Abstract
Bile acids (BAs) are produced from cholesterol in the liver and are termed primary BAs. Primary BAs are conjugated with glycine and taurine in the liver and then released into the intestine via the gallbladder. After the deconjugation of glycine or taurine by the gut microbiome, primary BAs are converted into secondary BAs by the gut microbiome through modifications such as dehydroxylation, oxidation, and epimerization. Most BAs in the intestine are reabsorbed and transported to the liver, where both primary and secondary BAs are conjugated with glycine or taurine and rereleased into the intestine. Thus, unconjugated primary Bas, as well as conjugated and unconjugated secondary BAs, have been modified by the gut microbiome. Some of the BAs reabsorbed from the intestine spill into the systemic circulation, where they bind to a variety of nuclear and cell-surface receptors in tissues, whereas some of the BAs are not reabsorbed and bind to receptors in the terminal ileum. BAs play crucial roles in the physiological regulation of various tissues. Furthermore, various factors, such as diet, age, and antibiotics influence BA composition. Here, we review recent findings regarding the physiological roles of BAs modified by the gut microbiome in the metabolic, immune, and nervous systems.
1. Introduction
Bile acids (BAs) are amphipathic steroid acids produced from cholesterol in the liver, and de novo synthesized bile acids in the liver are termed primary BAs. Primary BAs are conjugated with glycine and taurine in the liver and are then stored in the gallbladder. BAs are released from the gallbladder into the small intestine via food intake to facilitate the digestion and absorption of lipids and lipophilic vitamins by forming micelles in the small intestine. While most of the BAs are absorbed in the intestine, there is a significant deconjugation of conjugated BAs prior to reabsorption in the terminal ileum. BAs that are not absorbed in the intestine are converted to secondary BAs by the gut microbiome and these secondary BAs are then absorbed into the colon via passive diffusion [1,2]. After BAs reabsorbed from the gut are transported to the liver through the portal vein, they are conjugated with glycine and taurine in the liver and are secreted into the bile again for enterohepatic circulation (Figure 1).
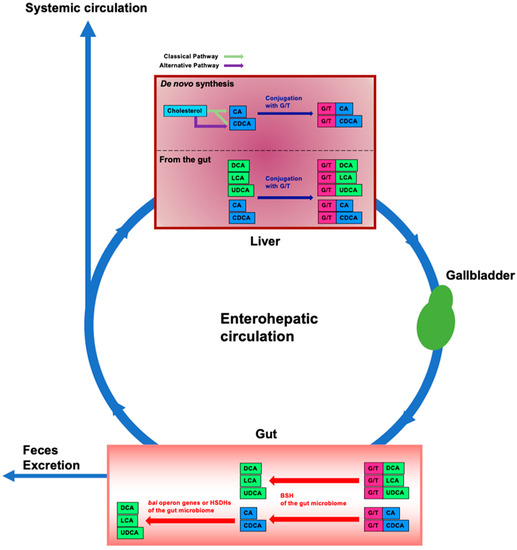
Figure 1.
Enterohepatic circulation and modification of bile acids in the intestine by the gut microbiome. In the liver, cholesterol is converted to cholic acid (CA) and chenodeoxycholic acid (CDCA) (de novo synthesis). CA and CDCA are then conjugated with glycine or taurine (G/T). Conjugated CA and CDCA are transferred to and stored in the gallbladder; food intake stimulates their release into the intestine. Conjugated BAs are deconjugated by bile salt hydrolases (BSHs) of gut bacteria in the gut microbiome. By bile acid-inducible (bai) operon genes of gut bacteria, CA and CDCA are then dehydrogenated and converted to deoxycholic acid (DCA) and lithocholic acid (LCA), respectively. CDCA is also converted to ursodeoxycholic acid (UDCA) by dehydrogenation and epimerization by hydroxysteroid dehydrogenases (HSDHs) of gut bacteria. Approximately 95% of BAs in the intestine are reabsorbed and transported to the liver, and the remainder is excreted in the feces. This circulation between the gut and the liver is termed enterohepatic circulation. Some of the BAs reabsorbed from the intestine spill into the systemic circulation. Unconjugated BAs from the gut are conjugated with G/T in the liver. Blue boxes represent primary BAs. Green boxes represent secondary BAs. Pink boxes represent glycine or taurine (G/T).
As BAs are amphipathic, they can affect cell-surface and intracellular membranes, including those of mitochondria and the endoplasmic reticulum (ER). Recent studies suggest that BAs are also hormones or signaling molecules because they can bind to several nuclear and cell-surface receptors, including farnesoid X receptor (FXR) and Takeda G protein receptor 5 (TGR5)—also known as G protein-coupled bile acid receptor 1 (GPBAR1) [3,4]. By activating these receptors, BAs can regulate BA, glucose, and lipid metabolism in various tissues, including the liver, pancreas, and both brown and white adipose tissue [5]. BAs also affect the immune system [6]. Furthermore, because more than 20 BAs have been detected in the brain of humans and rodents, BAs can affect the nervous system [7,8]. In this review, we address the current knowledge on the physiological role of BAs modified by the gut microbiome in the metabolic, immune, and nervous systems.
2. BA Production and Modification
2.1. BA Production in the Liver
BAs are principally produced in the liver via the classical or alternative pathways [4,9]. Over sixteen enzymes participate in the biosynthesis of BA from cholesterol in the liver [10]. The first step of the classical pathway is the conversion of cholesterol to 7α-hydroxycholesterol by cytochrome P450 (CYP) 7A1 (CYP7A1). CYP7A1 converts cholesterol to 7α-hydroxycholesterol, which is then converted to 7α-hydroxy-4-cholesten-3-one. CYP8B1 is involved in the generation of cholic acid (CA) from 7α-hydroxy-4-cholesten-3-one, which is also converted to chenodeoxycholic acid (CDCA) by CYP27A1. The first step of the alternative pathway is the conversion of cholesterol to (25R)-26-hydroxycholesterol by CYP27A1 [11]. (25R)-26-hydroxycholesterol is then converted to CDCA by CYP7B1. CA and CDCA are then conjugated with glycine or taurine by bile acid-CoA: amino acid N-acyltransferase (BAAT) [12]. The transportation of BAs from hepatocytes into the bile canaliculi is mediated by the bile salt export pump (BSEP) and multidrug resistance-associated protein 2 (MRP2). BSEP is the predominant transporter of BAs from hepatocytes to the bile canaliculi [3,13]. BAs are preserved in the gallbladder until food intake stimulates their release into the small intestine.
2.2. BA Modification by the Gut Microbiome
BAs are deconjugated, dehydroxylated, dehydrogenated, and epimerized by the gut microbiome (Figure 1). Conjugated BAs from the liver are mainly deconjugated in the small intestine by bile salt hydrolases (BSHs), which are found in gut bacteria [14], such as Lactobacillus spp. [15,16], Bifidobacterium spp. [16], Enterococcus spp. [17], Clostridium spp. [18,19], and Bacteroides spp. [20,21]. Most of these gut bacteria exist in the ileum and colon [22]. After glycine or taurine deconjugation, CA and CDCA are converted to deoxycholic acid (DCA) and lithocholic acid (LCA), respectively, by removing the 7α-hydroxy group. Only a few bacteria have been identified that are capable of 7α-dehydroxylation; these bacteria belong to Clostridium spp. [22]. The 7α-dehydroxylation of CA or CDCA is performed by several proteins encoded by the bile acid-inducible (bai) operon. BaiB ligates CoA to the unconjugated BA. BaiA oxidizes the 3α-hydroxyl group. BaiCD catalyzes the formation of the C4=C5 bond. BaiF hydrolyzes CoA. BaiE catalyzes the 7α-dehydration, which is a rate-limiting step. BaiH catalyzes the removal of the C6=C7 bond and BaiCD catalyzes the removal of the C4=C5 bond. BaiA converts the 3-oxo-intermediate to a secondary BA [2,23]. Furthermore, 7α-hydroxysteroid dehydrogenase (7α-HSDH) and 7β-HSDH dehydrogenate epimerize and convert CDCA to ursodeoxycholic acid (UDCA) [2,24,25]. HDSHs convert DCA and LCA to iso-DCA and iso-LCA, respectively [26]. Additionally, Eggerthella lenta strains also possess 7α-HSDH and produce 7-Oxo-DCA from CA [27]. 7-Oxo-DCA is converted to DCA by Clostridium spp. [23,28]. Eggerthella lenta also has 3α- and one 3β-HSDH for converting DCA to isoDCA. Since IsoDCA is less toxic to bacteria than DCA, the conversion of DCA to isoDCA leads to the growth of bacteria in the gut [29]. Additionally, a recent study has demonstrated that 25 strains in the gut microbiota conjugate glycine to DCA, CDCA, or CA, and 28 strains in the gut microbiota conjugate other amino acids, namely, alanine, arginine, aspartate, asparagine, glutamate, glutamine, histidine, lysine, methionine, serine, tryptophan, and valine [30]. Furthermore, CA conjugated with phenylalanine and tyrosine by Clostridium bolteae strains significantly activates FXR [31].
2.3. BA Circulation
Approximately 95% of BAs are reabsorbed from the gut, and the remainder is excreted in the feces [32]. Reabsorption primarily occurs in the small intestine and also at a low rate in the colon. Conjugated BAs are transported into the enterocytes via apical sodium-dependent bile acid transporters (ASBTs) in the apical brush border [1,33,34,35,36]. Ileal bile acid-binding protein (I-BABP) mediates the intracellular transport of BAs from the apical to the basolateral side of enterocytes [37]. Organic solute transporter (OST) α and β in the basolateral membrane of enterocytes transport BAs from the enterocytes into the portal circulation [38,39,40]. BAs are then translocated from the portal circulation to the liver via Na+-dependent and Na+-independent transporters. Na+-taurocholate co-transporting polypeptide (NTCP) transports BAs into the hepatocytes in a Na+-dependent manner [41,42,43]. Organic anion-transporting polypeptide 1B1 (OATP1B1) and OATP1B3 transport BAs into the hepatocytes in a Na+-independent manner [44]. OATP1B1 and OATP1B3 transport conjugated BAs more efficiently than unconjugated BAs [45]. The reabsorbed unconjugated BAs transported to the liver are also conjugated and released along with newly synthesized BAs (Figure 1). Therefore, unconjugated primary and secondary BAs and G/T conjugated secondary BAs are modified by the gut microbiome. This recycling process between the gut and the liver is termed enterohepatic circulation. However, some of the BAs reabsorbed from the intestine spill into the systemic circulation (Figure 1). The concentration of BAs in the portal vein is ~80 μM and the concentration of BAs in the systemic circulation is ~2–10 μM [46]. Thus, because BAs in the systemic circulation can affect various tissues, BAs play physiological roles.
2.4. Factors That Influence Bile Acid Composition
Specific diet components influence BA composition. High- and low-fat diets are associated with a decrease in the synthesis of primary BAs [47]. In feces, a high-fat diet increases DCA, LCA, and CDCA [48,49]. This increase in secondary bile acids, DCA, and LCA is related to cancer development [50]. Additionally, proteins affect the BA composition. High-protein diets increase the plasma concentration of DCA, CDCA, and CA [51]. Soybean protein isolate increases secondary BAs, the pool of BAs, and the secondary to primary BA ratio in the cecum and feces [52]. Milk protein intake increases total and primary BAs in the serum of obese, but not overweight, subjects [53]. As for carbohydrates, a whole grain diet increases the plasma concentrations of taurolithocholic acid (TLCA), taurocholic acid, glycocholic acid, taurochenodeoxycholic acid, and glycochenodeoxycholic acid compared with a refined grain diet [54].
