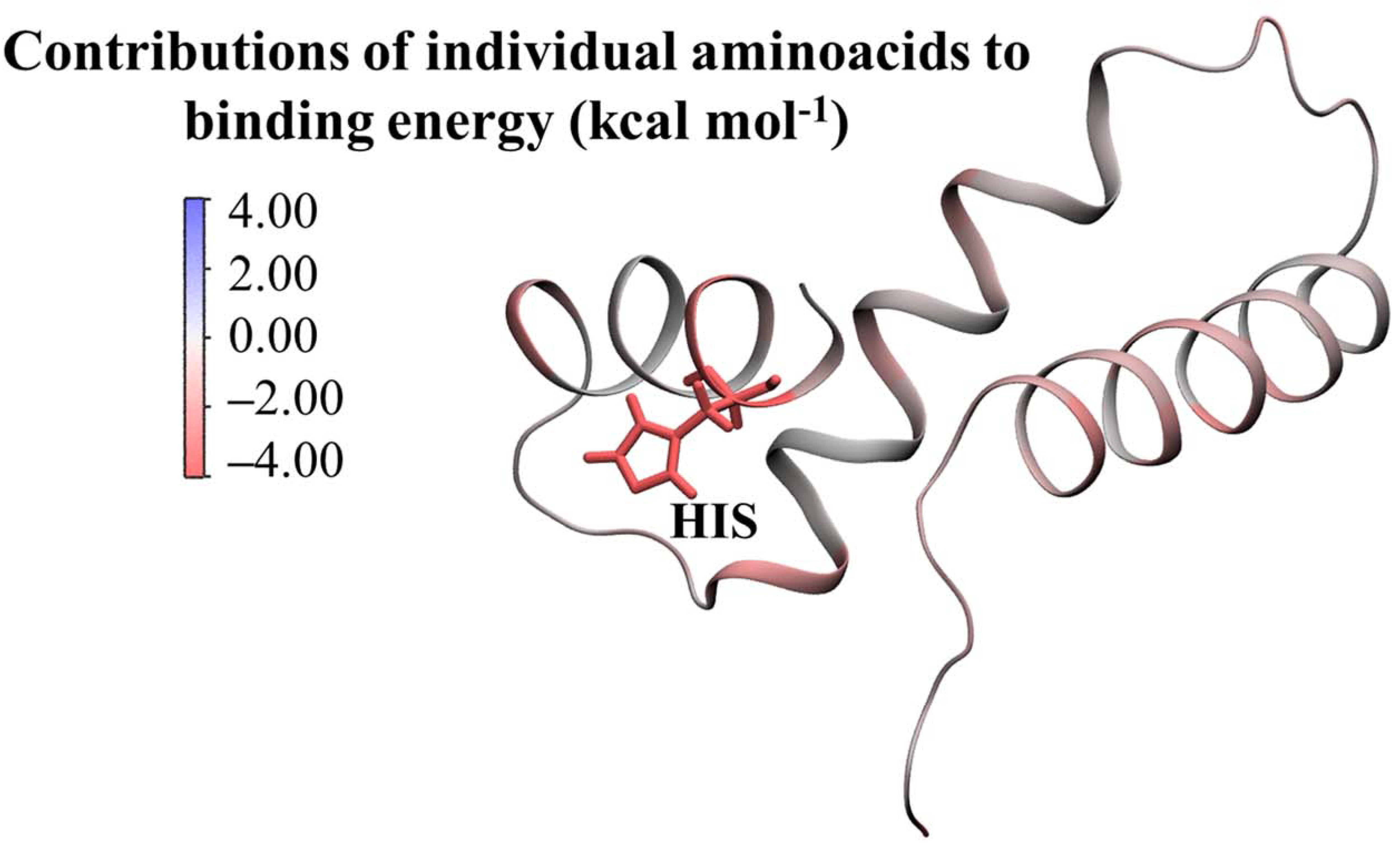
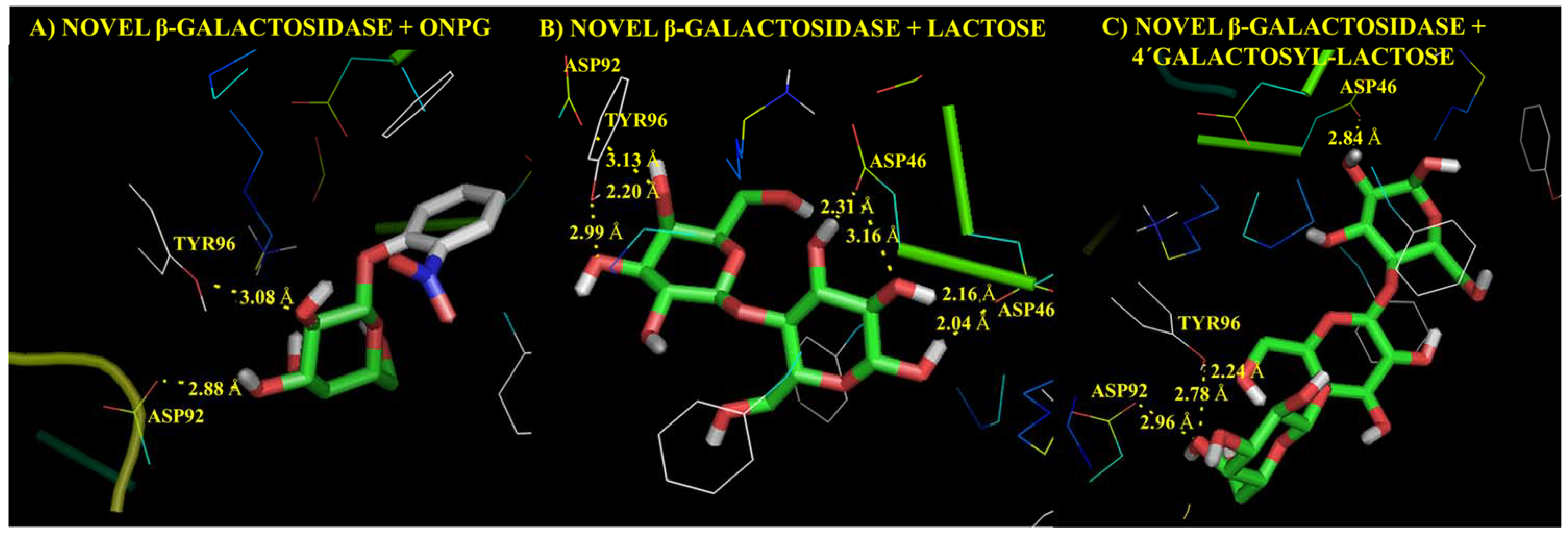
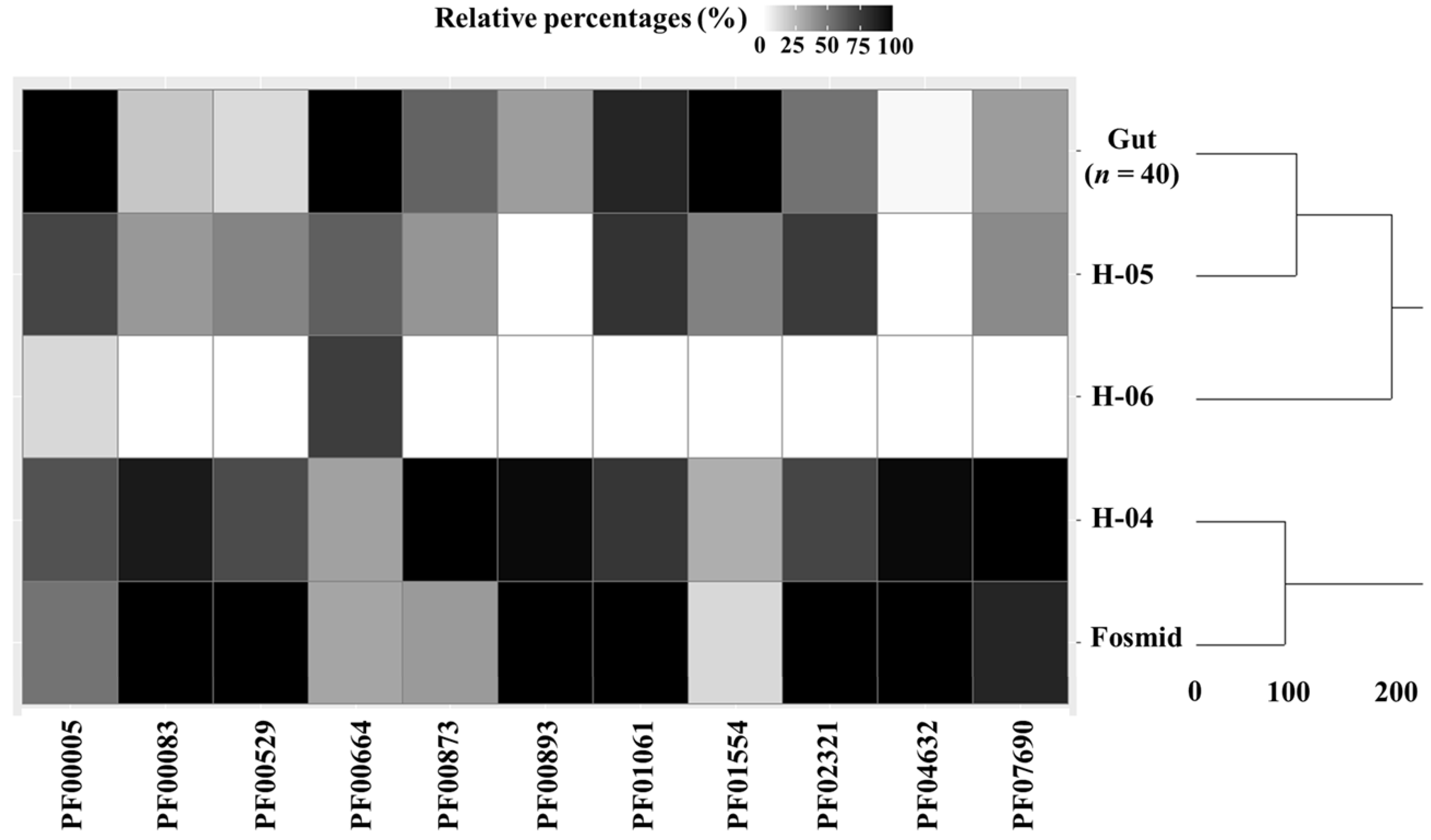
3.2. Functional Analysis of Bile Metagenome: Metagenomic Dark Matter
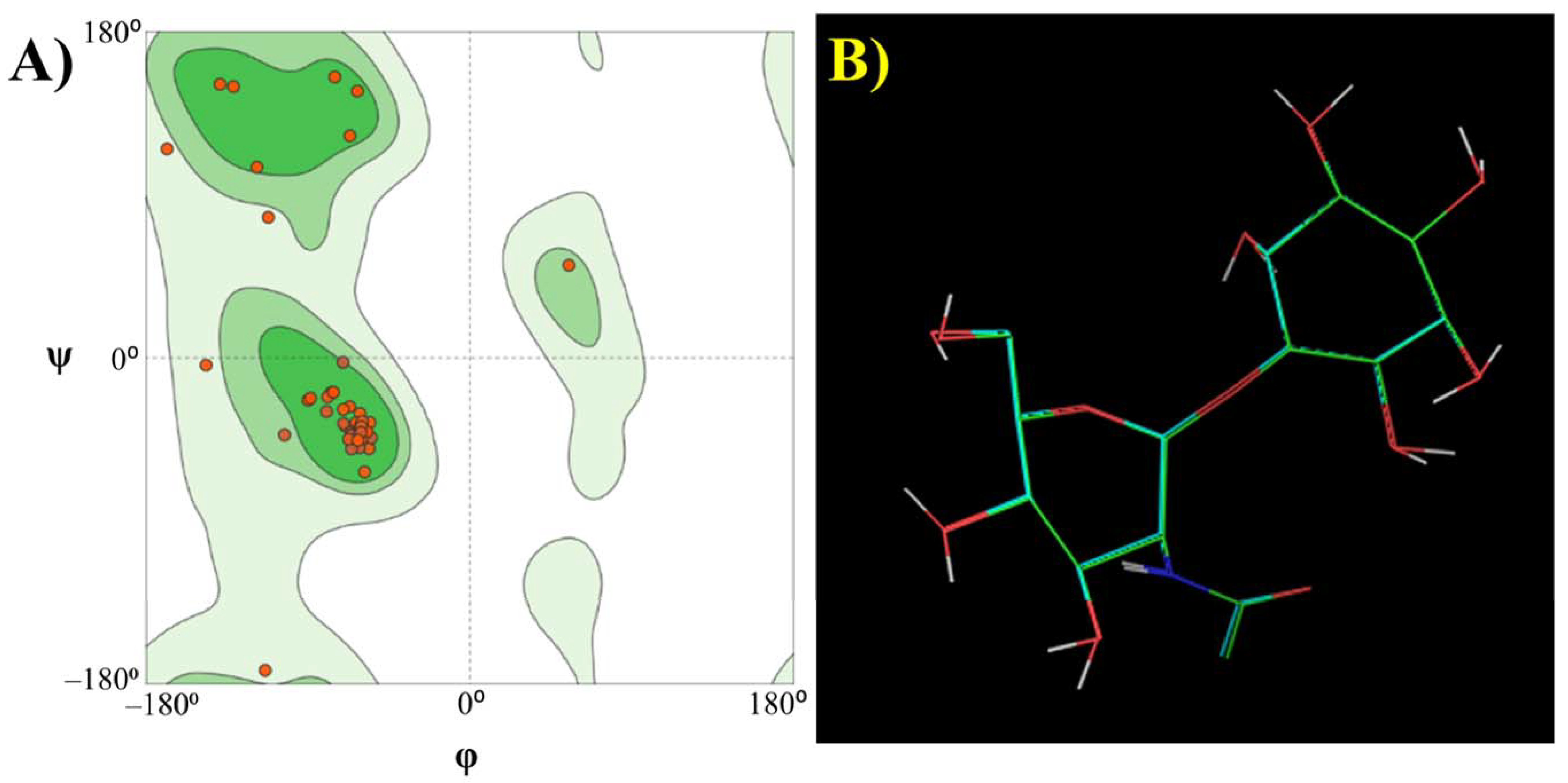
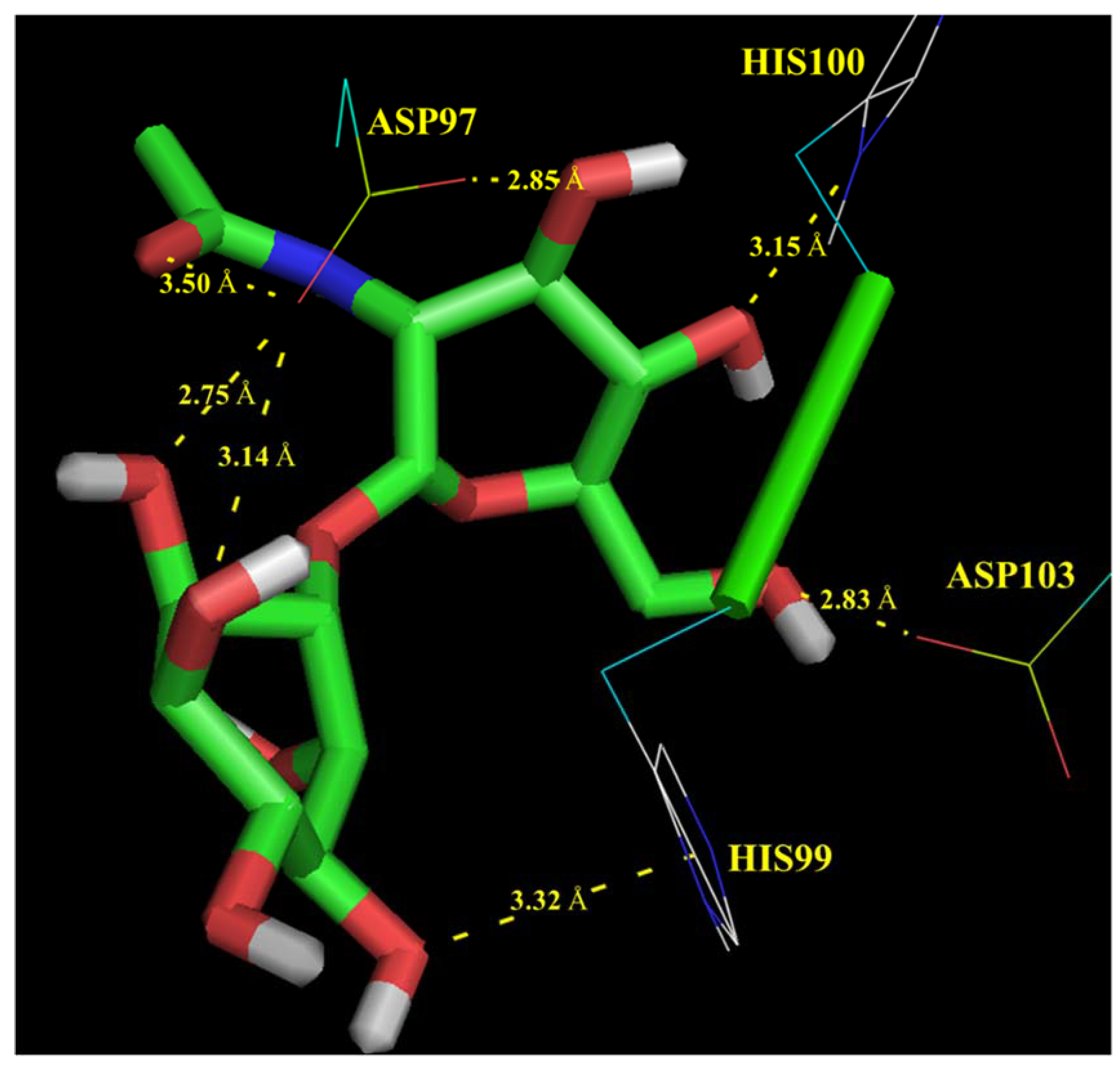
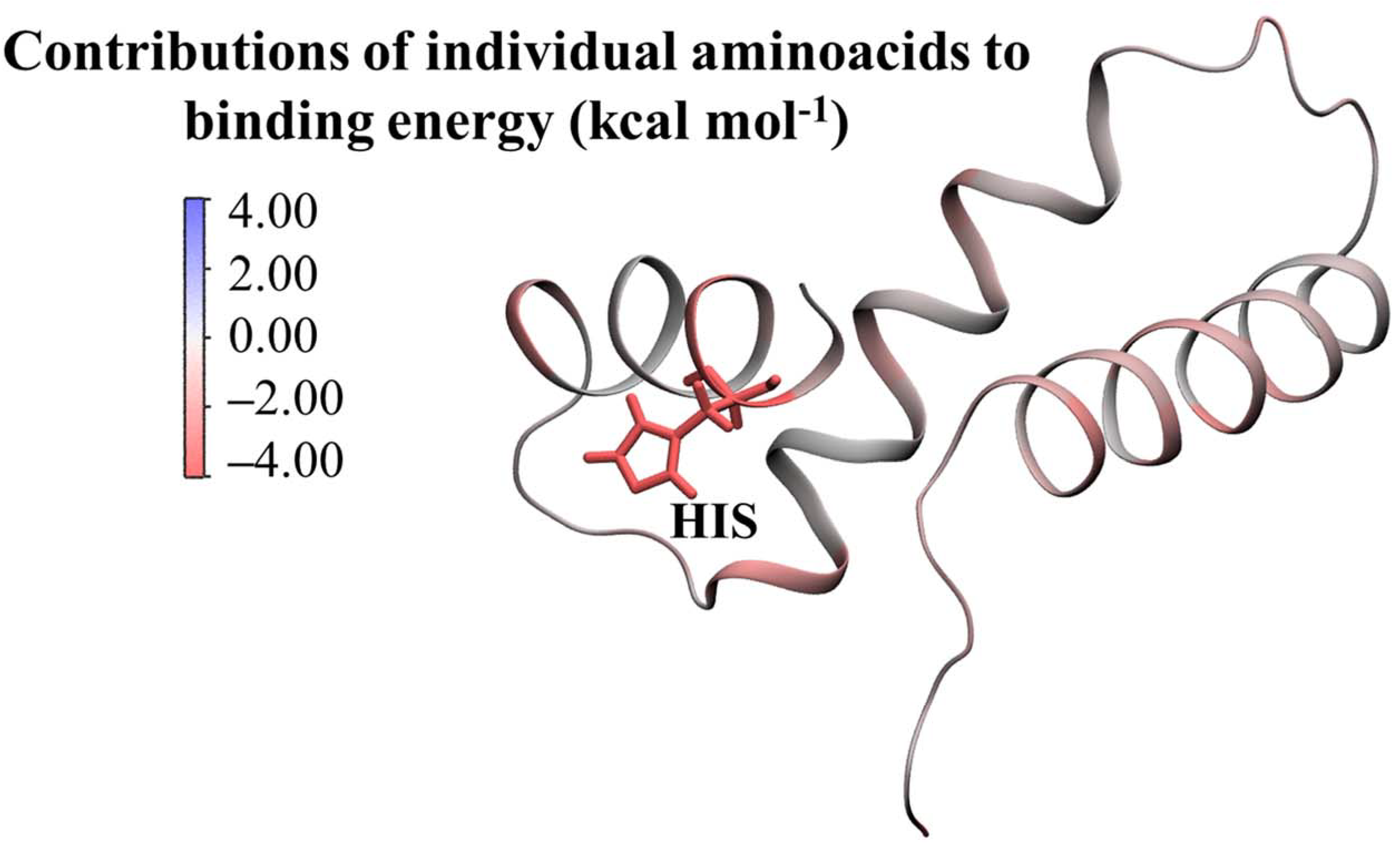
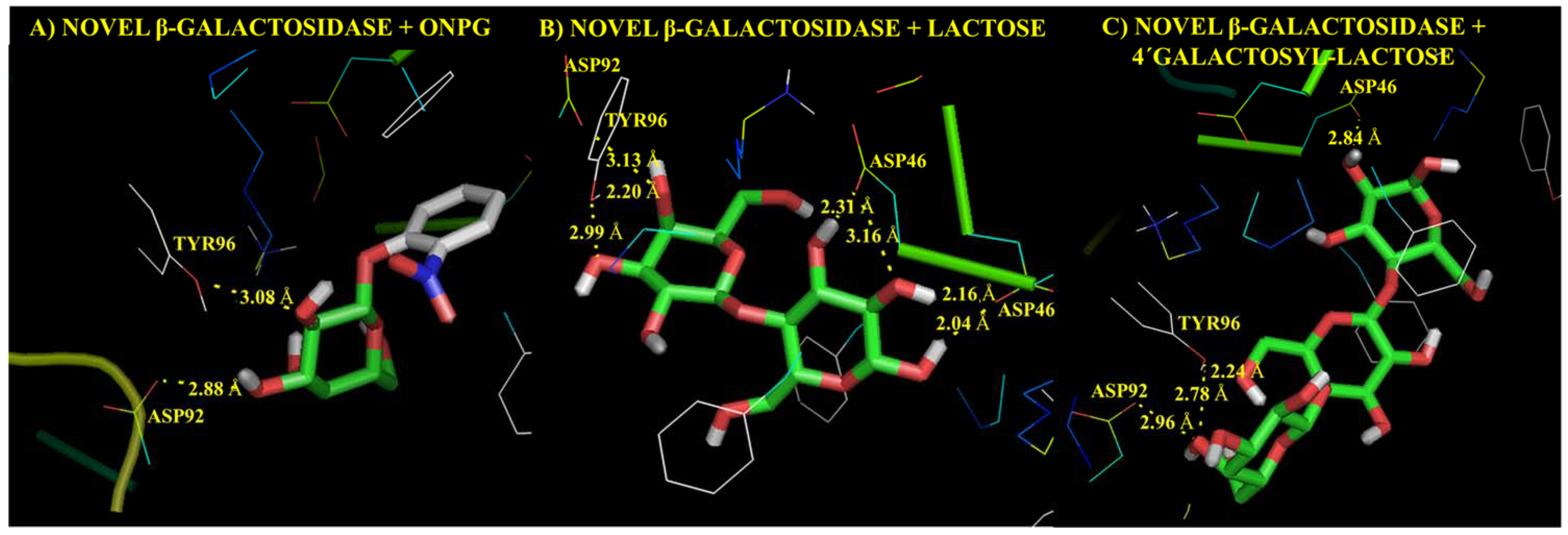
Molecular Modelling Techniques to Study Biological Functions of Metagenomic Dark Matter
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Molinero, N.; Ruiz, L.; Milani, C.; Gutiérrez-Díaz, I.; Sánchez, B.; Mangifesta, M.; Segura, J.; Cambero, I.; Campelo, A.B.; García-Bernardo, C.M.; et al. The human gallbladder microbiome is related to the physiological state and the biliary metabolic profile. Microbiome 2019, 7, 100. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Wang, X.A.; Hu, Y.; Li, H.; Ren, T.; Li, Y.; Liu, L.; Li, L.; Li, X.; Wang, Z.; et al. A metagenomic study of biliary microbiome change along the cholecystitis-carcinoma sequence. Clin. Transl. Med. 2020, 10, e97. [Google Scholar] [CrossRef]
- Shen, H.; Ye, F.; Xie, L.; Yang, J.; Li, Z.; Xu, P.; Meng, F.; Li, L.; Chen, Y.; Bo, X.; et al. Metagenomic sequencing of bile from gallstone patients to identify different microbial community patterns and novel biliary bacteria. Sci. Rep. 2015, 5, 17450. [Google Scholar] [CrossRef]
