Inhibition of Type III Interferon Expression in Intestinal Epithelial Cells—A Strategy Used by Coxsackie B Virus to Evade the Host’s Innate Immune Response at the Primary Site of Infection?

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Virus Stocks
2.2. Cell Lines and Human Pancreatic Islets
2.3. IFN Treatment of Cells
2.4. CVB3-V13 Infection of Cells
2.5. RNA Extraction and qRT-PCR
2.6. Western Blotting and Antibodies
2.7. Flow Cytometry
2.8. Poly (I:C) Treatment
2.9. Poly (I:C) Transfection
2.10. Statistical Analysis
3. Results
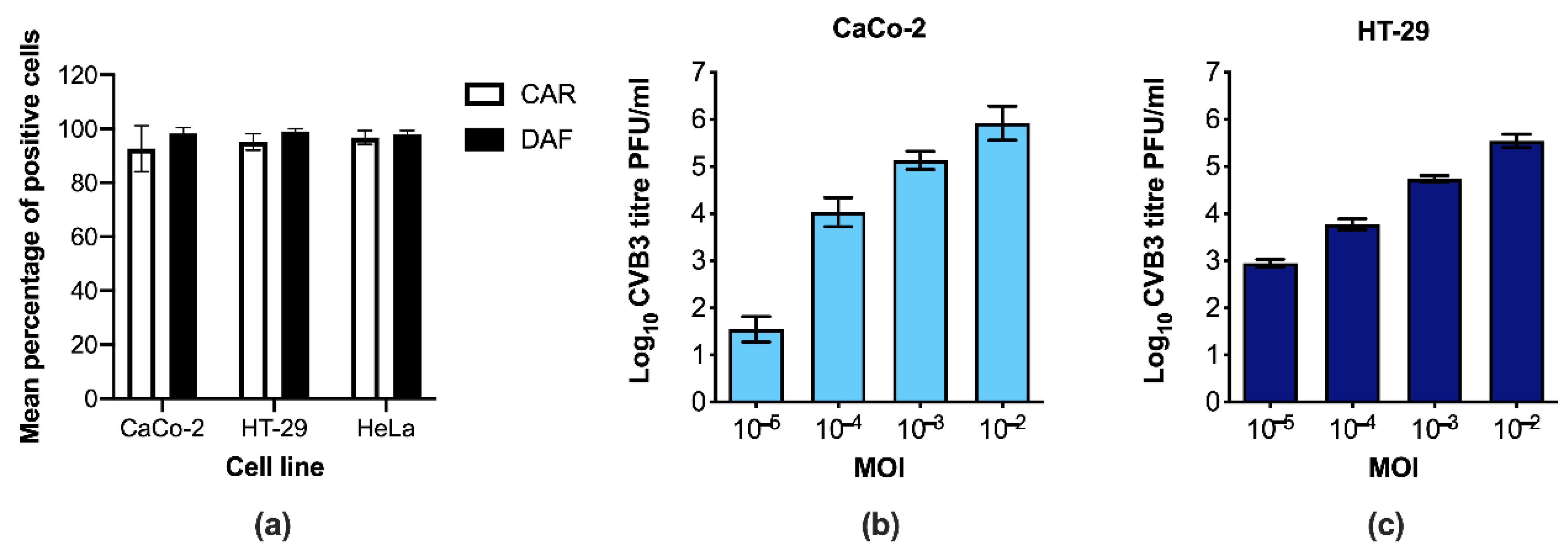
3.1. CaCo-2 and HT-29 Cells Are Permissive to CVB3 Infection
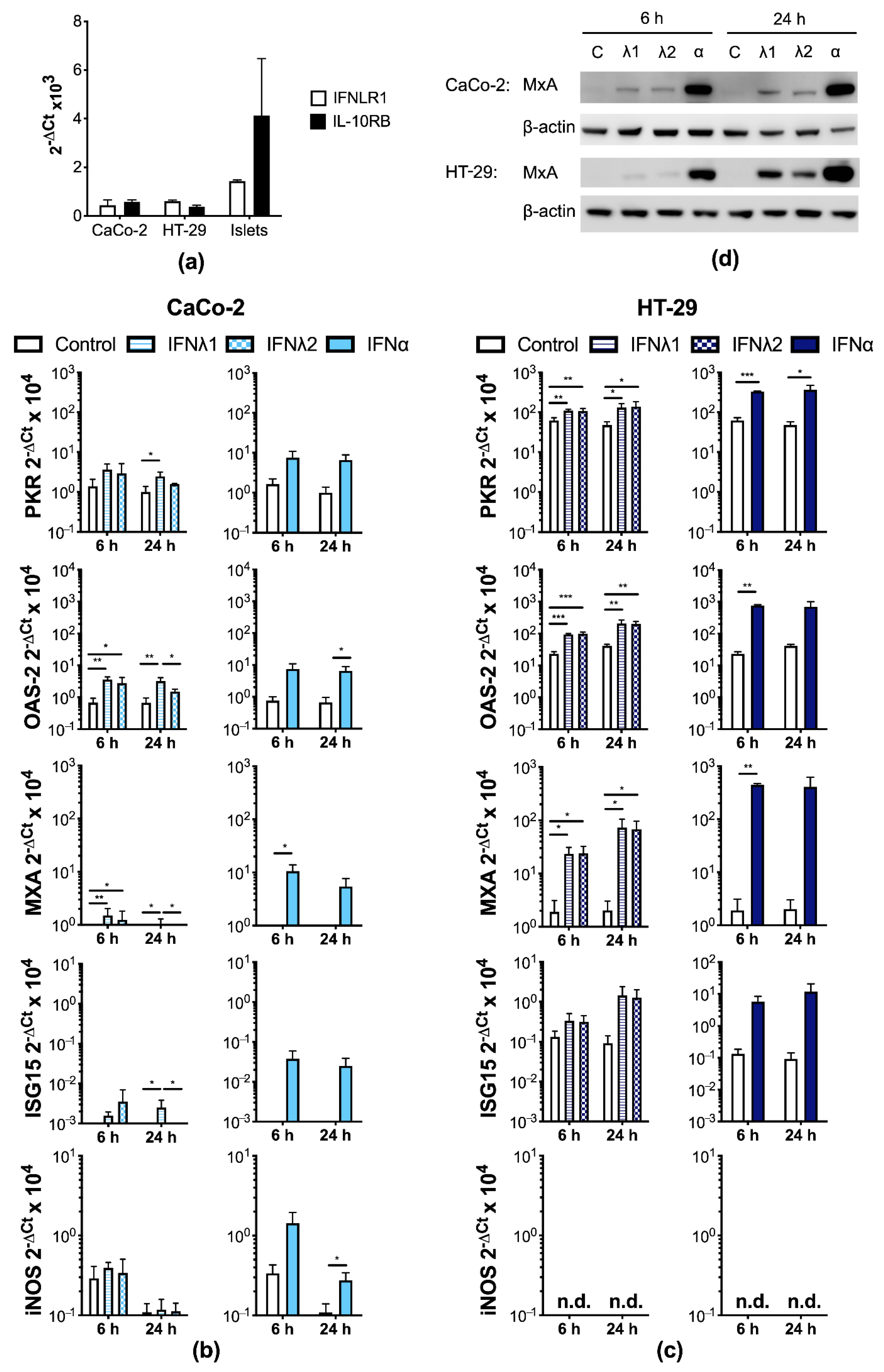
3.2. Human IECs Enter an Antiviral State after Type I and III IFN Treatment
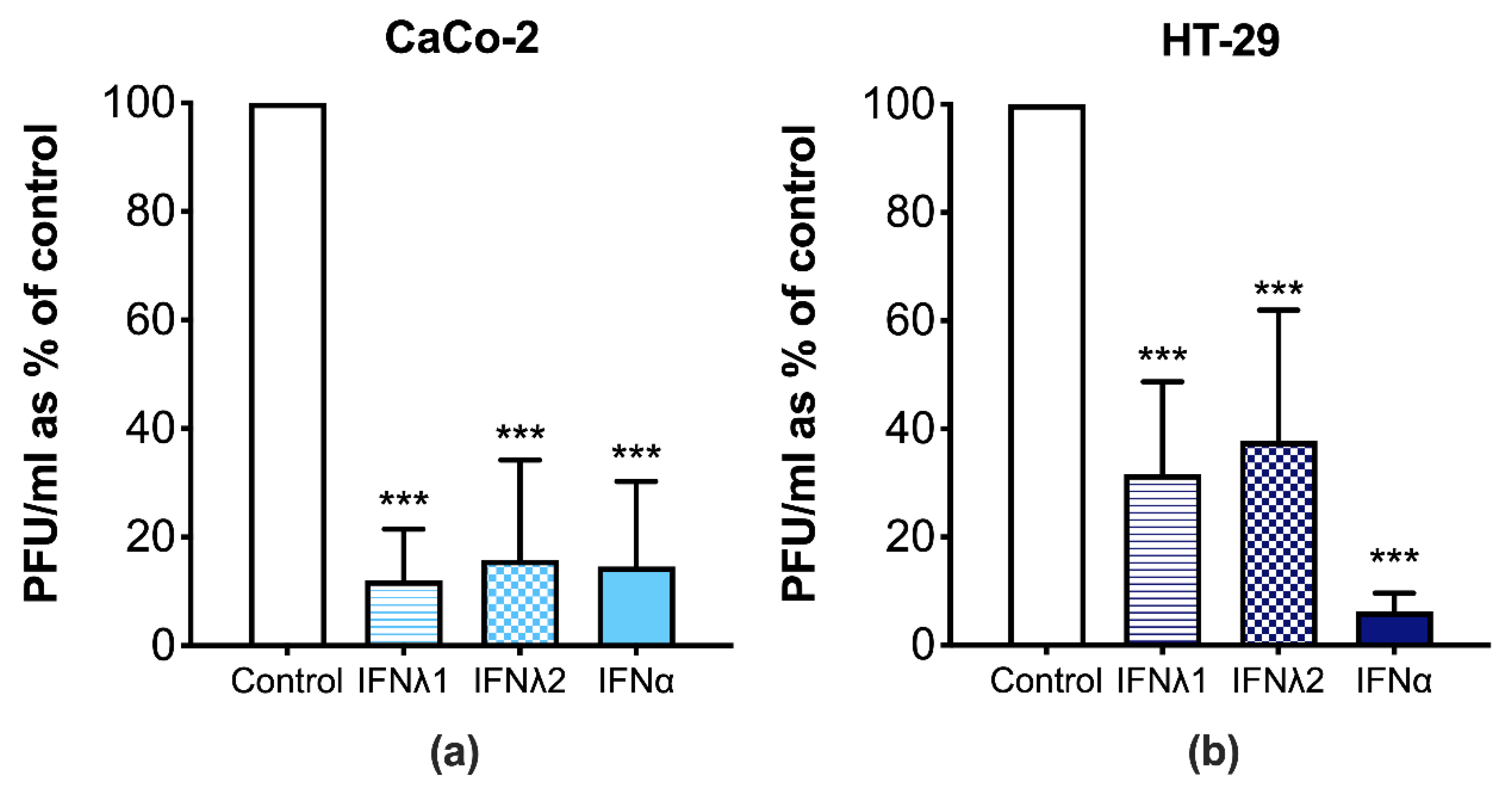
3.3. CVB3 Replication Is Perturbed in Human IECs Treated with Type I and III IFNs
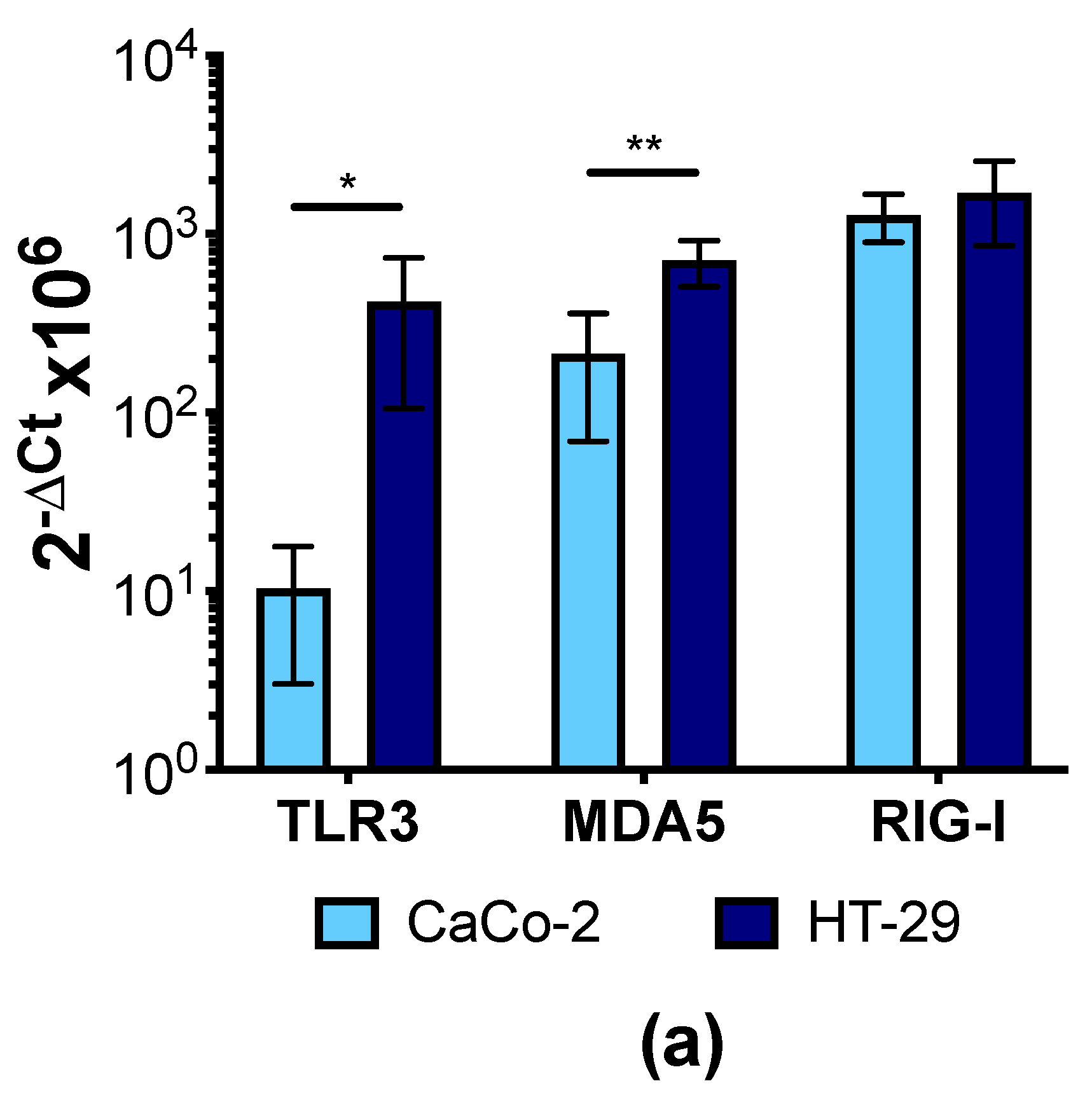
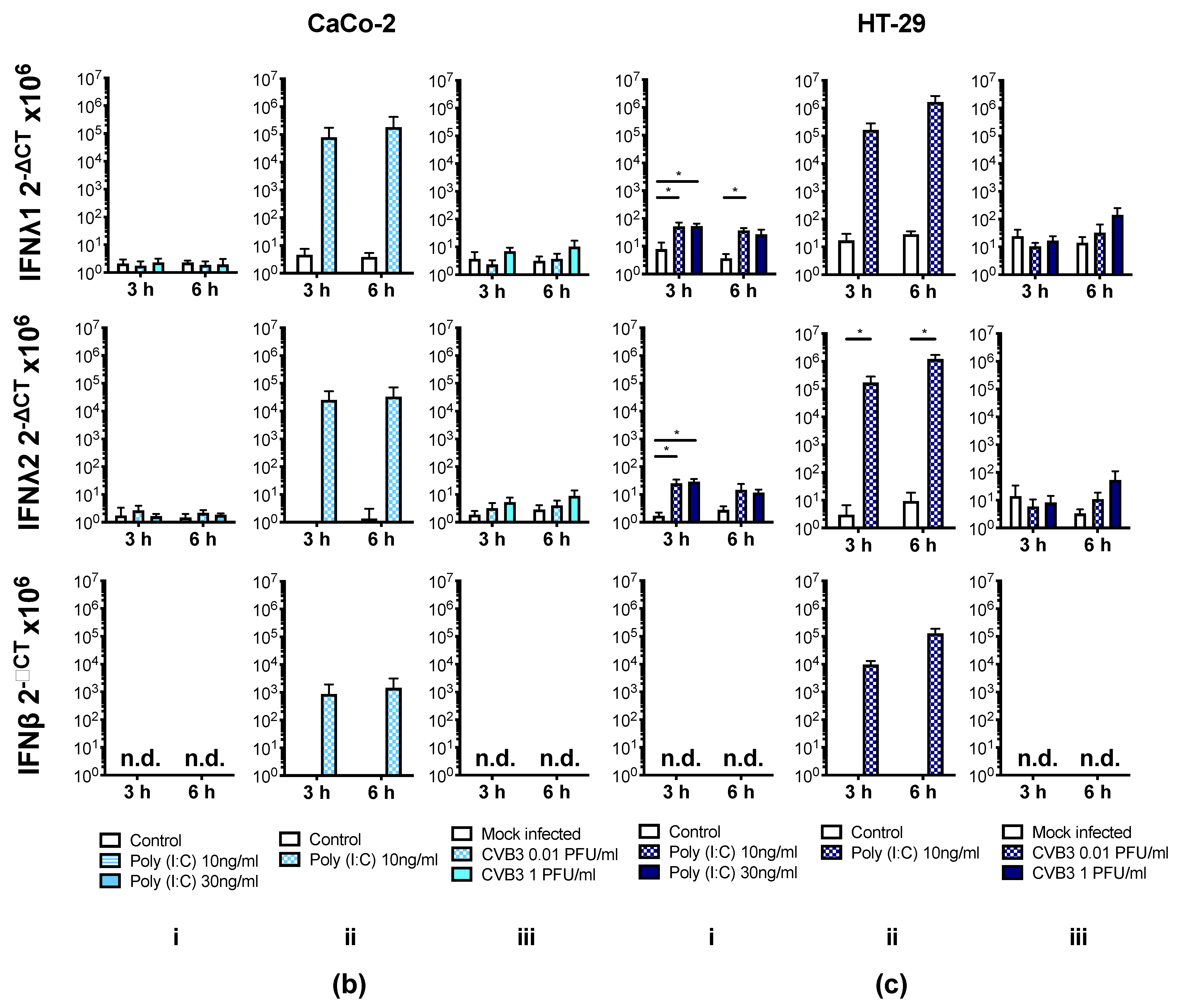