The plasma concentrations of most BAs decrease with age in men but not in women. A decrease in CDCA, GCDCA, TCDCA, and GUDCA is significantly related to age in men. The concentrations of DCA, GCA, UDCA, GDCA, and TDCA decrease with age in both men and women. Additionally, the concentration of DCA, GCA, and UDCA is significantly different between sexes [55]. Furthermore, the concentration of isoLCA, 3-oxoLCA, alloLCA, 3-oxoalloLCA, and isoalloLCA in the feces of centenarians is higher than in that of other age subjects [56].
Antibiotic treatment changes the gut microbiota by reducing specific species and modifying the bile acid composition [57,58]. Vancomycin and gentamicin reduce the total bacteria and intestinal microbiota diversity, leading to a change in the BA composition. These antibiotic treatments increase levels of conjugated primary BAs and decrease unconjugated primary and secondary BAs [59].
3. Physiological Role of BAs Modified by the Gut Microbiome
FXR and TGR5 are the most widely studied BA receptors [60]. BAs also regulate other nuclear and cell-surface receptors [4,61,62,63]. Nuclear receptors regulated by BAs include the constitutive androstane receptor (CAR), pregnane X receptor (PXR), vitamin D receptor (VDR), liver X receptor (LXR), and glucocorticoid receptor (GR). Cell-surface receptors regulated by BAs include sphingosine-1-phosphate receptor 2 (S1PR2), Mas-related G protein-coupled receptor X4 (MRGPRX4), and the M2 and M3 muscarinic receptors. Thus, BAs play a variety of physiological roles by regulating these receptors or other mechanisms.
3.1. The Role of Modified BAs in the Metabolic System
The main BA receptors are FXR and TGR5. BA binding to FXR and TGR5 regulates BA, glucose, and lipid levels. CDCA and DCA have a high affinity for FXR [64,65,66,67] and conjugated DCA acts as a ligand for FXR with NTCP—a BA transporter expressed in the cell [64].
FXR regulates the expression of various genes by binding to specific DNA sequences in the genome, either as a monomer or as a heterodimer with the retinoid X receptor (RXR). These specific DNA sequences are an inverted repeat of AGGTCA separated by one nucleotide (IR1), IR0, an everted repeat of AGGTCA separated by two or eight nucleotides (ER2 or 8), and a direct repeat of AGGTCA separated by one, four, or five nucleotides (DR1, 4, or 5) [68,69,70,71,72]. FXR also regulates epigenetic modifications. The activation of FXR recruits steroid receptor coactivator 1 (SRC1), which has histone acetyltransferase (HAT) activity in its C-terminal region, leading to the acetylation of histone H3 at the promoter of the glucagon-like peptide 1 (GLP-1) receptor [73,74,75]. In addition, FXR induces the expression of microRNA-29a, which downregulates DNA methyltransferases [76] and histone deacetylase (HDAC) 4 [77]. It has been shown that the percentage of common DNA binding sites of FXR between the liver and intestine is only 11% [69]. This suggests that the pattern of gene expression regulated by FXR is tissue-specific. Furthermore, FXR induces the expression of TGR5 [78].
TGR5 is a member of the G-protein-coupled receptor (GPCR) superfamily and couples to Gαs. Activated TGR5 leads to the activation of adenylyl cyclase, an increase in cyclic AMP (cAMP) concentration, and the activation of protein kinase A (PKA) and exchange proteins directly activated by cAMP (EPAC) [79]. LCA, DCA, CDCA, CA, and conjugated forms of these BAs activate TGR5. LCA and DCA, which are secondary BAs, and their conjugated forms were reported as the most effective TGR5 activators [80,81,82]. Additionally, UDCA is a weak activator of TGR5 [81,83].
3.1.1. BA Metabolism
FXR inhibits BA synthesis by inducing small heterodimer partner (SHP), a nuclear receptor that lacks a ligand-binding domain [84]. SHP interacts with and represses nuclear receptors and transcription factors, including PXR, CAR, LXR, GR, and FXR [85,86]. SHP also inhibits the activation of liver receptor homolog-1 (LRH-1), which induces CYP7A1, the key enzyme responsible for BA synthesis from cholesterol [87] (Figure 2A). Furthermore, FXR also inhibits the expression of CYP7A1 by inducing fibroblast growth factor (FGF) 15/19 (mouse FGF15 and human FGF19) and β-Klotho (βKL). βKL forms binary complexes with FGF receptor 4 (FGFR4) to function as a coreceptor of FGFR4 for FGF15/19 [88]. Transcription factor EB (TFEB) is activated by dephosphorylation and induces CYP7A1. After activating FGFR4/βKL by FGF15/19, the mechanistic target of rapamycin phosphorylates and inactivates TFEB, leading to CYP7A1 repression [89]. FGF15/19 also stabilizes SHP, which represses CYP7A1 and CYP8B1. CYP8B1 is a key enzyme for the synthesis of CA. The binding of FGF15 to FGFR4 activates the Hippo signaling pathway. In the Hippo signaling pathway, SHP is phosphorylated and stabilized by mammalian sterile 20-like kinase 1 (Mst1) and Mst2, homologs of the Hippo kinase in Drosophila [90] (Figure 2B). FXR also regulates BA transporters. FXR represses NTCP in the liver, leading to a reduction of the hepatic uptake of Bas [91]. On the other hand, FXR enhances BA efflux into the liver by inducing BSEP and OSTα/β [92,93]. Overall, FXR reduces excess Bas in the liver. Although TGR5 is weakly expressed in the liver, TGR5-knockout mice exhibit a decrease in total BA pool size [94]. Additionally, TGR5-knockout mice exhibit reduced levels of CYP7B1 and increased levels of secondary and hydrophobic Bas [95,96,97]. PXR is activated by LCA [98,99] and PXR regulates the expression of CYP3A and CYP2B, which convert LCA to hyocholic acid (HCA) and UDCA [100,101,102]. In addition, PXR indirectly suppresses the expression of CYP7A1 by inhibiting the activation of hepatocyte nuclear factor 4α, which upregulates the expression of CYP7A1 [103,104]. VDR is also activated by LCA and its metabolite, 3-keto LCA [105,106], and LCA induces the expression of CYP3A [106,107,108]. LXRα is weakly activated by hyodeoxycholic acid (HDCA) [109]. The BA pool size and its excretion are decreased in LXRα-knockout mice. LXRα-knockout mice also exhibit reduced CYP7A expression [110], but LXRβ-knockout mice do not [111].
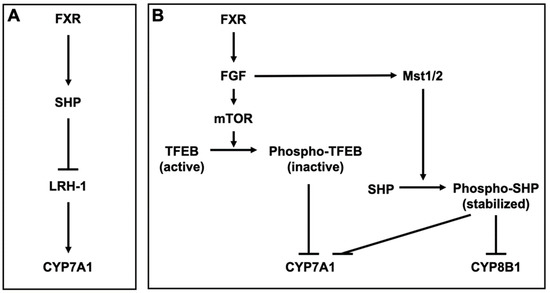
Figure 2.
Bile acid metabolism by FXR (A) Farnesoid X receptor (FXR) induces small heterodimer partner (SHP) and SHP inhibits liver receptor homolog-1 (LRH-1), which induces cytochrome P450 (CYP) 7A1 in the hepatocyte. (B) FXR induces fibroblast growth factor (FGF) 15/19 (mouse FGF15 and human FGF19) and FGF15/19 in the enterocytes. In the hepatocyte, FGF15/19 activates FGF receptor 4 (FGFR4) and its coreceptor β-Klotho (βKL), leading to the phosphorylation of transcription factor EB (TFEB) by the mechanistic target of rapamycin (mTOR). TFEB induces CYP7A1 and is inactivated by the phosphorylation. The binding of FGF15 to FGFR4 activates mammalian sterile 20-like kinases 1 (Mst1) and Mst2, homologs of the Hippo kinase in Drosophila. MST1 and 2 phosphorylate and stabilize SHP, which represses CYP7A1 and CYP8B1.
3.1.2. Glucose Metabolism
Glucose and insulin upregulate and downregulate FXR expression, respectively, and FXR is associated with glucose metabolism [112,113]. The association between FXR and glucose metabolism is differently affected by the fed and fasting states. In the fed state, treatment of wild-type mice with the FXR ligand decreases the expression of phosphoenolpyruvate carboxykinase (PEPCK) and glucose 6-phosphatase (G6Pase). PEPCK and G6Pase are essential for the upregulation of gluconeogenesis. In contrast, treatment of wild-type mice in the fasting state with FXR ligand increases the expression of PEPCK and G6Pase. FXR-knockout mice exhibit decreased blood glucose levels, and PEPCK and G6Pase expression are downregulated. The effects of FXR with the FXR ligand are mediated by GR [114]. The treatment of wild-type mice with CA reduces blood glucose levels and PEPCK and GP6ase expression. Furthermore, the treatment of SHP-knockout mice with CA increases blood glucose levels. Thus, the blood glucose-lowering effect of FXR is, at least in part, mediated by SHP [115]. FXR also inhibits PEPCK and G6Pase expression by inducing FGF15/19. FGF15/19 inactivates the cAMP regulatory element-binding protein (CREB), the transcription factor that induces peroxisome proliferator-activated receptor (PPAR)-γ coactivator-1α (PGC-1α) [116]. PGC-1α is a coactivator that induces the expression of genes involved in metabolic pathways and mitochondria by interacting with a variety of nuclear receptors [117,118]. FXR interacts with the carbohydrate responsive element binding protein (ChREBP) and suppresses the activity of ChREBP. ChREBP is a transcription factor that regulates the expression of genes controlling glycolysis and lipogenesis. ChREBP upregulates the expression of L-pyruvate kinase (L-PK), a key enzyme for glycolysis [119]. Activated FXR releases ChREBP and its coactivators (p300 and CREB-binding protein) from the L-PK promoter region and inhibits L-PK expression [120]. FXR and CDCA induce glucose transporter 4 expression in differentiated 3T3-L1 adipocytes and the hepatic cell line HepG2 [121]. FXR increases glucose-stimulated insulin secretion and expression by inducing the KLF11 transcription factor [113]. Although CDCA increases the glucose-stimulated secretion of insulin from wild-type mice, FXR-knockout mice are unaffected [122].