- Bernard, G.; Pathmanathan, J.S.; Lannes, R.; Lopez, P.; Bapteste, E. Microbial dark matter investigations: How microbial studies transform biological knowledge and empirically sketch a logic of scientific discovery. Genome Biol. Evol. 2018, 10, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Bowman, J.S. Identification of microbial dark matter in antarctic environments. Front. Microbiol. 2018, 9, 3165. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Byrd, A.L.; Deming, C.; Conlan, S.; Kong, H.H.; Segre, J.A. Biogeography and individuality shape function in the human skin metagenome. Nature 2014, 514, 59–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tirosh, O.; Conlan, S.; Deming, C.; Lee-Lin, S.Q.; Huang, X.; Su, H.C.; Freeman, A.F.; Segre, J.A.; Kong, H.H. Expanded skin virome in DOCK8-deficient patients. Nat. Med. 2018, 24, 1815–1821. [Google Scholar] [CrossRef]
- Zamkovaya, T.; Foster, J.S.; de Crécy-Lagard, V.; Conesa, A. A network approach to elucidate and prioritize microbial dark matter in microbial communities. ISME J. 2021, 15, 228–244. [Google Scholar] [CrossRef]
- Lobb, B.; Kurtz, D.A.; Moreno-Hagelsieb, G.; Doxey, A.C. Remote homology and the functions of metagenomic dark matter. Front. Genet. 2015, 6, 234. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Flores, A.; Pérez-Rueda, E.; Segovia, L. Protein homology detection and fold inference through multiple alignment entropy profiles. Proteins 2007, 70, 248–256. [Google Scholar] [CrossRef]
