
Recovery of Mycobacteria from Heavily Contaminated Environmental Matrices


Abstract
1. Introduction
1.1. Genus Mycobacterium
1.2. Direct Recovery of Mycobacteria—Methods in Microbiology
1.3. Biomolecular Methods
2. Materials and Methods
2.1. Samples’ Collection
2.2. Pre-Treatment Procedure
2.3. Decontamination with NaOH and TDAB
2.4. Isolates Identification
2.5. qPCR Method
2.6. Viability Testing by PMA Treated DNA
2.7. Statistical Analysis
3. Results
3.1. Culture Results
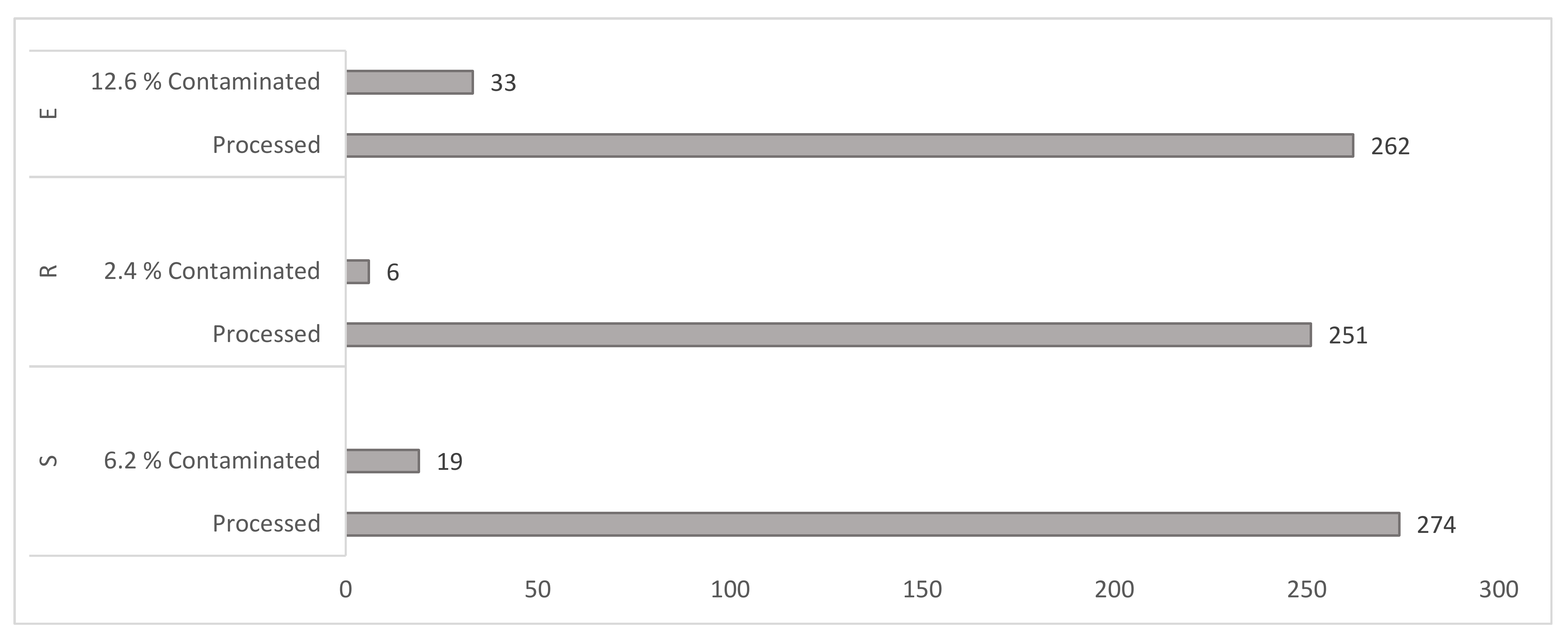
3.1.1. Culture Contamination Rates
3.1.2. Yield of Mycobacteria by Cultivation Method
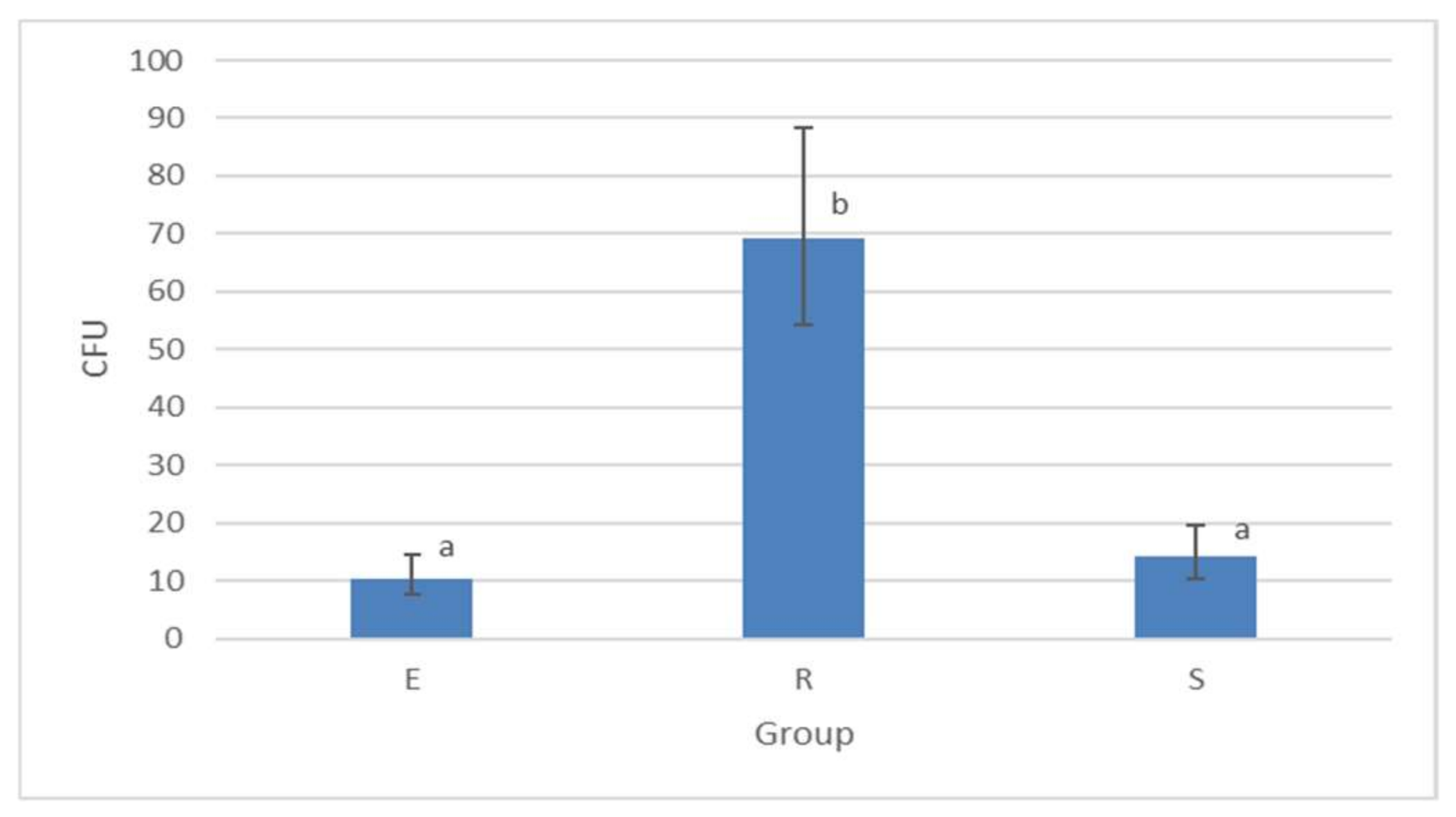
3.1.3. Colony-Forming Unit (CFU) Counts Detected by Culture Method
3.2. qPCR Results
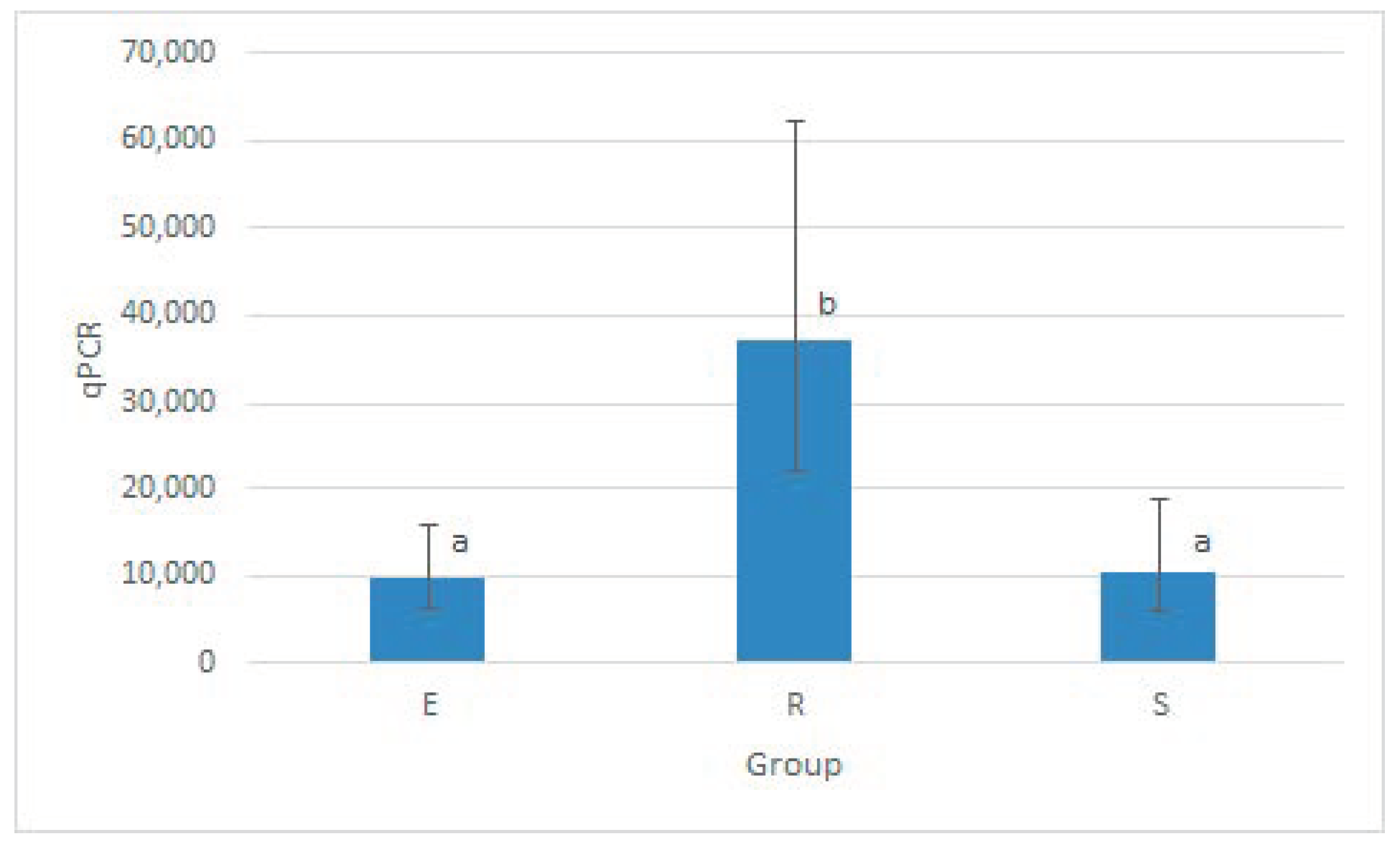
3.2.1. Mycobacteria Quantification in Different Matrices by qPCR
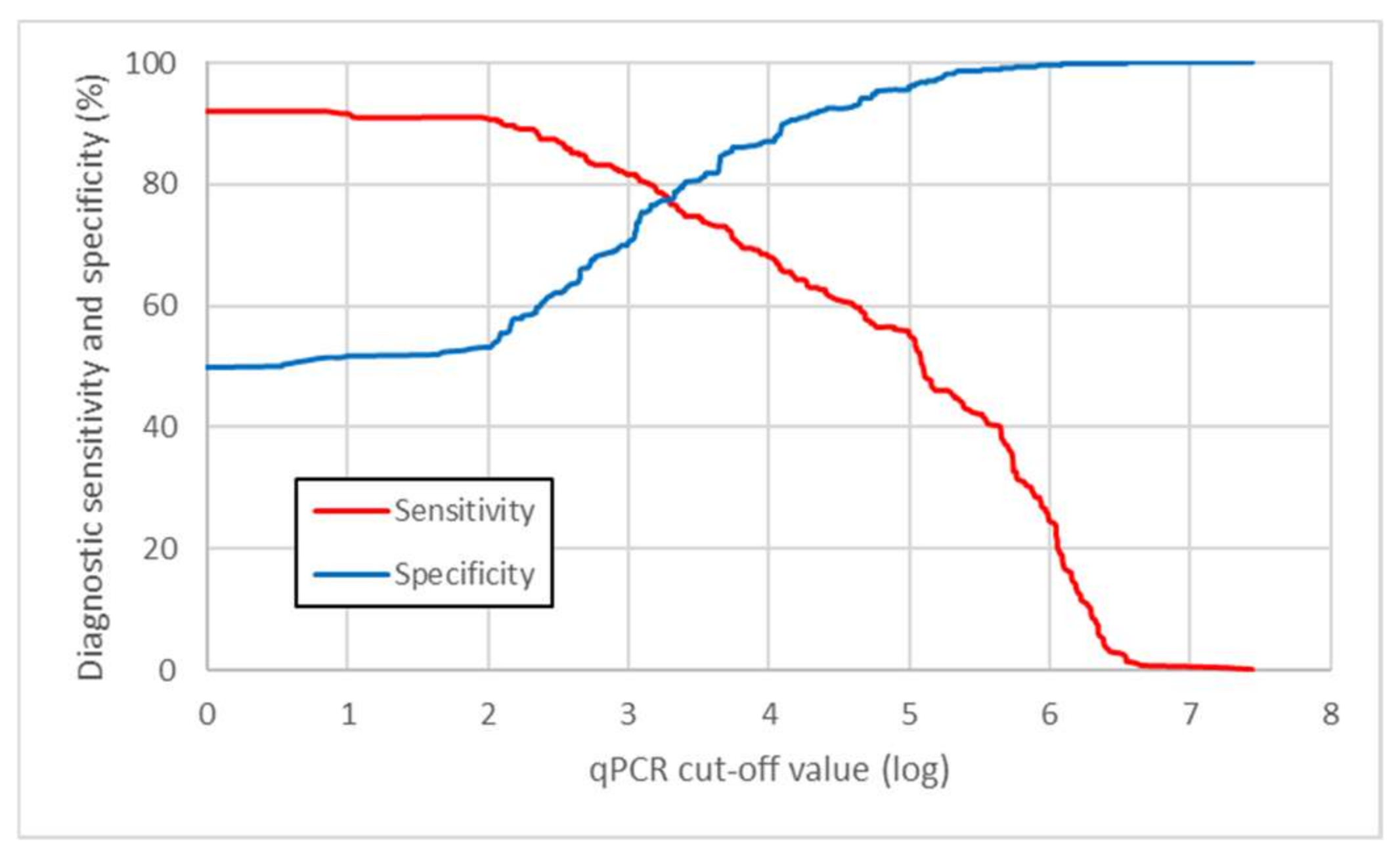
3.2.2. Parameters of the qPCR Method
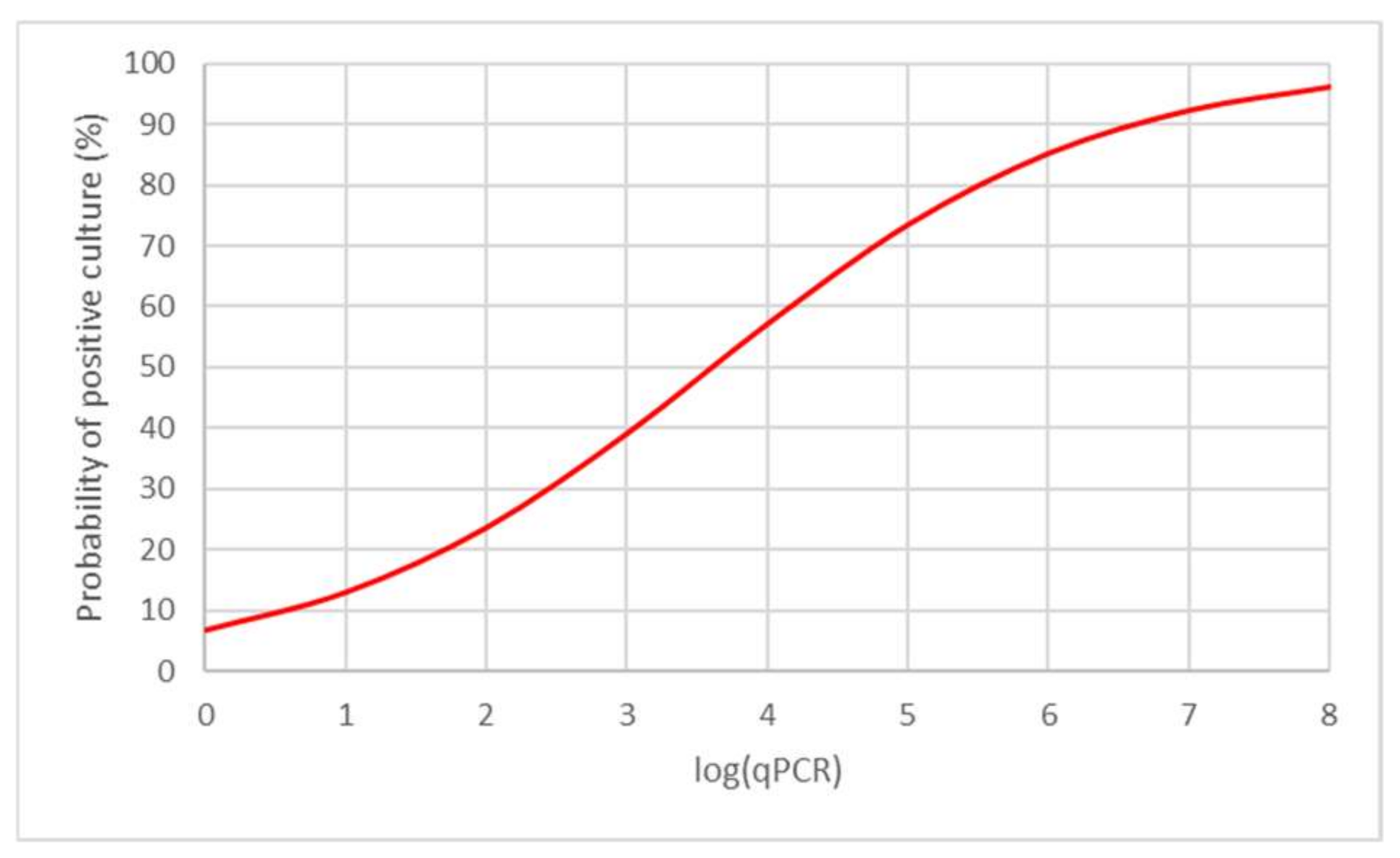
3.2.3. Model of Predictive Values of qPCR
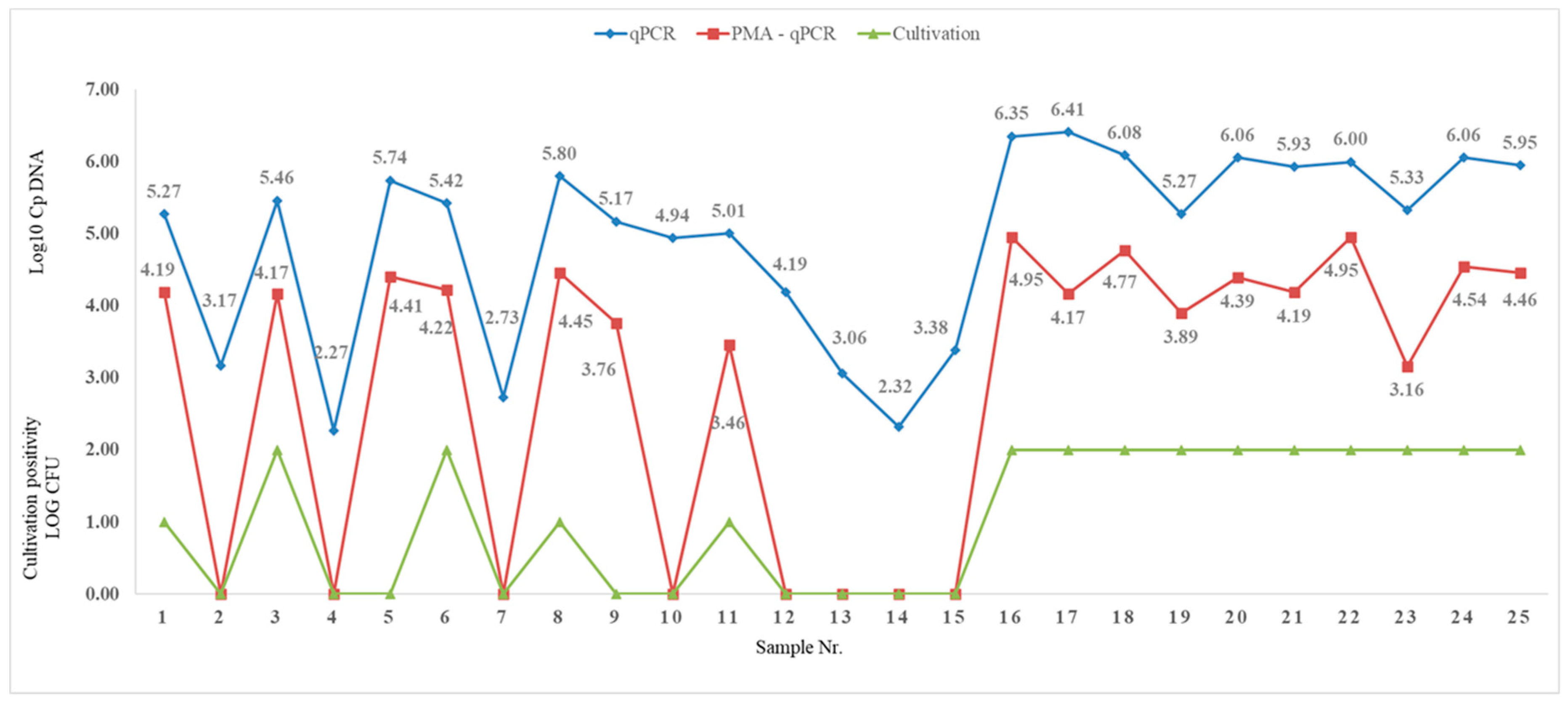
3.2.4. Evaluation of the Used PMA-qPCR Method
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- LPSN: List of Prokaryotic Names with Standing in Nomenclature. Available online: https://lpsn.dsmz.de/ (accessed on 6 October 2021).
- Forbes, B.A. Mycobacterial Taxonomy. J. Clin. Microbiol. 2017, 55, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Falkinham, J.O., 3rd. Surrounded by mycobacteria: Nontuberculous mycobacteria in the human environment. J. Appl. Microbiol. 2009, 107, 356–367. [Google Scholar] [CrossRef]
- Griffith, D.E.; Aksamit, T.; Brown-Elliott, B.A.; Catanzaro, A.; Daley, C.; Gordin, F.; Holland, S.M.; Horsburgh, R.; Huitt, G.; Iademarco, M.F.; et al. ATS Mycobacterial Diseases Subcommittee; American Thoracic Society; Infectious Disease Society of America. An official ATS/IDSA statement: Diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am. J. Respir. Crit. Care. Med. 2007, 175, 367–416. [Google Scholar] [CrossRef] [PubMed]
- Meissner, P.S.; Falkinham, J.O., 3rd. Plasmid DNA profiles as epidemiological markers for clinical and environmental isolates of Mycobacterium avium, Mycobacterium intracellulare and Mycobacterium scrofulaceum. J. Infect. Dis. 1986, 153, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Zewinger, S.; Meier, C.M.; Fliser, D.; Klingele, M. Mycobacterium fortuitum peritonitis in peritoneal dialysis and its effects on the peritoneum. Clin. Nephrol. 2014, 82, 341–346. [Google Scholar] [CrossRef]
- Nessar, R.; Cambau, E.; Reyrat, J.M.; Murray, A.; Gicquel, B. Mycobacterium abscessus: A new antibiotic nightmare. J. Antimicrob. Chemother. 2012, 67, 810–818. [Google Scholar] [CrossRef]
- DaCosta, A.; Jordan, C.L.; Giddings, O.; Lin, F.C.; Gilligan, P.; Esther, C.R., Jr. Outcomes associated with antibiotic regimens for treatment of Mycobacterium abscessus in cystic fibrosis patients. J. Cyst. Fibros. 2017, 16, 483–487. [Google Scholar] [CrossRef]