TGR5 activation increases the intestinal level of GLP-1 [123,124,125,126,127]. LCA, taurodeoxycholic acid (TDCA), and TLCA increases GLP-1 secretion via TGR5 activation [123,125]. GLP-1 release is mediated by TGR5 located in the basolateral membrane of L-cells [125]. Most active GLP-1 is degraded in the intestine and liver and 10–15% of secreted GLP-1 from the intestine reaches the pancreas via the systemic circulation [128]. GLP-1 induces the release and synthesis of insulin in β-cells in the pancreatic islets. The binding of GLP-1 to GLP-1 receptor activates adenylate cyclase, leading to increased cAMP levels. PKA and EPAC are then activated, which induces an increase in intracellular Ca2+ from the ER and extracellular sources via voltage-dependent Ca2+ channels [128,129]. Additionally, the activation of PKA by GLP-1 leads to the activation of pancreatic duodenal homeobox-1 protein (PDX-1), which binds to the promoter of the gene encoding insulin and induces the synthesis of insulin [128,130] (Figure 3). In contrast to TGR5, FXR suppresses GLP-1 production in L-cells by inhibiting the activity of ChREBP, which induces the expression and secretion of GLP-1 in L-cells [120,131]. Furthermore, the level of serum HCA is lower in diabetics and people with a high body mass index [132,133,134]. HCA induces GLP-1 by activating TGR5 and inhibiting FXR, and TGR5 activation increases serum insulin and decreases blood glucose levels [132].
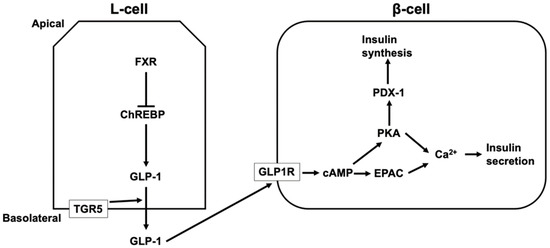
Figure 3.
The regulation of insulin secretion and synthesis via TGR5 and FXR. TGR5 located in the basolateral membrane of L-cells mediates glucagon-like peptide 1 (GLP-1) secretion from L-cells, and secreted GLP-1 from the intestine reaches pancreatic β-cells via the systemic circulation. The binding of GLP-1 to GLP-1 receptor (GLP1R) increases cyclic AMP (cAMP) levels, leading to the activation of protein kinase A (PKA) and exchange proteins directly activated by cAMP (EPAC), which induces an increase in intracellular Ca2+ and insulin secretion. In addition, the activation of PKA by GLP-1 leads to the activation of pancreatic duodenal homeobox-1 protein (PDX-1), which binds to the promoter of the gene encoding insulin and induces the synthesis of insulin. FXR suppresses GLP-1 production in L-cells by inhibiting the activity of a carbohydrate responsive element binding protein (ChREBP) that induces the expression and secretion of GLP-1 in L-cells.
3.1.3. Lipid Metabolism
Dietary lipids (triglycerides [TGs], cholesterol, and phospholipids) are absorbed and incorporated into chylomicrons in the intestine. Chylomicrons are then converted to very low-density lipoprotein (VLDL) in the liver. VLDL transports lipids from the liver to the systemic circulation. VLDL is then converted to HDL and returned to the liver [135]. In the process of lipogenesis, acetyl-CoA carboxylase (ACC) converts acetyl-CoA to malonyl-CoA, and fatty acid synthase (FAS) converts malonyl-CoA to palmitate. Stearoyl-CoA desaturase (SCD) mediates the conversion of saturated fatty acids (FAs) to monounsaturated FAs, and the newly-produced FAs are then stored as TGs. Sterol regulatory element-binding protein 1C (SREBP1C) is a transcription factor that induces lipogenic genes, including FAS, ACC, and SCD [136,137]. FXR-knockout mice exhibit an increase in SREBP1C, SCD-1, and FAS and also exhibit increased levels of serum and liver TGs, serum cholesterols, and serum free FAs (FFAs) [115]. Additionally, CA feeding decreases the serum levels of TGs, cholesterol, and FFAs in mice [115,138]. CA and CDCA decrease the expression of FAS, ACC, SCD, and SREBP1C [138]. SHP induced by FXR suppresses lipogenesis by suppressing lipogenic genes via the inhibition of LXR, which induces SREBP1C expression [138,139]. Furthermore, FXR induces the expression of carboxylesterase 1 (CES1), which reduces hepatic TGs by inducing TG hydrolysis to release FFAs. Released FFAs activate PPARα, leading to the enhancement of FA oxidation [140,141].
3.2. Role of Modified BAs in the Immune System
The liver in FXR-knockout mice exhibits an elevated expression of pro-inflammatory genes, such as interleukin-1 (IL-1), IL-6, interferon-γ (IFN-γ), inducible nitric oxide synthase, and cyclooxygenase-2. FXR activation inhibits the expression of these genes by inhibiting the activity of nuclear factor-κB (NF-κB), and also has an anti-inflammatory function [142]. Moreover, CDCA inhibits the production of IL-1, IL-6, and tumor necrosis factor α (TNFα) by human monocytes [143]. FXR activation by agonists leads to conjugation with small ubiquitin-like modifier (SUMO) 2. SUMOylated FXR interacts with NF-κB and nuclear receptor corepressor (NCOR) 1 or NCOR2, also called silencing mediator of retinoic acid and thyroid hormone receptor. NCORs are transcriptional corepressors that repress pro-inflammatory gene expression in the liver [144,145,146,147]. FXR also upregulates SHP. SHP overexpression inhibits NF-κB activation and leads to the downregulation of pro-inflammatory genes in the liver. In contrast, SHP deletion activates NF-κB. Thus, FXR can suppress NF-κB activation by inducing SHP [148]. The inhibitory effect of SHP on NF-κB is mediated by the binding of SHP to NF-κB [149].
Macrophages express both FXR and TGR5. Macrophages from FXR-knockout mice exhibit enhanced release of pro-inflammatory cytokines [147]. CDCA and FXR agonists reduce IL-6 expression in macrophages. However, IL-6 expression is unaffected by FXR agonists in FXR-knockout mice [150]. Activation of TGR5 leads to the suppression of pro-inflammatory cytokines from LSP-stimulated macrophages [151]. The activation of macrophage TGR5 by INT-777, a TGR5 agonist, inhibits pro-inflammatory cytokine production. This inhibition is mediated by cAMP signaling, followed by NF-κB inhibition [151]. Furthermore, TGR5 activation promotes macrophage polarization from M1 (pro-inflammatory phenotype) to M2 (anti-inflammatory phenotype) macrophages and reduces pro-inflammatory cytokines [152,153,154]. In contrast, macrophage differentiation is unaffected by FXR-knockout [150].
Dendritic cells are antigen-presenting cells that play an essential role in the adaptive immune system and express TGR5. DCA suppresses lipopolysaccharide (LPS)-induced expression of pro-inflammatory genes, including IL-1, IL-6, and TNFα in dendritic cells. Furthermore, TGR5-knockout mice exhibit a recovery of LPS-induced expression of pro-inflammatory genes. These inhibitory effects of TGR5 are mediated by the repression of NF-κB via TGR5–cAMP–PKA signaling [155].
Recent studies have shown that 3-oxoLCA and isoalloLCA regulate T cell differentiation. IsoalloLCA triggers oxidative phosphorylation in mitochondria, leading to the production of reactive oxygen species (ROS). ROS from the mitochondria, not from the cytoplasm, induces forkhead box P3 (FOXP3). FOXP3 is a transcription factor and a master regulator of the development of regulatory T cells (Tregs), which release immunosuppressive factors. This 3-oxoLCA acts as an inverse agonist of retinoid-related orphan receptor γt (RORγt), leading to the inhibition of the development of T helper 17 (Th17) cells, which release pro-inflammatory factors. Both 3-oxoLCA and isoalloLCA are not produced in germ-free mice [156,157]. It has been reported that Odoribacteraceae strains can produce isoalloLCA via 5α-reductase and 3β-HSDH. Moreover, isoalloLCA shows selective antibacterial activity against gram-positive multidrug-resistant pathogens [56].
3.3. The Role of Modified BAs in the Nervous System
Both conjugated and unconjugated BAs can be detected in human and rodent brains [7,8,158,159]. Although the origin of BAs in the brain has not been confirmed, BAs can be transported from the peripheral circulation by BA transporters or diffuse across the blood–brain barrier (BBB) [4,160]. As CA, CDCA, and DCA are lipophilic [161], these unconjugated BAs can pass through the BBB via passive diffusion. There is a correlation between the brain and serum levels of CA, CDCA, and DCA [162,163]. On the other hand, conjugated BAs are large molecules that are negatively charged at physiological pH, so these BAs rely on transporters to pass through the BBB [164,165]. Furthermore, BAs increase BBB permeability through the phosphorylation of occludin, which is involved in the formation of tight junctions in the BBB [166,167]. Thus, the brain communicates with the gut and gut microbiome through BAs. This is known as the microbiome–gut–brain axis or the gut–brain axis. N-methyl-D-aspartate (NMDA) receptors in the brain are crucial for learning and memory because they induce long-term potentiation and long-term depression [168,169,170]. In contrast to NMDA, γ-aminobutyric acid type A (GABAA) receptor activation inhibits neurotransmission [171,172]. CA, CDCA, and DCA hinder the activation of NMDA and GABAA receptors [173]. UDCA increases arousal by inhibiting GABAA receptor activation in neurons in the tuberomammillary nucleus of the hypothalamus [174]. G/T DCA and DCA activate M2 and M3 muscarinic acetylcholine receptors, respectively, and these receptors play an important role in cognitive function, memory, and learning [175,176,177]. Furthermore, tauroursodeoxycholic acid (TUDCA) induces neural stem cell proliferation and neural differentiation in the subventricular zone of the lateral ventricles in rats [178]. Additionally, the neuroprotective role of TUDCA, a taurine-conjugated secondary BA, has been demonstrated in models of neurodegenerative diseases, including Alzheimer’s, Parkinson’s, and Huntington’s diseases [179].
MRGPRX4, also known as sensory neuron-specific G-Protein coupled receptor 5/6, is involved in cholestatic itch [62,180,181]. DCA, CDCA, CA, and LCA evoke itching of human skin via the activation of MRGPRX4 expression in dorsal root ganglion (DRG) neurons [62]. Furthermore, DCA, TDCA, UDCA, and CDCA evoke scratching behavior in mice expressing human MRGPRX4 [63].
4. Concluding Remarks
BAs are synthesized in the liver as primary BAs, and a variety of secondary BAs are synthesized in the intestine through the involvement of various microorganisms. These BAs bind to multiple nuclear and cell-surface receptors. Therefore, BAs have been recognized as signaling molecules. Each BA has a different affinity for the receptor to which it can bind. The abundance ratio of primary and secondary BAs may affect short- and long-term health. Thus, factors such as food, aging, and antibiotics affect the gut microbiome and may change the BA composition and affect health. Currently, probiotics, prebiotics, synbiotics, and postbiotics are considered substances that maintain health by preventing metabolic diseases by controlling the composition of the gut microbiome [182]. A recent study demonstrated that centenarians have a unique gut microbiome that produces iso-, 3-oxo-, allo-, 3-oxoallo-, and isoalloLCA [56]. Further elucidation of the role of the gut microbiome and the modification of BAs by the gut microbiome will lead to the development of new strategies for treating disease and improving human health.