- Michalska, K.; Steen, A.D.; Chhor, G.; Endres, M.; Webber, A.T.; Bird, J.; Loyd, K.G.; Joachimiak, A. New aminopeptidase from “microbial dark matter” archaeon. FASEB J. 2015, 29, 4071–4079. [Google Scholar] [CrossRef] [Green Version]
- Sabater, C.; Blanco-Doval, A.; Margolles, A.; Corzo, N.; Montilla, A. Artichoke pectic oligosaccharide characterisation and virtual screening of prebiotic properties using in silico colonic fermentation. Carbohydr. Polym. 2021, 255, 117367. [Google Scholar] [CrossRef]
- Quijada, N.M.; Rodríguez-Lázaro, D.; Eiros, J.M.; Hernández, M. TORMES: An automated pipeline for whole bacterial genome analysis. Bioinformatics 2019, 35, 4207–4212. [Google Scholar] [CrossRef] [PubMed]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef] [PubMed]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef] [PubMed]
- Rho, M.; Tang, H.; Ye, Y. FragGeneScan: Predicting genes in short and error-prone reads. Nucleic Acids Res. 2010, 38, e191. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.; Tosatto, S.C.E.; Paladin, L.; Rai, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef] [Green Version]
- Saier Jr, M.H.; Reddy, V.S.; Moreno-Hagelsieb, G.; Hendargo, K.J.; Zhang, Y.; Iddamsetty, V.; Lam, K.J.K.; Tian, N.; Russum, S.; Wang, J.; et al. The Transporter Classification Database (TCDB): 2021 update. Nucleic Acids Res. 2021, 49, D461–D467. [Google Scholar] [CrossRef]
- Kovatcheva-Datchary, P.; Nilsson, A.; Akrami, R.; Lee, Y.S.; De Vadder, F.; Arora, T.; Hallen, A.; Martens, E.; Björck, I.; Bäckhed, F. Dietary fiber-induced improvement in glucose metabolism is associated with increased abundance of Prevotella. Cell Metab. 2015, 22, 971–982. [Google Scholar] [CrossRef] [Green Version]