- Zimmermann, P.; Finn, A.; Curtis, N. Does BCG vaccination protect against nontuberculous mycobacterial infection? A systematic review and meta-analysis. J. Infect. Dis. 2018, 218, 679–687. [Google Scholar] [CrossRef]
- Willemse, S.H.; Oomens, M.A.E.M.; De Lange, J.; Karssemakers, L.H.E. Diagnosing nontuberculous mycobacterial cervicofacial lymphadenitis in children: A systematic review. Int. J. Pediatr. Otorhinolaryngol. 2018, 112, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Delghandi, M.R.; El-Matbouli, M.; Menanteau-Ledouble, S. Mycobacteriosis and infections with non-tuberculous mycobacteria in aquatic organisms: A review. Microorganisms 2020, 8, 1368. [Google Scholar] [CrossRef] [PubMed]
- Mugetti, D.; Varello, K.; Pastorino, P.; Tomasoni, M.; Menconi, V.; Bozzetta, E.; Dondo, A.; Prearo, M. Investigation of potential reservoirs of non-tuberculous mycobacteria in a European Sea Bass (Dicentrarchus labrax). Farm. Pathogens 2021, 10, 1014. [Google Scholar] [CrossRef]
- Petroff, S.A. A new and rapid method for the isolation and cultivation of tubercle bacilli directly from the sputum and feces. J. Exp. Med. 1915, 21, 38–42. [Google Scholar] [CrossRef]
- Kubica, G.P.; Dye, W.E.; Cohn, M.L.; Middlebrook, G. Sputum digestion and decontamination with N-acetyl-L-cysteine-sodium hydroxide for culture of mycobacteria. Am. Rev. Respir. Dis. 1963, 87, 775–779. [Google Scholar]
- Whittier, S.; Hopfer, R.L.; Knowles, M.R.; Gilligan, P.H. Improved recovery of mycobacteria from respiratory secretions of patients with cystic fibrosis. J. Clin. Microbiol. 1993, 31, 861–864. [Google Scholar] [CrossRef] [PubMed]
- Wood, R.C.; Andama, A.; Hermansky, G.; Burkot, S.; Asege, L.; Job, M.; Katumba, D.; Nakaye, M.; Mwebe, S.Z.; Mulondo, J.; et al. Characterization of oral swab samples for diagnosis of pulmonary tuberculosis. PLoS ONE 2021, 16, e0251422. [Google Scholar] [CrossRef] [PubMed]
- Pena, J.A.; Ferraro, M.J.; Hoffman, C.G.; Branda, J.A. Growth detection failures by the nonradiometric Bactec MGIT 960 mycobacterial culture system. J. Clin. Microbiol. 2012, 50, 2092–2095. [Google Scholar] [CrossRef] [PubMed]
- Piersimoni, C.; Nista, D.; Bornigia, S.; Gherardi, G. Unreliable detection of Mycobacterium xenopi by the nonradiometric Bactec MGIT 960 culture system. J. Clin. Microbiol. 2009, 47, 804–806. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stephenson, D.; Perry, A.; Appleby, M.R.; Lee, D.; Davison, J.; Johnston, A.; Jones, A.L.; Nelson, A.; Bourke, S.J.; Thomas, M.F.; et al. An evaluation of methods for the isolation of nontuberculous mycobacteria from patients with cystic fibrosis, bronchiectasis and patients assessed for lung transplantation. BMC Pulm. Med. 2019, 19, 19. [Google Scholar] [CrossRef] [PubMed]
- Radomski, N.; Cambau, E.; Moulin, L.; Haenn, S.; Moilleron, R.; Lucas, F.S. Comparison of culture methods for isolation of nontuberculous mycobacteria from surface waters. Appl. Environ. Microbiol. 2010, 76, 3514–3520. [Google Scholar] [CrossRef]