Author Contributions
Y.K. conceptualized and wrote this manuscript. H.N. revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
The APC was funded by Tokushima Bunri University.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dawson, P.A.; Karpen, S.J. Intestinal transport and metabolism of bile acids. J. Lipid Res. 2015, 56, 1085–1099. [Google Scholar] [CrossRef]
- Doden, H.; Ridlon, J. Microbial Hydroxysteroid Dehydrogenases: From Alpha to Omega. Microorganisms 2021, 9, 469. [Google Scholar] [CrossRef]
- Thomas, C.; Pellicciari, R.; Pruzanski, M.; Auwerx, J.; Schoonjans, K. Targeting bile-acid signalling for metabolic diseases. Nat. Rev. Drug Discov. 2008, 7, 678–693. [Google Scholar] [CrossRef]
- Kiriyama, Y.; Nochi, H. The Biosynthesis, Signaling, and Neurological Functions of Bile Acids. Biomolecules 2019, 9, 232. [Google Scholar] [CrossRef]
- Molinaro, A.; Wahlström, A.; Marschall, H.-U. Role of Bile Acids in Metabolic Control. Trends Endocrinol. Metab. 2018, 29, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Bromke, M.A.; Krzystek-Korpacka, M. Bile Acid Signaling in Inflammatory Bowel Disease. Int. J. Mol. Sci. 2021, 22, 9096. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, T.; Zhao, A.; Wang, X.; Xie, G.; Huang, F.; Liu, J.; Zhao, Q.; Wang, S.; Wang, C.; et al. The Brain Metabolome of Male Rats across the Lifespan. Sci. Rep. 2016, 6, 24125. [Google Scholar] [CrossRef]
- Baloni, P.; Funk, C.C.; Yan, J.; Yurkovich, J.T.; Kueider-Paisley, A.; Nho, K.; Heinken, A.; Jia, W.; Mahmoudiandehkordi, S.; Louie, G.; et al. Metabolic Network Analysis Reveals Altered Bile Acid Synthesis and Metabolism in Alzheimer’s Disease. Cell Rep. Med. 2020, 1, 100138. [Google Scholar] [CrossRef]
- Chiang, J.Y.; Ferrell, J.M. Up to date on cholesterol 7 alpha-hydroxylase (CYP7A1) in bile acid synthesis. Liver Res. 2020, 4, 47–63. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.W. Fifty years of advances in bile acid synthesis and metabolism. J. Lipid Res. 2009, 50, S120–S125. [Google Scholar] [CrossRef]
- Fakheri, R.J.; Javitt, N.B. 27-Hydroxycholesterol, does it exist? On the nomenclature and stereochemistry of 26-hydroxylated sterols. Steroids 2012, 77, 575–577. [Google Scholar] [CrossRef] [PubMed]
- Styles, N.A.; Shonsey, E.M.; Falany, J.L.; Guidry, A.L.; Barnes, S.; Falany, C.N. Carboxy-terminal mutations of bile acid CoA: N-acyltransferase alter activity and substrate specificity. J. Lipid Res. 2016, 57, 1133–1143. [Google Scholar] [CrossRef]
- Trauner, M.; Boyer, J.L. Bile Salt Transporters: Molecular Characterization, Function, and Regulation. Physiol. Rev. 2003, 83, 633–671. [Google Scholar] [CrossRef] [PubMed]
- Daly, J.; Keely, S.; Gahan, C. Functional and Phylogenetic Diversity of BSH and PVA Enzymes. Microorganisms 2021, 9, 732. [Google Scholar] [CrossRef] [PubMed]
- Foley, M.H.; O’Flaherty, S.; Allen, G.; Rivera, A.J.; Stewart, A.K.; Barrangou, R.; Theriot, C.M. Lactobacillus bile salt hydrolase substrate specificity governs bacterial fitness and host colonization. Proc. Natl. Acad. Sci. USA 2021, 118, e2017709118. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Zhao, J.; Zhang, H.; Zhai, Q.; Chen, W. Mining genome traits that determine the different gut colonization potential of Lactobacillus and Bifidobacterium species. Microb. Genom. 2021, 7, 000581. [Google Scholar] [CrossRef] [PubMed]
- Bhagwat, A.; Annapure, U.S. In vitro assessment of metabolic profile of Enterococcus strains of human origin. J. Genet. Eng. Biotechnol. 2019, 17, 11. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.P.; Hudson, L.L. Cloning and characterization of a conjugated bile acid hydrolase gene from Clostridium perfringens. Appl. Environ. Microbiol. 1995, 61, 2514–2520. [Google Scholar] [CrossRef] [PubMed]
- Gopal-Srivastava, R.; Hylemon, P.B. Purification and characterization of bile salt hydrolase from Clostridium perfringens. J. Lipid Res. 1988, 29, 1079–1085. [Google Scholar] [CrossRef]
- Yao, L.; Seaton, S.C.; Ndousse-Fetter, S.; Adhikari, A.A.; DiBenedetto, N.; Mina, A.; Banks, A.S.; Bry, L.; Devlin, A.S. A selective gut bacterial bile salt hydrolase alters host metabolism. eLife 2018, 7, e37182. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, A.A.; Seegar, T.C.M.; Ficarro, S.B.; McCurry, M.D.; Ramachandran, D.; Yao, L.; Chaudhari, S.N.; Ndousse-Fetter, S.; Banks, A.S.; Marto, J.A.; et al. Development of a covalent inhibitor of gut bacterial bile salt hydrolases. Nat. Chem. Biol. 2020, 16, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef]
- Funabashi, M.; Grove, T.L.; Wang, M.; Varma, Y.; McFadden, M.E.; Brown, L.C.; Guo, C.; Higginbottom, S.; Almo, S.C.; Fischbach, M.A. A metabolic pathway for bile acid dehydroxylation by the gut microbiome. Nat. Cell Biol. 2020, 582, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Lepercq, P.; Gerard, P.; Beguet, F.; Raibaud, P.; Grill, J.P.; Relano, P.; Cayuela, C.; Juste, C. Epimerization of chenodeoxycholic acid to ursodeoxycholic acid by Clostridium baratii isolated from human feces. FEMS Microbiol. Lett. 2004, 235, 65–72. [Google Scholar] [CrossRef]
- Heinken, A.; Ravcheev, D.A.; Baldini, F.; Heirendt, L.; Fleming, R.M.T.; Thiele, I. Systematic assessment of secondary bile acid metabolism in gut microbes reveals distinct metabolic capabilities in inflammatory bowel disease. Microbiome 2019, 7, 75. [Google Scholar] [CrossRef]
- Reed, A.; Theriot, C. Contribution of Inhibitory Metabolites and Competition for Nutrients to Colonization Resistance against Clostridioides difficile by Commensal Clostridium. Microorganisms 2021, 9, 371. [Google Scholar] [CrossRef]
- Harris, S.C.; Devendran, S.; Mendez-Garcia, C.; Mythen, S.M.; Wright, C.L.; Fields, C.J.; Hernandez, A.G.; Cann, I.; Hylemon, P.B.; Ridlon, J.M. Bile acid oxidation by Eggerthella lenta strains C592 and DSM 2243(T). Gut Microbes 2018, 9, 523–539. [Google Scholar]
- Doden, H.L.; Wolf, P.G.; Gaskins, H.R.; Anantharaman, K.; Alves, J.M.P.; Ridlon, J.M. Completion of the gut microbial epi-bile acid pathway. Gut Microbes 2021, 13, 1907271. [Google Scholar] [CrossRef]
- Devlin, A.S.; Fischbach, M.A. A biosynthetic pathway for a prominent class of microbiota-derived bile acids. Nat. Chem. Biol. 2015, 11, 685–690. [Google Scholar] [CrossRef]
- Lucas, L.N.; Barrett, K.; Kerby, R.L.; Zhang, Q.; Cattaneo, L.E.; Stevenson, D.; Rey, F.E.; Amador-Noguez, D. Dominant Bacterial Phyla from the Human Gut Show Widespread Ability to Transform and Conjugate Bile Acids. mSystems 2021, 6, e00805-21. [Google Scholar] [CrossRef] [PubMed]
- Quinn, R.A.; Melnik, A.V.; Vrbanac, A.; Fu, T.; Patras, K.A.; Christy, M.P.; Bodai, Z.; Belda-Ferre, P.; Tripathi, A.; Chung, L.K.; et al. Global chemical effects of the microbiome include new bile-acid conjugations. Nature 2020, 579, 123–129. [Google Scholar] [CrossRef]
- Chiang, J.Y.L. Bile Acid Metabolism and Signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [CrossRef]
- Oelkers, P.; Kirby, L.C.; Heubi, J.E.; Dawson, P.A. Primary bile acid malabsorption caused by mutations in the ileal sodium-dependent bile acid transporter gene (SLC10A2). J. Clin. Investig. 1997, 99, 1880–1887. [Google Scholar] [CrossRef] [PubMed]
- Meier, Y.; Eloranta, J.J.; Darimont, J.; Ismair, M.G.; Hiller, C.; Fried, M.; Kullak-Ublick, G.A.; Vavricka, S.R. Regional Distribution of Solute Carrier mRNA Expression Along the Human Intestinal Tract. Drug Metab. Dispos. 2007, 35, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Shneider, B.L.; Dawson, P.A.; Christie, D.M.; Hardikar, W.; Wong, M.H.; Suchy, F.J. Cloning and molecular characterization of the ontogeny of a rat ileal sodium-dependent bile acid transporter. J. Clin. Investig. 1995, 95, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Christie, D.M.; Dawson, P.A.; Thevananther, S.; Shneider, B.L. Comparative analysis of the ontogeny of a sodium-dependent bile acid transporter in rat kidney and ileum. Am. J. Physiol. Content 1996, 271, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Kramer, W.; Wilson, F. Identification of cytosolic and microsomal bile acid-binding proteins in rat ileal enterocytes. J. Biol. Chem. 1990, 265, 14986–14995. [Google Scholar] [CrossRef]
- Christian, W.V.; Li, N.; Hinkle, P.M.; Ballatori, N. beta-Subunit of the Ostalpha-Ostbeta organic solute transporter is required not only for heterodimerization and trafficking but also for function. J. Biol. Chem. 2012, 287, 21233–21243. [Google Scholar] [CrossRef] [PubMed]
- Ballatori, N.; Christian, W.V.; Lee, J.Y.; Dawson, P.A.; Soroka, C.J.; Boyer, J.L.; Madejczyk, M.S.; Li, N. OSTalpha-OSTbeta: A major basolateral bile acid and steroid transporter in human intestinal, renal, and biliary epithelia. Hepatology 2005, 42, 1270–1279. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.A.; Hubbert, M.; Haywood, J.; Craddock, A.L.; Zerangue, N.; Christian, W.V.; Ballatori, N. The heteromeric organic solute transporter alpha-beta, Ostalpha-Ostbeta, is an ileal basolateral bile acid transporter. J. Biol. Chem. 2005, 280, 6960–6968. [Google Scholar] [CrossRef] [PubMed]