- Sabater, C.; Ruiz, L.; Margolles, A. A Machine Learning Approach to Study Glycosidase Activities from Bifidobacterium. Microorganisms 2021, 9, 1034. [Google Scholar] [CrossRef] [PubMed]
- Uritskiy, G.V.; DiRuggiero, J.; Taylor, J. MetaWRAP—A flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 2018, 6, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Steinegger, M.; Meier, M.; Mirdita, M.; Vöhringer, H.; Haunsberger, S.J.; Söding, J. HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinform. 2019, 20, 473. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina. Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Kumari, R.; Kumar, R.; Open Source Drug Discovery Consortium; Lynn, A. g_mmpbsa-A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Ahn, J. Assessment of conjugal transfer of antibiotic resistance genes in Salmonella Typhimurium exposed to bile salts. J. Microbiol. 2014, 52, 716–719. [Google Scholar] [CrossRef]
- Gipson, K.S.; Nickerson, K.P.; Drenkard, E.; Llanos-Chea, A.; Dogiparthi, S.K.; Lanter, B.B.; Hibbler, R.M.; Yonker, L.M.; Hurley, B.P.; Faherty, C.S. The great ESKAPE: Exploring the crossroads of bile and antibiotic resistance in bacterial pathogens. Infect. Immun. 2020, 88, e00865-19. [Google Scholar] [CrossRef]