- Pileggi, S.M.; Jordan, H.; Clennon, J.A.; Whitney, E.; Benbow, M.E.; Merritt, R.; McIntosh, M.; Kimbirauskas, R.; Small, P.; Boakye, D.; et al. Landscape and environmental influences on Mycobacterium ulcerans distribution among aquatic sites in Ghana. PLoS ONE 2017, 12, e0176375. [Google Scholar] [CrossRef]
- Parashar, D.; Das, R.; Chauhan, D.S.; Sharma, V.D.; Lavania, M.; Yadav, V.S.; Chauhan, S.V.; Katoch, V.M. Identification of environmental mycobacteria isolated from Agra, north India by conventional & molecular approaches. Ind. J. Med. Res. 2009, 129, 424–431. [Google Scholar]
- Cortesia, C.; Lopez, G.J.; de Waard, J.H.; Takiff, H.E. The use of quaternary ammonium disinfectants selects for persisters at high frequency from some species of non-tuberculous mycobacteria and may be associated with outbreaks of soft tissue infections. J. Antimicrob. Chemother. 2010, 65, 2574–2581. [Google Scholar] [CrossRef]
- Gerba, C.P. Quaternary ammonium biocides: Efficacy in application. Appl. Envir. Microbiol. 2015, 81, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Vaerewijck, M.J.M.; Huys, G.; Palomino, J.C.; Swings, J.; Portaels, F. Recovery of non-tuberculous mycobacteria from water is influenced by phenotypic characteristics and decontamination methods. FEMS Microbiol. Rev. 2005, 29, 911–934. [Google Scholar] [CrossRef]
- Fernandes, H.M.Z.; Conceicao, E.C.; Gomes, K.M.; da Silva, M.G.; Dias, R.C.S.; Duarte, R.S. Comparison of culture methods for isolation of nontuberculous mycobacteria from surface waters. Curr. Microbiol. 2020, 77, 621–631. [Google Scholar] [CrossRef]
- Rasanen, N.H.J.; Rintala, H.; Miettinen, I.T.; Torvinen, E. Comparison of culture and qPCR methods in detection of mycobacteria from drinking waters. Can. J. Microbiol. 2013, 59, 208–286. [Google Scholar] [CrossRef] [PubMed]
- Mohajeri, P.; Yazdani, L.; Shahraki, A.H.; Alvandi, A.; Atashi, S.; Farahani, A.; Almasi, A.; Rezaei, M. Verification of frequency in species of nontuberculous mycobacteria in Kermanshah drinking water supplies using the PCR-sequencing method. Microb. Drug Resist. 2017, 23, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Oriani, A.S.; Marfil, M.J.; Zumarraga, M.J.; Baldini, M.D. Prevalence and species diversity of nontuberculous mycobacteria in drinking water supply system of Bahía Blanca City, Argentina. Int. J. Mycobacteriol. 2019, 8, 138–145. [Google Scholar]
- Kaelin, M.B.; Kuster, S.P.; Hasse, B.; Schulthess, B.; Imkamp, F.; Halbe, M.; Sander, P.; Sax, H.; Schreiber, P.W. Diversity of nontuberculous mycobacteria in heater-cooler devices: Results from prospective surveillance. J. Hosp. Infect. 2020, 105, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Akkaya, O.; Kurtoglu, M.G. Comparison of conventional and molecular methods used for diagnosis of Mycobacterium tuberculosis in clinical samples. Clin. Lab. 2019, 65. [Google Scholar] [CrossRef]