- Ananthanarayanan, M.; Ng, O.C.; Boyer, J.L.; Suchy, F.J. Characterization of cloned rat liver Na(+)-bile acid cotransporter using peptide and fusion protein antibodies. Am. J. Physiol. Content 1994, 267, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Stieger, B.; Hagenbuch, B.; Landmann, L.; Hochli, M.; Schroeder, A.; Meier, P.J. In situ localization of the hepatocytic Na+/Taurocholate cotransporting polypeptide in rat liver. Gastroenterology 1994, 107, 1781–1787. [Google Scholar] [CrossRef]
- Hagenbuch, B.; Scharschmidt, B.F.; Meier, P.J. Effect of antisense oligonucleotides on the expression of hepatocellular bile acid and organic anion uptake systems in Xenopus laevis oocytes. Biochem. J. 1996, 316, 901–904. [Google Scholar] [CrossRef]
- Nigam, S.K.; Bush, K.T.; Martovetsky, G.; Ahn, S.-Y.; Liu, H.C.; Richard, E.; Bhatnagar, V.; Wu, W. The Organic Anion Transporter (OAT) Family: A Systems Biology Perspective. Physiol. Rev. 2015, 95, 83–123. [Google Scholar] [CrossRef] [PubMed]
- Suga, T.; Yamaguchi, H.; Sato, T.; Maekawa, M.; Goto, J.; Mano, N. Preference of Conjugated Bile Acids over Unconjugated Bile Acids as Substrates for OATP1B1 and OATP1B3. PLoS ONE 2017, 12, e0169719. [Google Scholar] [CrossRef] [PubMed]
- Vallim, T.Q.D.A.; Tarling, E.J.; Edwards, P.A. Pleiotropic Roles of Bile Acids in Metabolism. Cell Metab. 2013, 17, 657–669. [Google Scholar] [CrossRef]
- Bisschop, P.H.; Bandsma, R.H.J.; Stellaard, F.; Ter Harmsel, A.; Meijer, A.J.; Sauerwein, H.P.; Kuipers, F.; Romijn, J.A. Low-fat, high-carbohydrate and high-fat, low-carbohydrate diets decrease primary bile acid synthesis in humans. Am. J. Clin. Nutr. 2004, 79, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Stenman, L.K.; Holma, R.; Korpela, R. High-fat-induced intestinal permeability dysfunction associated with altered fecal bile acids. World J. Gastroenterol. 2012, 18, 923–929. [Google Scholar] [CrossRef]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Wang, D.Q.-H.; Molina, E.M.; Baccetto, R.; Calamita, G.; Palmieri, V.O.; Portincasa, P. Bile Acids and Cancer: Direct and Environmental-Dependent Effects. Ann. Hepatol. 2017, 16, S87–S105. [Google Scholar] [CrossRef] [PubMed]
- Bortolotti, M.; Kreis, R.; Debard, C.; Cariou, B.; Faeh, D.; Chetiveaux, M.; Ith, M.; Vermathen, P.; Stefanoni, N.; Lê, K.-A.; et al. High protein intake reduces intrahepatocellular lipid deposition in humans. Am. J. Clin. Nutr. 2009, 90, 1002–1010. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Igarashi, M.; Li, X.; Nakatani, A.; Miyamoto, J.; Inaba, Y.; Sutou, A.; Saito, T.; Sato, T.; Tachibana, N.; et al. Dietary soybean protein ameliorates high-fat diet-induced obesity by modifying the gut microbiota-dependent biotransformation of bile acids. PLoS ONE 2018, 13, e0202083. [Google Scholar] [CrossRef] [PubMed]
- Weickert, M.O.; Hattersley, J.G.; Kyrou, I.; Arafat, A.M.; Rudovich, N.; Roden, M.; Nowotny, P.; Von Loeffelholz, C.; Matysik, S.; Schmitz, G.; et al. Effects of supplemented isoenergetic diets varying in cereal fiber and protein content on the bile acid metabolic signature and relation to insulin resistance. Nutr. Diabetes 2018, 8, 11. [Google Scholar] [CrossRef]
- Ginos, B.N.; Navarro, S.L.; Schwarz, Y.; Gu, H.; Wang, D.; Randolph, T.W.; Shojaie, A.; Hullar, M.A.; Lampe, P.D.; Kratz, M.; et al. Circulating bile acids in healthy adults respond differently to a dietary pattern characterized by whole grains, legumes and fruits and vegetables compared to a diet high in refined grains and added sugars: A randomized, controlled, crossover feeding study. Metabolism 2018, 83, 197–204. [Google Scholar] [CrossRef]
- Frommherz, L.; Bub, A.; Hummel, E.; Rist, M.J.; Roth, A.; Watzl, B.; Kulling, S.E. Age-Related Changes of Plasma Bile Acid Concentrations in Healthy Adults—Results from the Cross-Sectional KarMeN Study. PLoS ONE 2016, 11, e0153959. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Atarashi, K.; Plichta, D.R.; Arai, Y.; Sasajima, S.; Kearney, S.M.; Suda, W.; Takeshita, K.; Sasaki, T.; Okamoto, S.; et al. Novel bile acid biosynthetic pathways are enriched in the microbiome of centenarians. Nat. Cell Biol. 2021, 599, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Willing, B.P.; Russell, S.L.; Finlay, B.B. Shifting the balance: Antibiotic effects on host–microbiota mutualism. Nat. Rev. Genet. 2011, 9, 233–243. [Google Scholar] [CrossRef]
- Ramirez, J.; Guarner, F.; Fernandez, L.B.; Maruy, A.; Sdepanian, V.L.; Cohen, H. Antibiotics as Major Disruptors of Gut Microbiota. Front. Cell. Infect. Microbiol. 2020, 10, 572912. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Zhou, K.; Lu, Y.; Yan, W.; Cai, W.; Wang, Y. Administration of antibiotics contributes to cholestasis in pediatric patients with intestinal failure via the alteration of FXR signaling. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef]
- Chiang, J.Y.L.; Ferrell, J.M. Bile acid receptors FXR and TGR5 signaling in fatty liver diseases and therapy. Am. J. Physiol. Liver Physiol. 2020, 318, G554–G573. [Google Scholar] [CrossRef] [PubMed]
- Ticho, A.; Malhotra, P.; Dudeja, P.K.; Gill, R.K.; Alrefai, W.A. Bile acid receptors and gastrointestinal functions. Liver Res. 2019, 3, 31–39. [Google Scholar] [CrossRef]
- Yu, H.; Zhao, T.; Liu, S.; Wu, Q.; Johnson, O.; Wu, Z.; Zhuang, Z.; Shi, Y.; Peng, L.; He, R.; et al. MRGPRX4 is a bile acid receptor for human cholestatic itch. eLife 2019, 8, e48431. [Google Scholar] [CrossRef]
- Meixiong, J.; Vasavda, C.; Snyder, S.H.; Dong, X. MRGPRX4 is a G protein-coupled receptor activated by bile acids that may contribute to cholestatic pruritus. Proc. Natl. Acad. Sci. USA 2019, 116, 10525–10530. [Google Scholar] [CrossRef]
- Wang, H.; Chen, J.; Hollister, K.; Sowers, L.C.; Forman, B.M. Endogenous Bile Acids Are Ligands for the Nuclear Receptor FXR/BAR. Mol. Cell 1999, 3, 543–553. [Google Scholar] [CrossRef]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a Nuclear Receptor for Bile Acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef]
- Parks, D.J.; Blanchard, S.G.; Bledsoe, R.K.; Chandra, G.; Consler, T.G.; Kliewer, S.A.; Stimmel, J.B.; Willson, T.M.; Zavacki, A.M.; Moore, D.D.; et al. Bile Acids: Natural Ligands for an Orphan Nuclear Receptor. Science 1999, 284, 1365–1368. [Google Scholar] [CrossRef]
- Distrutti, E.; Santucci, L.; Cipriani, S.; Renga, B.; Schiaroli, E.; Ricci, P.; Donini, A.; Fiorucci, S. Bile acid activated receptors are targets for regulation of integrity of gastrointestinal mucosa. J. Gastroenterol. 2015, 50, 707–719. [Google Scholar] [CrossRef]
- Seol, W.; Choi, H.S.; Moore, D.D. Isolation of proteins that interact specifically with the retinoid X receptor: Two novel orphan receptors. Mol. Endocrinol. 1995, 9, 72–85. [Google Scholar] [CrossRef]
- Thomas, A.M.; Hart, S.; Kong, B.; Fang, J.; Zhong, X.-B.; Guo, G.L. Genome-wide tissue-specific farnesoid X receptor binding in mouse liver and intestine. Hepatology 2009, 51, 1410–1419. [Google Scholar] [CrossRef]
- Laffitte, B.A.; Kast, H.R.; Nguyen, C.M.; Zavacki, A.M.; Moore, D.D.; Edwards, P.A. Identification of the DNA Binding Specificity and Potential Target Genes for the Farnesoid X-activated Receptor. J. Biol. Chem. 2000, 275, 10638–10647. [Google Scholar] [CrossRef]
- Kast, H.R.; Goodwin, B.; Tarr, P.T.; Jones, S.A.; Anisfeld, A.M.; Stoltz, C.M.; Tontonoz, P.; Kliewer, S.; Willson, T.M.; Edwards, P.A. Regulation of Multidrug Resistance-associated Protein 2 (ABCC2) by the Nuclear Receptors Pregnane X Receptor, Farnesoid X-activated Receptor, and Constitutive Androstane Receptor. J. Biol. Chem. 2002, 277, 2908–2915. [Google Scholar] [CrossRef] [PubMed]
- Edwards, P.A.; Kast, H.R.; Anisfeld, A.M. BAREing it all: The adoption of LXR and FXR and their roles in lipid homeostasis. J. Lipid Res. 2002, 43, 2–12. [Google Scholar] [CrossRef]
- Xu, J.; Wu, R.-C.; O’Malley, B.W. Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. Nat. Rev. Cancer 2009, 9, 615–630. [Google Scholar] [CrossRef]
- Kong, X.; Feng, L.; Yan, D.; Li, B.; Yang, Y.; Ma, X. FXR-mediated epigenetic regulation of GLP-1R expression contributes to enhanced incretin effect in diabetes after RYGB. J. Cell. Mol. Med. 2021, 1–12. [Google Scholar] [CrossRef]
- Kong, X.; Li, B.; Deng, Y.; Ma, X. FXR Mediates Adenylyl Cyclase 8 Expression in Pancreatic beta-Cells. J. Diabetes Res. 2019, 2019, 8915818. [Google Scholar] [CrossRef]