- Grattagliano, I.; Ciampi, S.A.; Portincasa, P. Gallbladder disease: Relevance of oxidative stress. In Gastrointestinal Tissue; Gracia-Sancho, J., Salvadó, J., Eds.; Academic Press: London, UK, 2017; Volume 34, pp. 187–194. [Google Scholar] [CrossRef]
- Singh, S.; Verma, N.; Taneja, N. The human gut resistome: Current concepts & future prospects. Indian J. Med. Res. 2019, 150, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Elkins, C.A.; Nikaido, H. Substrate specificity of the RND-type multidrug efflux pumps AcrB and AcrD of Escherichia coli is determined predominately by two large periplasmic loops. J. Bacteriol. 2002, 184, 6490–6498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Wang, Y.; Li, J.; Mao, L.; Nguyen, S.H.; Duarte, T.; Coin, L.; Bond, P.; Yuan, Z.; Guo, J. Triclosan at environmentally relevant concentrations promotes horizontal transfer of multidrug resistance genes within and across bacterial genera. Environ. Int. 2018, 121, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Sistrunk, J.R.; Nickerson, K.P.; Chanin, R.B.; Rasko, D.A.; Faherty, C.S. Survival of the fittest: How bacterial pathogens utilize bile to enhance infection. Clin. Microbiol. Rev. 2016, 29, 819–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Kocabas, E.; Hernick, M. The activity and cofactor preferences of N-acetyl-1-D-myo-inosityl-2-amino-2-deoxy-α-D-glucopyranoside deacetylase (MshB) change depending on environmental conditions. J. Biol. Chem. 2011, 286, 20275–20282. [Google Scholar] [CrossRef] [Green Version]