- Kralik, P.; Slana, I.; Kralova, A.; Babak, V.; Whitlock, R.H.; Pavlik, I. Development of a predictive model for detection of Mycobacterium avium subsp. paratuberculosis in faeces by quantitative real time PCR. Vet. Microbiol. 2011, 149, 133–138. [Google Scholar] [CrossRef]
- Klanicova, B.; Vondruskova, H.; Slana, I.; Kaevska, M.; Pavlik, I. Real-time quantitative PCR detection of Mycobacterium avium subspecies in meat products. J. Food Prot. 2011, 74, 636–640. [Google Scholar] [CrossRef]
- Slany, M.; Jezek, P.; Fiserova, V.; Bodnarova, M.; Stork, J.; Havelkova, M.; Kalat, F.; Pavlik, I. Mycobacterium marinum infections in humans and tracing of its possible environmental sources. Can. J. Microbiol. 2012, 58, 39–44. [Google Scholar] [CrossRef]
- Klanicova, B.; Lorencova, A.; Makovcova, J.; Vlkova, H.; Kralik, P.; Pavlik, I.; Slany, M. Survival of three Mycobacterium avium subsp. hominissuis isolates in fish products after hot smoking and frying. Int. J. Food Sci. Technol. 2013, 48, 533–538. [Google Scholar] [CrossRef]
- Moravkova, M.; Lamka, J.; Slany, M.; Pavlik, I. Genetic IS901 diversity among Mycobacterium avium subsp. avium isolates from four pheasant flocks. J. Vet. Sci. 2013, 14, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Slana, I.; Kralik, P.; Kralova, A.; Pavlik, I. On-farm spread of Mycobacterium avium subsp. paratuberculosis in raw milk studied by IS900 and F57 competitive real time quantitative PCR and culture examination. Int. J. Food Microbiol. 2008, 128, 250–257. [Google Scholar] [CrossRef]
- Slana, I.; Liapi, M.; Moravkova, M.; Kralova, A.; Pavlik, I. Mycobacterium avium subsp. paratuberculosis in cow bulk tank milk in Cyprus detected by culture and quantitative IS900 and F57 real–time PCR. Prev. Vet. Med. 2009, 89, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Slana, I.; Kaevska, M.; Kralik, P.; Horvathova, A.; Pavlik, I. Distribution of Mycobacterium avium subsp. avium and M. a. hominissuis in artificially infected pigs studied by culture and IS901 and IS1245 quantitative real time PCR. Vet. Microbiol. 2010, 144, 437–443. [Google Scholar] [CrossRef]
- Hruska, K.; Slana, I.; Kralik, P.; Pavlik, I. Mycobacterium avium subsp. paratuberculosis in powdered infant milk: F57 competitive real time PCR. Vet. Med. Czech. 2011, 56, 226–230. [Google Scholar] [CrossRef]
- Lorencova, A.; Klanicova, B.; Makovcova, J.; Slana, I.; Vojkovska, H.; Babak, V.; Pavlik, I.; Slany, M. Nontuberculous mycobacteria in freshwater fish and fish products intended for human consumption. Foodborne Pathog. Dis. 2013, 10, 573–576. [Google Scholar] [CrossRef] [PubMed]
- Kazda, J.; Pavlik, I.; Falkinham, J.; Hruska, K. The Ecology of Mycobacteria: Impact on Animal’s and Human’s Health, 1st ed.; Springer: Dordrecht, The Netherlands, 2009; p. 520. [Google Scholar]
- Nkuipou-Kenfack, E.; Engel, H.; Fakih, S.; Nocker, A. Improving efficiency of viability-PCR for selective detection of live cells. J. Microbiol. Meth. 2013, 93, 20–24. [Google Scholar] [CrossRef]