- Yang, Y.-L.; Kuo, H.-C.; Wang, F.-S.; Huang, Y.-H. MicroRNA-29a Disrupts DNMT3b to Ameliorate Diet-Induced Non-Alcoholic Steatohepatitis in Mice. Int. J. Mol. Sci. 2019, 20, 1499. [Google Scholar] [CrossRef]
- Huang, Y.-H.; Tiao, M.-M.; Huang, L.-T.; Chuang, J.-H.; Kuo, K.-C.; Yang, Y.-L.; Wang, F.-S. Activation of Mir-29a in Activated Hepatic Stellate Cells Modulates Its Profibrogenic Phenotype through Inhibition of Histone Deacetylases 4. PLoS ONE 2015, 10, e0136453. [Google Scholar] [CrossRef]
- Pathak, P.; Liu, H.; Boehme, S.; Xie, C.; Krausz, K.W.; Gonzalez, F.; Chiang, J.Y.L. Farnesoid X receptor induces Takeda G-protein receptor 5 cross-talk to regulate bile acid synthesis and hepatic metabolism. J. Biol. Chem. 2017, 292, 11055–11069. [Google Scholar] [CrossRef]
- Kumar, D.P.; Asgharpour, A.; Mirshahi, F.; Park, S.H.; Liu, S.; Imai, Y.; Nadler, J.L.; Grider, J.R.; Murthy, K.S.; Sanyal, A.J. Activation of Transmembrane Bile Acid Receptor TGR5 Modulates Pancreatic Islet α Cells to Promote Glucose Homeostasis. J. Biol. Chem. 2016, 291, 6626–6640. [Google Scholar] [CrossRef] [PubMed]
- Duboc, H.; Taché, Y.; Hofmann, A.F. The bile acid TGR5 membrane receptor: From basic research to clinical application. Dig. Liver Dis. 2014, 46, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, Y.; Fujii, R.; Hosoya, M.; Harada, M.; Yoshida, H.; Miwa, M.; Fukusumi, S.; Habata, Y.; Itoh, T.; Shintani, Y.; et al. A G Protein-coupled Receptor Responsive to Bile Acids. J. Biol. Chem. 2003, 278, 9435–9440. [Google Scholar] [CrossRef]
- Maruyama, T.; Miyamoto, Y.; Nakamura, T.; Tamai, Y.; Okada, H.; Sugiyama, E.; Nakamura, T.; Itadani, H.; Tanaka, K. Identification of membrane-type receptor for bile acids (M-BAR). Biochem. Biophys. Res. Commun. 2002, 298, 714–719. [Google Scholar] [CrossRef]
- De Marino, S.; Carino, A.; Masullo, D.; Finamore, C.; Marchianò, S.; Cipriani, S.; Di Leva, F.S.; Catalanotti, B.; Novellino, E.; Limongelli, V.; et al. Hyodeoxycholic acid derivatives as liver X receptor α and G-protein-coupled bile acid receptor agonists. Sci. Rep. 2017, 7, srep43290. [Google Scholar] [CrossRef]
- Huang, C.; Wang, J.; Hu, W.; Wang, C.; Lu, X.; Tong, L.; Wu, F.; Zhang, W. Identification of functional farnesoid X receptors in brain neurons. FEBS Lett. 2016, 590, 3233–3242. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hagedorn, C.H.; Wang, L. Role of nuclear receptor SHP in metabolism and cancer. Biochim. Biophys. Acta 2011, 1812, 893–908. [Google Scholar] [CrossRef] [PubMed]
- Chanda, D.; Park, J.-H.; Choi, H.-S. Molecular Basis of Endocrine Regulation by Orphan Nuclear Receptor Small Heterodimer Partner. Endocr. J. 2008, 55, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, B.; Jones, S.A.; Price, R.R.; Watson, M.A.; McKee, D.D.; Moore, L.B.; Galardi, C.; Wilson, J.G.; Lewis, M.C.; Roth, M.E.; et al. A Regulatory Cascade of the Nuclear Receptors FXR, SHP-1, and LRH-1 Represses Bile Acid Biosynthesis. Mol. Cell 2000, 6, 517–526. [Google Scholar] [CrossRef]
- Fu, T.; Kim, Y.C.; Byun, S.; Kim, D.H.; Seok, S.; Suino-Powell, K.; Xu, H.E.; Kemper, B.; Kemper, J.K. FXR Primes the Liver for Intestinal FGF15 Signaling by Transient Induction of beta-Klotho. Mol. Endocrinol. 2016, 30, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gunewardena, S.; Li, F.; Matye, D.J.; Chen, C.; Chao, X.; Jung, T.; Zhang, Y.; Czerwinski, M.; Ni, H.M.; et al. An FGF15/19-TFEB regulatory loop controls hepatic cholesterol and bile acid homeostasis. Nat. Commun. 2020, 11, 3612. [Google Scholar] [CrossRef]
- Ji, S.; Liu, Q.; Zhang, S.; Chen, Q.; Wang, C.; Zhang, W.; Xiao, C.; Li, Y.; Nian, C.; Li, J.; et al. FGF15 Activates Hippo Signaling to Suppress Bile Acid Metabolism and Liver Tumorigenesis. Dev. Cell 2019, 48, 460–474.e9. [Google Scholar] [CrossRef]
- Denson, L.A.; Sturm, E.; Echevarria, W.; Zimmerman, T.L.; Makishima, M.; Mangelsdorf, D.; Karpen, S.J. The Orphan Nuclear Receptor, shp, Mediates Bile Acid-Induced Inhibition of the Rat Bile Acid Transporter, ntcp. Gastroenterology 2001, 121, 140–147. [Google Scholar] [CrossRef]
- Zollner, G.; Wagner, M.; Fickert, P.; Geier, A.; Fuchsbichler, A.; Silbert, D.; Gumhold, J.; Zatloukal, K.; Kaser, A.; Tilg, H.; et al. Role of nuclear receptors and hepatocyte-enriched transcription factors for Ntcp repression in biliary obstruction in mouse liver. Am. J. Physiol. Liver Physiol. 2005, 289, G798–G805. [Google Scholar] [CrossRef][Green Version]
- Ijssennagger, N.; Janssen, A.W.; Milona, A.; Pittol, J.M.R.; Hollman, D.A.; Mokry, M.; Betzel, B.; Berends, F.; Janssen, I.M.; van Mil, S.W.; et al. Gene expression profiling in human precision cut liver slices in response to the FXR agonist obeticholic acid. J. Hepatol. 2016, 64, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Tanaka, K.; Suzuki, J.; Miyoshi, H.; Harada, N.; Nakamura, T.; Miyamoto, Y.; Kanatani, A.; Tamai, Y. Targeted disruption of G protein-coupled bile acid receptor 1 (Gpbar1/M-Bar) in mice. J. Endocrinol. 2006, 191, 197–205. [Google Scholar] [CrossRef]
- Spatz, M.; Ciocan, D.; Merlen, G.; Rainteau, D.; Humbert, L.; Gomes-Rochette, N.; Hugot, C.; Trainel, N.; Mercier-Nomé, F.; Domenichini, S.; et al. Bile acid-receptor TGR5 deficiency worsens liver injury in alcohol-fed mice by inducing intestinal microbiota dysbiosis. JHEP Rep. 2021, 3, 100230. [Google Scholar] [CrossRef]
- Donepudi, A.C.; Boehme, S.; Li, F.; Chiang, J.Y. G-protein-coupled bile acid receptor plays a key role in bile acid metabolism and fasting-induced hepatic steatosis in mice. Hepatology 2017, 65, 813–827. [Google Scholar] [CrossRef]
- Bidault-Jourdainne, V.; Merlen, G.; Glénisson, M.; Doignon, I.; Garcin, I.; Péan, N.; Boisgard, R.; Ursic-Bedoya, J.; Serino, M.; Ullmer, C.; et al. TGR5 controls bile acid composition and gallbladder function to protect the liver from bile acid overload. JHEP Rep. 2021, 3, 100214. [Google Scholar] [CrossRef] [PubMed]
- Staudinger, J.L.; Goodwin, B.; Jones, S.A.; Hawkins-Brown, D.; MacKenzie, K.I.; LaTour, A.; Liu, Y.; Klaassen, C.D.; Brown, K.K.; Reinhard, J.; et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc. Natl. Acad. Sci. USA 2001, 98, 3369–3374. [Google Scholar] [CrossRef] [PubMed]
- Schuster, D.; Langer, T. The Identification of Ligand Features Essential for PXR Activation by Pharmacophore Modeling. J. Chem. Inf. Model. 2005, 45, 431–439. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Goodwin, B.; Willson, T.M. The Nuclear Pregnane X Receptor: A Key Regulator of Xenobiotic Metabolism. Endocr. Rev. 2002, 23, 687–702. [Google Scholar] [CrossRef]
- Xie, W.; Barwick, J.L.; Simon, C.M.; Pierce, A.M.; Safe, S.; Blumberg, B.; Guzelian, P.S.; Evans, R.M. Reciprocal activation of Xenobiotic response genes by nuclear receptors SXR/PXR and CAR. Genes Dev. 2000, 14, 3014–3023. [Google Scholar] [CrossRef]
- Willson, T.M.; Kliewer, S.A. Pxr, car and drug metabolism. Nat. Rev. Drug Discov. 2002, 1, 259–266. [Google Scholar] [CrossRef]
- Li, T.; Chiang, J. Mechanism of rifampicin and pregnane X receptor inhibition of human cholesterol 7α-hydroxylase gene transcription. Am. J. Physiol. Liver Physiol. 2005, 288, G74–G84. [Google Scholar] [CrossRef]
- Bhalla, S.; Ozalp, C.; Fang, S.; Xiang, L.; Kemper, J.K. Ligand-activated Pregnane X Receptor Interferes with HNF-4 Signaling by Targeting a Common Coactivator PGC-1α. J. Biol. Chem. 2004, 279, 45139–45147. [Google Scholar] [CrossRef]
- Masuno, H.; Ikura, T.; Morizono, D.; Orita, I.; Yamada, S.; Shimizu, M.; Ito, N. Crystal structures of complexes of vitamin D receptor ligand-binding domain with lithocholic acid derivatives. J. Lipid Res. 2013, 54, 2206–2213. [Google Scholar] [CrossRef]
- Makishima, M.; Lu, T.T.; Xie, W.; Whitfield, G.K.; Domoto, H.; Evans, R.M.; Haussler, M.R.; Mangelsdorf, D.J. Vitamin D Receptor As an Intestinal Bile Acid Sensor. Science 2002, 296, 1313–1316. [Google Scholar] [CrossRef]
- Pavek, P.; Pospechova, K.; Svecova, L.; Syrova, Z.; Stejskalova, L.; Blazkova, J.; Dvorak, Z.; Blahoš, J. Intestinal cell-specific vitamin D receptor (VDR)-mediated transcriptional regulation of CYP3A4 gene. Biochem. Pharmacol. 2010, 79, 277–287. [Google Scholar] [CrossRef]
- Matsubara, T.; Yoshinari, K.; Aoyama, K.; Sugawara, M.; Sekiya, Y.; Nagata, K.; Yamazoe, Y. Role of Vitamin D Receptor in the Lithocholic Acid-Mediated CYP3A Induction in Vitro and in Vivo. Drug Metab. Dispos. 2008, 36, 2058–2063. [Google Scholar] [CrossRef]
- Song, C.; Hiipakka, R.A.; Liao, S. Selective activation of liver X receptor alpha by 6alpha-hydroxy bile acids and analogs. Steroids 2000, 65, 423–427. [Google Scholar] [CrossRef]
- Peet, D.J.; Turley, S.D.; Ma, W.; Janowski, B.A.; Lobaccaro, J.M.; Hammer, R.E.; Mangelsdorf, D.J. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR alpha. Cell 1998, 93, 693–704. [Google Scholar] [CrossRef]