- Newton, G.L.; Buchmeier, N.; Fahey, R.C. Biosynthesis and functions of mycothiol, the unique protective thiol of Actinobacteria. Microbiol. Mol. Biol. Rev. 2008, 72, 471–494. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.J.; Wang, B.J.; Jiang, C.Y.; Luo, Y.M.; Jin, J.H.; Liu, S.J. Identification and quantification of mycothiol in Actinobacteria by a novel enzymatic method. Appl. Microbiol. Biotechnol. 2010, 88, 1393–1401. [Google Scholar] [CrossRef]
- Jothivasan, V.K.; Hamilton, C.J. Mycothiol: Synthesis, biosynthesis and biological functions of the major low molecular weight thiol in actinomycetes. Nat. Prod. Rep. 2008, 25, 1091–1117. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.L.; Piel, J.; Sunagawa, S. A roadmap for metagenomic enzyme discovery. Nat. Prod. Rep. 2021, d1np00006c. [Google Scholar] [CrossRef]
- Huang, X.; Hernick, M. Automated docking studies provide insights into molecular determinants of ligand recognition by N-acetyl-1-d-myo-inosityl-2-amino-2-deoxy-α-d-glucopyranoside deacetylase (MshB). Biopolymers 2014, 101, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Lamprecht, D.A.; Muneri, N.O.; Eastwood, H.; Naidoo, K.J.; Strauss, E.; Jardine, A. An enzyme-initiated Smiles rearrangement enables the development of an assay of MshB, the GlcNAc-Ins deacetylase of mycothiol biosynthesis. Org. Biomol. Chem. 2012, 10, 5278–5288. [Google Scholar] [CrossRef] [PubMed]