- Elizaquivel, P.; Aznar, R.; Sánchez, G. Recent developments in the use of viability dyes and quantitative PCR in the food microbiology field. J. Appl. Microbiol. 2014, 116, 1–13. [Google Scholar] [CrossRef]
- Codony, F.; Dinh-Thanh, M.; Agusti, G. Key factors for removing bias in viability PCR-based methods: A review. Curr. Microbiol. 2020, 77, 682–687. [Google Scholar] [CrossRef]
- Cao, Y.; Zhou, D.; Li, R.; Yu, Y.; Xiao, X.; Zhou, A.; Liu, D.; Li, X. Molecular monitoring of disinfection efficacy of E. coli O157: H7 in bottled purified drinking water by quantitative PCR with a novel dye. J. Food Process. Preserv. 2019, 43, e13875. [Google Scholar] [CrossRef]
- Nocker, A.; Cheung, C.Y.; Camper, A.K. Comparison of propidium monoazide with ethidium monoazide for differentiation of live vs. dead bacteria by selective removal of DNA from dead cells. J. Microbiol. Meth. 2006, 67, 310–320. [Google Scholar] [CrossRef]
- Kralik, P.; Nocker, A.; Pavlik, I. Mycobacterium avium subsp. paratuberculosis viability determination using f57 quantitative PCR in combination with propidium monoazide treatment. Int. J. Food Microbiol. 2010, 141, S80–S86. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Lee, S.M.; Park, B.K.; Kim, S.S.; Yi, J.; Kim, H.H.; Lee, E.Y.; Chang, C.L. Evaluation of propidium monoazide real-time PCR for early detection of viable Mycobacterium tuberculosis in clinical respiratory specimens. Ann. Lab. Med. 2014, 34, 203–209. [Google Scholar] [CrossRef]
- Kayigire, X.A.; Friedrich, S.O.; Karinja, M.N.; van der Merwe, L.; Martinson, N.A.; Diacon, A.H. Propidium monoazide and Xpert MTB/RIF to quantify Mycobacterium tuberculosis cells. Tuberculosis 2016, 101, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Dorn-In, S.; Gareis, M.; Schwaiger, K. Differentiation of live and dead Mycobacterium tuberculosis complex in meat samples using PMA qPCR. Food Microbiol. 2019, 84, 103275. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Zheng, H.; Chu, P.; Han, S.; Yang, H.; Wang, Z.; Shi, J.; Yang, Z. Direct detection from clinical sputum samples to differentiate live and dead Mycobacterium tuberculosis. J. Clin. Lab. Anal. 2019, 33, e22716. [Google Scholar] [CrossRef]
- Ditommaso, S.; Giacomuzzi, M.; Memoli, G.; Cavallo, R.; Curtoni, A.; Avolio, M.; Silvestre, C.; Zotti, C.M. Reduction of turnaround time for non-tuberculous mycobacteria detection in heater-cooler units by propidium monoazide-real-time polymerase chain reaction. J. Hosp. Infect. 2020, 104, 365–373. [Google Scholar] [CrossRef]
- Nossa, C.W.; Oberdorf, W.E.; Yang, L.; Aas, J.A.; Paster, B.J.; Desantis, T.Z.; Brodie, E.L.; Malamud, D.; Poles, M.A.; Pei, Z. Design of 16S rRNA gene primers for 454 pyrosequencing of the human foregut microbiome. World J. Gastroenterol. 2010, 16, 4135–4144. [Google Scholar] [CrossRef] [PubMed]