- Alberti, S.; Schuster, G.; Parini, P.; Feltkamp, D.; Diczfalusy, U.; Rudling, M.; Angelin, B.; Bjorkhem, I.; Pettersson, S.; Gustafsson, J.A. Hepatic cholesterol metabolism and resistance to dietary cholesterol in LXRbeta-deficient mice. J. Clin. Investig. 2001, 107, 565–573. [Google Scholar] [CrossRef]
- Duran-Sandoval, D.; Mautino, G.; Martin, G.; Percevault, F.; Barbier, O.; Fruchart, J.-C.; Kuipers, F.; Staels, B. Glucose Regulates the Expression of the Farnesoid X Receptor in Liver. Diabetes 2004, 53, 890–898. [Google Scholar] [CrossRef]
- Renga, B.; Mencarelli, A.; Vavassori, P.; Brancaleone, V.; Fiorucci, S. The bile acid sensor FXR regulates insulin transcription and secretion. Biochim. Biophys. Acta 2010, 1802, 363–372. [Google Scholar] [CrossRef]
- Renga, B.; Mencarelli, A.; D’Amore, C.; Cipriani, S.; Baldelli, F.; Zampella, A.; Distrutti, E.; Fiorucci, S. Glucocorticoid receptor mediates the gluconeogenic activity of the farnesoid X receptor in the fasting condition. FASEB J. 2012, 26, 3021–3031. [Google Scholar] [CrossRef]
- Ma, K.; Saha, P.K.; Chan, L.; Moore, D.D. Farnesoid X receptor is essential for normal glucose homeostasis. J. Clin. Investig. 2006, 116, 1102–1109. [Google Scholar] [CrossRef]
- Potthoff, M.J.; Boney-Montoya, J.; Choi, M.; He, T.; Sunny, N.E.; Satapati, S.; Suino-Powell, K.; Xu, H.E.; Gerard, R.D.; Finck, B.N.; et al. FGF15/19 regulates hepatic glucose metabolism by inhibiting the CREB-PGC-1alpha pathway. Cell Metab. 2011, 13, 729–738. [Google Scholar] [CrossRef]
- Kiriyama, Y.; Nochi, H. Intra- and Intercellular Quality Control Mechanisms of Mitochondria. Cells 2017, 7, 1. [Google Scholar] [CrossRef]
- Rowe, G.C.; Arany, Z. Genetic models of PGC-1 and glucose metabolism and homeostasis. Rev. Endocr. Metab. Disord. 2013, 15, 21–29. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Takenoshita, M.; Kabashima, T.; Uyeda, K. Glucose and cAMP regulate the L-type pyruvate kinase gene by phosphorylation/dephosphorylation of the carbohydrate response element binding protein. Proc. Natl. Acad. Sci. USA 2001, 98, 13710–13715. [Google Scholar] [CrossRef]
- Caron, S.; Samanez, C.H.; Dehondt, H.; Ploton, M.; Briand, O.; Lien, F.; Dorchies, E.; Dumont, J.; Postic, C.; Cariou, B.; et al. Farnesoid X Receptor Inhibits the Transcriptional Activity of Carbohydrate Response Element Binding Protein in Human Hepatocytes. Mol. Cell. Biol. 2013, 33, 2202–2211. [Google Scholar] [CrossRef]
- Shen, H.; Zhang, Y.; Ding, H.; Wang, X.; Chen, L.; Jiang, H.; Shen, X. Farnesoid X Receptor Induces GLUT4 Expression Through FXR Response Element in the GLUT4 Promoter. Cell. Physiol. Biochem. 2008, 22, 1–14. [Google Scholar] [CrossRef]
- Seyer, P.; Vallois, D.; Poitry-Yamate, C.; Schutz, F.; Metref, S.; Tarussio, D.; Maechler, P.; Staels, B.; Lanz, B.; Grueter, R.; et al. Hepatic glucose sensing is required to preserve beta cell glucose competence. J. Clin. Investig. 2013, 123, 1662–1676. [Google Scholar] [CrossRef]
- Katsuma, S.; Hirasawa, A.; Tsujimoto, G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem. Biophys. Res. Commun. 2005, 329, 386–390. [Google Scholar] [CrossRef]
- Thomas, C.; Gioiello, A.; Noriega, L.; Strehle, A.; Oury, J.; Rizzo, G.; Macchiarulo, A.; Yamamoto, H.; Mataki, C.; Pruzanski, M.; et al. TGR5-Mediated Bile Acid Sensing Controls Glucose Homeostasis. Cell Metab. 2009, 10, 167–177. [Google Scholar] [CrossRef]
- Brighton, C.; Rievaj, J.; Kuhre, R.E.; Glass, L.; Schoonjans, K.; Holst, J.J.; Gribble, F.; Reimann, F. Bile Acids Trigger GLP-1 Release Predominantly by Accessing Basolaterally Located G Protein–Coupled Bile Acid Receptors. Endocrinology 2015, 156, 3961–3970. [Google Scholar] [CrossRef]
- Christiansen, C.B.; Trammell, S.; Albrechtsen, N.J.W.; Schoonjans, K.; Albrechtsen, R.; Gillum, M.; Kuhre, R.E.; Holst, J.J. Bile acids drive colonic secretion of glucagon-like-peptide 1 and peptide-YY in rodents. Am. J. Physiol. Liver Physiol. 2019, 316, G574–G584. [Google Scholar] [CrossRef] [PubMed]
- Bala, V.; Rajagopal, S.; Kumar, D.P.; Nalli, A.D.; Mahavadi, S.; Sanyal, A.J.; Grider, J.R.; Murthy, K.S. Release of GLP-1 and PYY in response to the activation of G protein-coupled bile acid receptor TGR5 is mediated by Epac/PLC-epsilon pathway and modulated by endogenous H2S. Front. Physiol. 2014, 5, 420. [Google Scholar] [CrossRef]
- Muller, T.D.; Finan, B.; Bloom, S.R.; D’Alessio, D.; Drucker, D.J.; Flatt, P.R.; Fritsche, A.; Gribble, F.; Grill, H.J.; Habener, J.F.; et al. Glucagon-like peptide 1 (GLP-1). Mol. Metab. 2019, 30, 72–130. [Google Scholar] [CrossRef]
- Stožer, A.; Leitgeb, E.P.; Pohorec, V.; Dolenšek, J.; Bombek, L.K.; Gosak, M.; Klemen, M.S. The Role of cAMP in Beta Cell Stimulus–Secretion and Intercellular Coupling. Cells 2021, 10, 1658. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhou, J.; Doyle, M.E.; Egan, J.M. Glucagon-like peptide-1 causes pancreatic duodenal homeobox-1 protein translocation from the cytoplasm to the nucleus of pancreatic beta-cells by a cyclic adenosine monophosphate/protein kinase A-dependent mechanism. Endocrinology 2001, 142, 1820–1827. [Google Scholar] [CrossRef]
- Trabelsi, M.-S.; Daoudi, M.; Prawitt, J.; Ducastel, S.; Touche, V.; Sayin, S.I.; Perino, A.; Brighton, C.A.; Sebti, Y.; Kluza, J.; et al. Farnesoid X receptor inhibits glucagon-like peptide-1 production by enteroendocrine L cells. Nat. Commun. 2015, 6, 7629. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, T.; Jiang, R.; Zhao, A.; Wu, Q.; Kuang, J.; Sun, D.; Ren, Z.; Li, M.; Zhao, M.; et al. Hyocholic acid species improve glucose homeostasis through a distinct TGR5 and FXR signaling mechanism. Cell Metab. 2021, 33, 791–803.e7. [Google Scholar] [CrossRef] [PubMed]
- Chávez-Talavera, O.; Wargny, M.; Pichelin, M.; Descat, A.; Vallez, E.; Kouach, M.; Bigot-Corbel, E.; Joliveau, M.; Goossens, J.-F.; Le May, C.; et al. Bile acids associate with glucose metabolism, but do not predict conversion from impaired fasting glucose to diabetes. Metabolism 2020, 103, 154042. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, T.; Zhao, A.; Ning, Z.; Kuang, J.; Wang, S.; You, Y.; Bao, Y.; Ma, X.; Yu, H.; et al. Hyocholic acid species as novel biomarkers for metabolic disorders. Nat. Commun. 2021, 12, 1487. [Google Scholar] [CrossRef]
- Wang, T.Y.; Liu, M.; Portincasa, P.; Wang, D.Q.-H. New insights into the molecular mechanism of intestinal fatty acid absorption. Eur. J. Clin. Investig. 2013, 43, 1203–1223. [Google Scholar] [CrossRef]
- Wang, Y.; Viscarra, J.; Kim, S.-J.; Sul, H.S. Transcriptional regulation of hepatic lipogenesis. Nat. Rev. Mol. Cell Biol. 2015, 16, 678–689. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Houten, S.M.; Wang, L.; Moschetta, A.; Mangelsdorf, D.J.; Heyman, R.A.; Moore, D.D.; Auwerx, J. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J. Clin. Investig. 2004, 113, 1408–1418. [Google Scholar] [CrossRef]
- Dixon, E.; Nardo, A.; Claudel, T.; Trauner, M. The Role of Lipid Sensing Nuclear Receptors (PPARs and LXR) and Metabolic Lipases in Obesity, Diabetes and NAFLD. Genes 2021, 12, 645. [Google Scholar] [CrossRef]
- Sapiro, J.M.; Mashek, M.T.; Greenberg, A.S.; Mashek, D.G. Hepatic triacylglycerol hydrolysis regulates peroxisome proliferator-activated receptor α activity. J. Lipid Res. 2009, 50, 1621–1629. [Google Scholar] [CrossRef]
- Xu, J.; Li, Y.; Chen, W.-D.; Xu, Y.; Yin, L.; Ge, X.; Jadhav, K.; Adorini, L.; Zhang, Y. Hepatic carboxylesterase 1 is essential for both normal and farnesoid X receptor-controlled lipid homeostasis. Hepatology 2014, 59, 1761–1771. [Google Scholar] [CrossRef]
- Wang, Y.D.; Chen, W.D.; Wang, M.; Yu, D.; Forman, B.M.; Huang, W. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology 2008, 48, 1632–1643. [Google Scholar] [CrossRef] [PubMed]
- Calmus, Y.; Guechot, J.; Podevin, P.; Bonnefis, M.-T.; Giboudeau, J.; Poupon, R. Differential effects of chenodeoxycholic and ursodeoxycholic acids on interleukin 1, interleukin 6 and tumor necrosis factor–α production by monocytes. Hepatology 1992, 16, 719–723. [Google Scholar] [CrossRef]
- Jung, H.; Chen, J.; Hu, X.; Sun, H.; Wu, S.-Y.; Chiang, C.-M.; Kemper, B.; Chen, L.-F.; Kemper, J.K. BRD4 inhibition and FXR activation, individually beneficial in cholestasis, are antagonistic in combination. JCI Insight 2021, 6, e141640. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-H.; Xiao, Z.; Kwon, S.; Sun, X.; Ryerson, D.; Tkac, D.; Ma, P.; Wu, S.-Y.; Chiang, C.-M.; Zhou, E.; et al. A dysregulated acetyl/SUMO switch of FXR promotes hepatic inflammation in obesity. EMBO J. 2015, 34, 184–199. [Google Scholar] [CrossRef] [PubMed]
- Mottis, A.; Mouchiroud, L.; Auwerx, J. Emerging roles of the corepressors NCoR1 and SMRT in homeostasis. Genes Dev. 2013, 27, 819–835. [Google Scholar] [CrossRef]