- Metaferia, B.B.; Fetterolf, B.J.; Shazad-ul-Hussan, S.; Moravec, M.; Smith, J.A.; Ray, S.; Gutierrez-Lugo, M.T.; Bewley, C.A. Synthesis of natural product-inspired inhibitors of Mycobacterium tuberculosis mycothiol-associated enzymes: The first inhibitors of GlcNAc-Ins deacetylase. J. Med. Chem. 2007, 50, 6326–6336. [Google Scholar] [CrossRef]
- Rogers, I.L.; Gammon, D.W.; Naidoo, K.J. Conformational preferences of plumbagin with phenyl-1-thioglucoside conjugates in solution and bound to MshB determined by aromatic association. Carbohydr. Res. 2013, 371, 52–60. [Google Scholar] [CrossRef]
- Lordan, C.; Thapa, D.; Ross, R.P.; Cotter, P.D. Potential for enriching next-generation health-promoting gut bacteria through prebiotics and other dietary components. Gut Microbes 2020, 11, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Wu, J.; Wu, D. Rational design of the beta-galactosidase from Aspergillus oryzae to improve galactooligosaccharide production. Food Chem. 2019, 286, 362–367. [Google Scholar] [CrossRef]
- Zhu, J.; Sun, J.; Tang, Y.; Xie, J.; Wei, D. Expression, characterization and structural profile of a heterodimeric β-galactosidase from the novel strain Lactobacillus curieae M2011381. Process Biochem. 2020, 97, 87–95. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dark Matter Functional Domain | Number of Samples Containing Each Domain / Total Number of Samples | ||||
|---|---|---|---|---|---|
| H-06 (n = 1) | H-05 (n = 1) | H-04 (n = 1) | Fosmid (n = 1) | Gut Metagenome (n = 40) | |
| Acetyltransferase | - | - | - | 1/1 | - |
| Deacetylase | - | - | - | 1/1 | - |