- Telenti, A.; Marchesi, F.; Balz, M.; Bally, F.; Bottger, E.C.; Bodmer, T. Rapid identification of mycobacteria to the species level by polymerase chain reaction and restriction enzyme analysis. J. Clin. Microbiol. 1993, 31, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Turankar, R.P.; Singh, V.; Gupta, H.; Pathak, V.K.; Ahuja, M.; Singh, I.; Lavania, M.; Dinda, A.K.; Sengupta, U. Association of non-tuberculous mycobacteria with Mycobacterium leprae in environment of leprosy endemic regions in India. Infect. Genet. Evol. 2019, 72, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Casini, B.; Tuvo, B.; Totaro, M.; Baggiani, A.; Privitera, G. Detection and decontamination of Mycobacterium chimaera and other non-tuberculosis mycobacteria in heater-cooler devices used in cardiopulmonary bypass: A Manufacturer and National Guidelines Summary, and a potential resolution to the problem requiring further investigation. Perfusion 2020, 35, 190–196. [Google Scholar] [PubMed]
- Palomino, J.C.; Portaels, F. Effects of decontamination methods and culture conditions on viability of Mycobacterium ulcerans in the BACTEC system. J. Clin. Microbiol. 1998, 36, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Portaels, F.; De Muynec, A.; Sylla, M.P. Selective isolation of mycobacteria from soil: A statistical analysis approach. J. Gen. Microbiol. 1988, 134, 849–855. [Google Scholar] [CrossRef]
- Allen, B.W. Comparison of three methods for decontamination of faeces for isolation of Mycobacterium tuberculosis. Tubercle 1991, 72, 214–217. [Google Scholar] [CrossRef]
- Sattar, A.; Zakaria, Z.; Abu, J.; Aziz, S.A.; Gabriel, R.P. Evaluation of six decontamination procedures for isolation of Mycobacterium avium complex from avian feces. PLoS ONE 2018, 13, e0202034. [Google Scholar] [CrossRef]
- Neumann, M.; Schulze-Robbecke, R.; Hagenau, C.; Behringer, K. Comparison of methods for isolation of mycobacteria from water. Appl. Environ. Microbiol. 1997, 63, 547–552. [Google Scholar] [CrossRef]
- Iivanainen, E.; Martikainen, P.J.; Vaananen, P.; Katila, M.L. Environmental factors affecting the occurrence of mycobacteria in brook sediments. J. Appl. Microbiol. 1999, 86, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Falkinham, J.O., 3rd. Current epidemiologic trends of the nontuberculous mycobacteria (NTM). Curr. Environ. Health Rep. 2016, 3, 161–167. [Google Scholar] [CrossRef]
- Honda, J.R.; Hess, T.; Carlson, R.; Kandasamy, P.; Nieto Ramirez, L.M.; Norton, G.J.; Virdi, R.; Islam, M.N.; Mehaffy, C.; Hasan, N.A.; et al. Nontuberculous mycobacteria show differential infectivity and use phospholipids to antagonize LL-37. Am. J. Respir. Cell. Mol. Biol. 2020, 62, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, D.; Perry, A.; Nelson, A.; Robb, A.E.; Thomas, M.F.; Bourke, S.J.; Perry, J.D.; Jones, A.L. Decontamination strategies used for AFB culture significantly reduce the viability of Mycobacterium abscessus complex in sputum samples from patients with cystic fibrosis. Microorganisms 2021, 9, 1597. [Google Scholar] [CrossRef]