- Vavassori, P.; Mencarelli, A.; Renga, B.; Distrutti, E.; Fiorucci, S. The Bile Acid Receptor FXR Is a Modulator of Intestinal Innate Immunity. J. Immunol. 2009, 183, 6251–6261. [Google Scholar] [CrossRef]
- Zou, A.; Magee, N.; Deng, F.; Lehn, S.; Zhong, C.; Zhang, Y. Hepatocyte nuclear receptor SHP suppresses inflammation and fibrosis in a mouse model of nonalcoholic steatohepatitis. J. Biol. Chem. 2018, 293, 8656–8671. [Google Scholar] [CrossRef]
- Yuk, J.-M.; Shin, D.-M.; Lee, H.-M.; Kim, J.-J.; Kim, S.-W.; Jin, H.S.; Yang, C.-S.; Park, K.A.; Chanda, D.; Kim, D.-K.; et al. The orphan nuclear receptor SHP acts as a negative regulator in inflammatory signaling triggered by Toll-like receptors. Nat. Immunol. 2011, 12, 742–751. [Google Scholar] [CrossRef]
- Cao, S.; Meng, X.; Li, Y.; Sun, L.; Jiang, L.; Xuan, H.; Chen, X. Bile Acids Elevated in Chronic Periaortitis Could Activate Farnesoid-X-Receptor to Suppress IL-6 Production by Macrophages. Front. Immunol. 2021, 12, 632864. [Google Scholar] [CrossRef]
- Pols, T.W.; Nomura, M.; Harach, T.; Sasso, G.L.; Oosterveer, M.H.; Thomas, C.; Rizzo, G.; Gioiello, A.; Adorini, L.; Pellicciari, R.; et al. TGR5 Activation Inhibits Atherosclerosis by Reducing Macrophage Inflammation and Lipid Loading. Cell Metab. 2011, 14, 747–757. [Google Scholar] [CrossRef]
- Boutilier, A.; Elsawa, S. Macrophage Polarization States in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 6995. [Google Scholar] [CrossRef]
- Shi, Y.; Su, W.; Zhang, L.; Shi, C.; Zhou, J.; Wang, P.; Wang, H.; Shi, X.; Wei, S.; Wang, Q.; et al. TGR5 Regulates Macrophage Inflammation in Nonalcoholic Steatohepatitis by Modulating NLRP3 Inflammasome Activation. Front. Immunol. 2021, 11, 609060. [Google Scholar] [CrossRef]
- Biagioli, M.; Carino, A.; Cipriani, S.; Francisci, D.; Marchiano, S.; Scarpelli, P.; Sorcini, D.; Zampella, A.; Fiorucci, S. The Bile Acid Receptor GPBAR1 Regulates the M1/M2 Phenotype of Intestinal Macrophages and Activation of GPBAR1 Rescues Mice from Murine Colitis. J. Immunol. 2017, 199, 718–733. [Google Scholar] [CrossRef]
- Hu, J.; Wang, C.; Huang, X.; Yi, S.; Pan, S.; Zhang, Y.; Yuan, G.; Cao, Q.; Ye, X.; Li, H. Gut microbiota-mediated secondary bile acids regulate dendritic cells to attenuate autoimmune uveitis through TGR5 signaling. Cell Rep. 2021, 36, 109726. [Google Scholar] [CrossRef] [PubMed]
- Hang, S.; Paik, D.; Yao, L.; Kim, E.; Trinath, J.; Lu, J.; Ha, S.; Nelson, B.N.; Kelly, S.P.; Wu, L.; et al. Bile acid metabolites control TH17 and Treg cell differentiation. Nature 2019, 576, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, X.; Zhu, Y.; Liu, X.; Gu, Y.; Dai, X.; Li, B. Transcriptional and posttranslational regulation of Th17/Treg balance in health and disease. Eur. J. Immunol. 2021, 51, 2137–2150. [Google Scholar] [CrossRef] [PubMed]
- Mano, N.; Goto, T.; Uchida, M.; Nishimura, K.; Ando, M.; Kobayashi, N.; Goto, J. Presence of protein-bound unconjugated bile acids in the cytoplasmic fraction of rat brain. J. Lipid Res. 2004, 45, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Elliott, C.T.; McGuinness, B.; Passmore, P.; Kehoe, P.G.; Hölscher, C.; McClean, P.L.; Graham, S.F.; Green, B.D. Metabolomic Profiling of Bile Acids in Clinical and Experimental Samples of Alzheimer’s Disease. Metabolites 2017, 7, 28. [Google Scholar] [CrossRef]
- Monteiro-Cardoso, V.F.; Corlianò, M.; Singaraja, R.R. Bile Acids: A Communication Channel in the Gut-Brain Axis. Neuromol. Med. 2021, 23, 99–117. [Google Scholar] [CrossRef]
- Wang, D.Q.-H.; Tazuma, S.; Cohen, D.E.; Carey, M.C. Feeding natural hydrophilic bile acids inhibits intestinal cholesterol absorption: Studies in the gallstone-susceptible mouse. Am. J. Physiol. Liver Physiol. 2003, 285, G494–G502. [Google Scholar] [CrossRef]
- Higashi, T.; Watanabe, S.; Tomaru, K.; Yamazaki, W.; Yoshizawa, K.; Ogawa, S.; Nagao, H.; Minato, K.; Maekawa, M.; Mano, N. Unconjugated bile acids in rat brain: Analytical method based on LC/ESI-MS/MS with chemical derivatization and estimation of their origin by comparison to serum levels. Steroids 2017, 125, 107–113. [Google Scholar] [CrossRef]
- Kamp, F.; Hamilton, J. Movement of fatty acids, fatty acid analogs, and bile acids across phospholipid bilayers. Biochemistry 1993, 32, 11074–11085. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, A.; Di Sario, A.; Marucci, L.; Svegliati-Baroni, G.; Schteingart, C.D.; Ton-Nu, H.T.; Hofmann, A.F. Carrier-mediated transport of conjugated bile acids across the basolateral membrane of biliary epithelial cells. Am. J. Physiol. Content 1997, 272, G1416–G1424. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, M.; Kullak-Ublick, G.; Hagenbuch, B.; Meier, P. Transport of bile acids in hepatic and non-hepatic tissues. J. Exp. Biol. 2001, 204, 1673–1686. [Google Scholar] [CrossRef]
- Rao, R. Occludin Phosphorylation in Regulation of Epithelial Tight Junctions. Ann. N. Y. Acad. Sci. 2009, 1165, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Quinn, M.; McMillin, M.; Galindo, C.; Frampton, G.; Pae, H.Y.; DeMorrow, S. Bile acids permeabilize the blood brain barrier after bile duct ligation in rats via Rac1-dependent mechanisms. Dig. Liver Dis. 2014, 46, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Kiriyama, Y.; Nochi, H. D-Amino Acids in the Nervous and Endocrine Systems. Scientifica 2016, 2016, 6494621. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A. NMDA Receptor Function During Senescence: Implication on Cognitive Performance. Front. Neurosci. 2015, 9, 473. [Google Scholar] [CrossRef]
- Baez, M.V.; Cercato, M.C.; Jerusalinsky, D.A. NMDA Receptor Subunits Change after Synaptic Plasticity Induction and Learning and Memory Acquisition. Neural Plast. 2018, 2018, 5093048. [Google Scholar] [CrossRef]
- Çiçek, S.S. Structure-Dependent Activity of Natural GABA(A) Receptor Modulators. Molecules 2018, 23, 1512. [Google Scholar] [CrossRef] [PubMed]
- Evenseth, L.S.M.; Gabrielsen, M.; Sylte, I. The GABAB Receptor—Structure, Ligand Binding and Drug Development. Molecules 2020, 25, 3093. [Google Scholar] [CrossRef]
- Schubring, S.R.; Fleischer, W.; Lin, J.S.; Haas, H.L.; Sergeeva, O.A. The bile steroid chenodeoxycholate is a potent antagonist at NMDA and GABA(A) receptors. Neurosci. Lett. 2012, 506, 322–326. [Google Scholar] [CrossRef]
- Yanovsky, Y.; Schubring, S.R.; Yao, Q.; Zhao, Y.; Li, S.; May, A.; Haas, H.L.; Lin, J.-S.; Sergeeva, O.A. Waking Action of Ursodeoxycholic Acid (UDCA) Involves Histamine and GABAA Receptor Block. PLoS ONE 2012, 7, e42512. [Google Scholar] [CrossRef]
- Poulin, B.; Butcher, A.; McWilliams, P.; Bourgognon, J.-M.; Pawlak, R.; Kong, K.C.; Bottrill, A.; Mistry, S.; Wess, J.; Rosethorne, E.M.; et al. The M3-muscarinic receptor regulates learning and memory in a receptor phosphorylation/arrestin-dependent manner. Proc. Natl. Acad. Sci. USA 2010, 107, 9440–9445. [Google Scholar] [CrossRef] [PubMed]
- Seeger, T.; Fedorova, I.; Zheng, F.; Miyakawa, T.; Koustova, E.; Gomeza, J.; Basile, A.S.; Alzheimer, C.; Wess, J. M2 Muscarinic Acetylcholine Receptor Knock-Out Mice Show Deficits in Behavioral Flexibility, Working Memory, and Hippocampal Plasticity. J. Neurosci. 2004, 24, 10117–10127. [Google Scholar] [CrossRef]
- Wess, J.; Eglen, R.M.; Gautam, D. Muscarinic acetylcholine receptors: Mutant mice provide new insights for drug development. Nat. Rev. Drug Discov. 2007, 6, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Soares, R.; Ribeiro, F.F.; Xapelli, S.; Genebra, T.; Ribeiro, M.F.; Sebastião, A.M.; Rodrigues, C.M.P.; Solá, S. Tauroursodeoxycholic Acid Enhances Mitochondrial Biogenesis, Neural Stem Cell Pool, and Early Neurogenesis in Adult Rats. Mol. Neurobiol. 2017, 55, 3725–3738. [Google Scholar] [CrossRef] [PubMed]
- Zangerolamo, L.; Vettorazzi, J.F.; Rosa, L.R.; Carneiro, E.M.; Barbosa, H.C. The bile acid TUDCA and neurodegenerative disorders: An overview. Life Sci. 2021, 272, 119252. [Google Scholar] [CrossRef]
- Cao, C.; Kang, H.J.; Singh, I.; Chen, H.; Zhang, C.; Ye, W.; Hayes, B.W.; Liu, J.; Gumpper, R.H.; Bender, B.J.; et al. Structure, function and pharmacology of human itch GPCRs. Nature 2021, 600, 170–175. [Google Scholar] [CrossRef]
- Lembo, P.M.; Grazzini, E.; Groblewski, T.; O’Donnell, D.; Roy, M.-O.; Zhang, J.; Hoffert, C.; Cao, J.; Schmidt, R.; Pelletier, M.; et al. Proenkephalin A gene products activate a new family of sensory neuron–specific GPCRs. Nat. Neurosci. 2002, 5, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-Y.; Zhou, D.-D.; Gan, R.-Y.; Huang, S.-Y.; Zhao, C.-N.; Shang, A.; Xu, X.-Y.; Li, H.-B. Effects and Mechanisms of Probiotics, Prebiotics, Synbiotics, and Postbiotics on Metabolic Diseases Targeting Gut Microbiota: A Narrative Review. Nutrients 2021, 13, 3211. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).