| RNA repair and recombination protein | - | - | 1/1 | - | - |
| Outer membrane protein | - | - | 1/1 | - | - |
| Outer membrane ion transport | - | - | 1/1 | - | - |
| Outer membrane protein assembly factor | - | 1/1 | - | - | 34/40 |
| Response regulator aspartate phosphatase | - | - | 1/1 | - | - |
| Uncharacterised two-domain protein structure | - | - | 1/1 | - | 1/40 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sabater, C.; Molinero, N.; Ferrer, M.; Bernardo, C.M.G.; Delgado, S.; Margolles, A. Functional Characterisation of Bile Metagenome: Study of Metagenomic Dark Matter. Microorganisms 2021, 9, 2201. https://doi.org/10.3390/microorganisms9112201
Sabater C, Molinero N, Ferrer M, Bernardo CMG, Delgado S, Margolles A. Functional Characterisation of Bile Metagenome: Study of Metagenomic Dark Matter. Microorganisms. 2021; 9(11):2201. https://doi.org/10.3390/microorganisms9112201
Chicago/Turabian StyleSabater, Carlos, Natalia Molinero, Manuel Ferrer, Carmen María García Bernardo, Susana Delgado, and Abelardo Margolles. 2021. "Functional Characterisation of Bile Metagenome: Study of Metagenomic Dark Matter" Microorganisms 9, no. 11: 2201. https://doi.org/10.3390/microorganisms9112201
APA StyleSabater, C., Molinero, N., Ferrer, M., Bernardo, C. M. G., Delgado, S., & Margolles, A. (2021). Functional Characterisation of Bile Metagenome: Study of Metagenomic Dark Matter. Microorganisms, 9(11), 2201. https://doi.org/10.3390/microorganisms9112201