- Alexander, K.J.; Furlong, J.L.; Baron, J.L.; Rihs, J.D.; Stephenson, D.; Perry, J.D.; Stout, J.E. Evaluation of a new culture medium for isolation of nontuberculous mycobacteria from environmental water samples. PLoS ONE 2021, 16, e0247166. [Google Scholar] [CrossRef]










{kind=link}
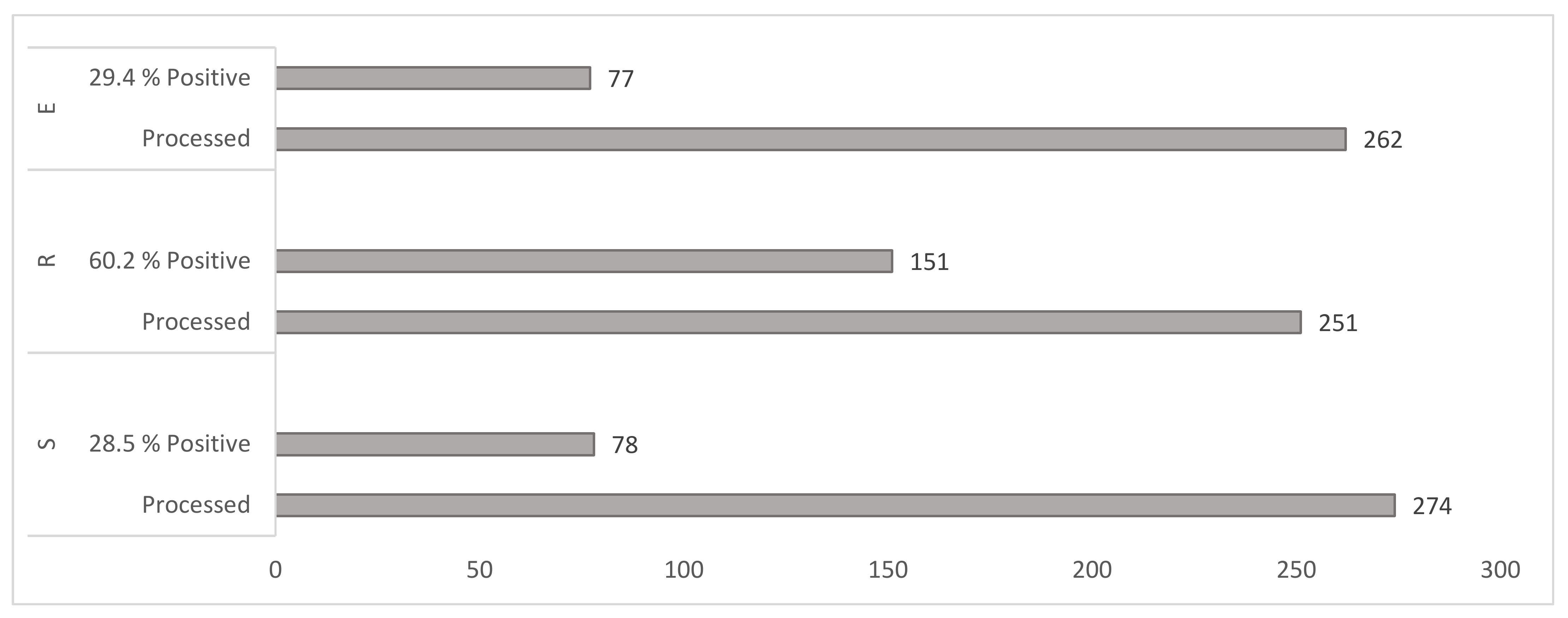{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Growth Characteristics | No. of Isolates in Cluster | Total No. of Isolates | ||
|---|---|---|---|---|---|
| S | R | E | |||
| M. algericum | RG, 25–42 °C (opt. 37 °C) | 0 | 0 | 1 | 1 |
| M. arupense | RG, 25–37 °C (opt. 30 °C) | 4 | 0 | 2 | 6 |
| M. aurum | RG, 25–37 °C (opt. 30 °C) | 2 | 0 | 0 | 2 |
| M. avium ssp. hominissuis | SG, 25–42 °C (opt. 37 °C) | 20 | 103 | 24 | 155 |
| M. bohemicum | SG, 25–42 °C (opt. 37 °C) | 1 | 0 | 0 | 1 |
| M. duvalii | RG, 25–37 °C (opt. 30 °C) | 1 | 0 | 0 | 1 |
| M. europaeum | SG, 25–42 °C (opt. 37 °C) | 0 | 1 | 0 | 1 |
| M. fallax | RG, 25–37 °C (opt. 30 °C) | 1 | 0 | 0 | 1 |
| M. flavescens | SG, 25–37 °C (opt. 30 °C) | 2 | 0 | 0 | 2 |
| M. fortuitum | RG, 25–42 °C (opt. 37 °C) | 13 | 9 | 17 | 39 |
| M. goodii | RG, 25–42 °C (opt. 37 °C) | 0 | 2 | 0 | 2 |
| M. gordonae | SG, 25–42 °C (opt. 37 °C) | 5 | 1 | 1 | 7 |
| M. hassiacum | RG, 30–45 °C (opt. 42 °C) | 1 | 0 | 0 | 1 |
| M. heckeshornense | SG, 37–45 °C (opt. 40 °C) | 1 | 0 | 0 | 1 |
| M. hiberniae | SG, 20–37 °C (opt. 30 °C) | 0 | 0 | 5 | 5 |
| M. chelonae | RG, 25–42 °C (opt. 30 °C) | 0 | 0 | 1 | 1 |
| M. interjectum | SG, 20–37 °C (opt. 30 °C) | 0 | 1 | 0 | 1 |
| M. intracellulare | SG, 25–42 °C (opt. 37 °C) | 3 | 7 | 0 | 10 |
| M. kumamotonense | SG, 25–42 °C (opt. 37 °C) | 0 | 0 | 2 | 2 |
| M. malmoense | SG, 25–42 °C (opt. 37 °C) | 1 | 38 | 0 | 39 |
| M. mucogenicum | RG, 25–37 °C (opt. 30 °C) | 1 | 0 | 0 | 1 |
| M. nebraskense | SG, 25–35 °C (opt. 30 °C) | 1 | 0 | 0 | 1 |
| M. numidimassilliense | SG, 28–42 °C (opt. 37 °C) | 4 | 1 | 0 | 5 |
| M. paragordonae | SG, 25–30 °C (opt. 25 °C) | 8 | 1 | 0 | 9 |
| M. parmense | RG, 25–37 °C (opt. 30 °C) | 0 | 6 | 0 | 6 |
| M. peregrinum | RG, 25–37 °C (opt. 30 °C) | 4 | 0 | 5 | 9 |
| M. porcinum | RG, 25–42 °C (opt. 37 °C) | 1 | 0 | 0 | 1 |
| M. saskatchewanense | SG, 25–42 °C (opt. 37 °C) | 0 | 2 | 0 | 2 |
| M. scrofulaceum | SG, 25–42 °C (opt. 37 °C) | 5 | 14 | 0 | 19 |
| M. septicum | RG, 25–37 °C (opt. 30 °C) | 7 | 5 | 2 | 14 |
| M. simiae | SG, 25–42 °C (opt. 37 °C) | 0 | 1 | 0 | 1 |
| M. terrae | SG, 25–42 °C (opt. 37 °C) | 2 | 0 | 4 | 6 |
| M. terrae complex | SG, 25–42 °C (opt. 37 °C) | 1 | 3 | 5 | 9 |
| M. timonense | SG, 25–42 °C (opt. 37 °C) | 0 | 0 | 0 | 0 |
| M. triviale | SG, 25–42 °C (opt. 37 °C) | 5 | 0 | 1 | 6 |
| M. xenopi | SG, 37–45 °C (opt. 40 °C) | 0 | 37 | 0 | 37 |
| Mycobacerium sp. | SG, 20–37 °C | 22 | 51 | 29 | 102 |
| Total no. of isolates | 124 | 283 | 99 | 506 | |
| Species diversity | 25 | 18 | 14 | 38 | |
| Cluster | n | Cult + ve | %Cult + ve | Median | G. Mean | 95% CI of G. Mean | |
|---|---|---|---|---|---|---|---|
| S | 255 | 78 | 30.59 | 15 | 14.21 | 10.36 | 19.49 |
| R | 245 | 151 | 61.63 | 200 | 69.20 | 54.15 | 88.42 |
| E | 229 | 77 | 33.62 | 10 | 10.48 | 7.57 | 14.50 |
| Total/Means | 729 | 306 | 41.98 | 30 | 28.74 | 23.72 | 34.83 |
| Cluster | n | Cult + ve | %Cult + ve | Median | G. Mean | 95% CI of G. Mean | |
|---|---|---|---|---|---|---|---|
| S | 255 | 146 | 57.25 | 6524 | 10,516 | 5884 | 18,796 |
| R | 245 | 180 | 73.47 | 55,486 | 37,110 | 22,084 | 62,361 |
| E | 229 | 168 | 73.36 | 8800 | 9839 | 6190 | 15,639 |
| Total/Means | 729 | 494 | 67.76 | 12,468 | 16,277 | 12,021 | 22,039 |
| Cult − ve | No. of CFU | |||
|---|---|---|---|---|
| 1–10 | 11–100 | >100 | ||
| Number of qPCR positive samples | 212 | 92 | 92 | 98 |
| Minimum | 2.46 × 100 | 8.03 × 100 | 1.25 × 102 | 1.03 × 102 |
| Maximum | 3.60 × 106 | 2.40 × 106 | 4.24 × 106 | 2.80 × 107 |
| Median | 1.25 × 103 | 2.56 × 104 | 1.48 × 105 | 9.13 × 105 |
| 95% CI of median (lower bound) | 1.12 × 103 | 1.10 × 104 | 1.00 × 105 | 5.54 × 105 |
| 95% CI of median (upper bound) | 2.15 × 103 | 1.13 × 105 | 3.60 × 105 | 1.13 × 106 |
| Lower quartile (Q0.25) | 3.45 × 102 | 2.27 × 103 | 1.91 × 104 | 1.12 × 105 |
| Upper quartile (Q0.75) | 1.12 × 104 | 2.65 × 105 | 9.26 × 105 | 1.98 × 106 |
| Interquartile range (Q0.75–Q0.25) | 1.09 × 104 | 2.63 × 105 | 9.07 × 105 | 1.86 × 106 |
| Geometric mean | 1.85 × 103 | 2.15 × 104 | 9.91 × 104 | 2.52 × 105 |
| 95% CI of geom. mean (lower bound) | 1.31 × 103 | 1.13 × 104 | 5.85 × 104 | 1.37 × 105 |
| 95% CI of geom. mean (upper bound) | 2.63 × 103 | 4.09 × 104 | 1.68 × 105 | 4.63 × 105 |
| qPCR Result | Cult + ve | Cult − ve | Total | Diagnostic Sensitivity | Diagnostic Specificity |
|---|---|---|---|---|---|
| qPCR-positive a | 241 | 96 | 337 | 78.8% | 77.3% |
| qPCR-negative b | 65 | 327 | 392 | (73.8–83.0%) c | (73.1–81.0%) c |
| Total | 306 | 423 | 729 |
| qPCR Result | Cult + ve | Cult − ve | Total | Diagnostic Sensitivity | Diagnostic Specificity |
|---|---|---|---|---|---|
| qPCR-positive | 15 | 2 | 17 | 100% | 80% |
| qPCR-negative | 0 | 8 | 8 | (78.20–100%) a | (44.39–97.48%) a |
| Total | 306 | 423 | 729 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ulmann, V.; Modrá, H.; Babak, V.; Weston, R.T.; Pavlik, I. Recovery of Mycobacteria from Heavily Contaminated Environmental Matrices. Microorganisms 2021, 9, 2178. https://doi.org/10.3390/microorganisms9102178
Ulmann V, Modrá H, Babak V, Weston RT, Pavlik I. Recovery of Mycobacteria from Heavily Contaminated Environmental Matrices. Microorganisms. 2021; 9(10):2178. https://doi.org/10.3390/microorganisms9102178
Chicago/Turabian StyleUlmann, Vit, Helena Modrá, Vladimir Babak, Ross Tim Weston, and Ivo Pavlik. 2021. "Recovery of Mycobacteria from Heavily Contaminated Environmental Matrices" Microorganisms 9, no. 10: 2178. https://doi.org/10.3390/microorganisms9102178
APA StyleUlmann, V., Modrá, H., Babak, V., Weston, R. T., & Pavlik, I. (2021). Recovery of Mycobacteria from Heavily Contaminated Environmental Matrices. Microorganisms, 9(10), 2178. https://doi.org/10.3390/microorganisms9102178