3.4. Human IECs Do Not Up-Regulate the Expression of Type I and III IFNs in Response to CVB3 Infection
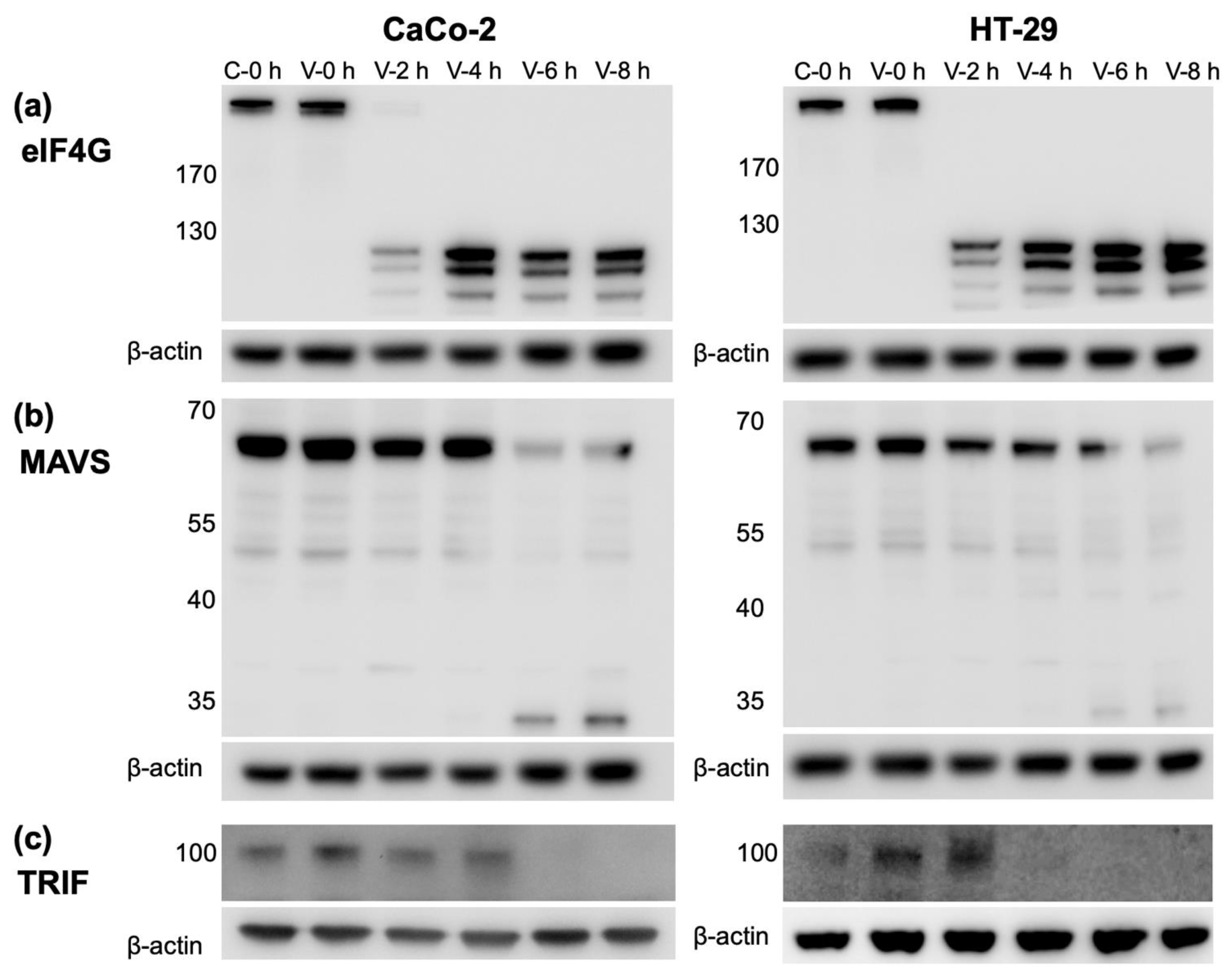
3.5. CVB3 Infection Results in the Cleavage of Proteins Involved in IFN Production
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Broggi, A.; Granucci, F.; Zanoni, I. Type III interferons: Balancing tissue tolerance and resistance to pathogen invasion. J. Exp. Med. 2020, 217, e20190295. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity 2019, 50, 907–923. [Google Scholar] [CrossRef] [PubMed]
- Ingle, H.; Peterson, S.T.; Baldridge, M.T. Distinct Effects of Type I and III Interferons on Enteric Viruses. Viruses 2018, 10, 46. [Google Scholar] [CrossRef] [PubMed]
- Wells, A.I.; Coyne, C.B. Type III Interferons in Antiviral Defenses at Barrier Surfaces. Trends Immunol. 2018, 39, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Schnepf, D.; Staeheli, P. Interferon-lambda orchestrates innate and adaptive mucosal immune responses. Nat. Rev. Immunol. 2019, 19, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Kemball, C.C.; Alirezaei, M.; Whitton, J.L. Type B coxsackieviruses and their interactions with the innate and adaptive immune systems. Future Microbiol. 2010, 5, 1329–1347. [Google Scholar] [CrossRef]
- Richardson, S.J.; Morgan, N.G. Enteroviral infections in the pathogenesis of type 1 diabetes: New insights for therapeutic intervention. Curr. Opin. Pharmacol. 2018, 43, 11–19. [Google Scholar] [CrossRef]
- Blanter, M.; Sork, H.; Tuomela, S.; Flodstrom-Tullberg, M. Genetic and Environmental Interaction in Type 1 Diabetes: A relationship between genetic risk alleles and molecular traits of enterovirus infection? Curr. Diab. Rep. 2019, 19, 82. [Google Scholar] [CrossRef]
- Lind, K.; Huhn, M.H.; Flodstrom-Tullberg, M. Immunology in the clinic review series; focus on type 1 diabetes and viruses: The innate immune response to enteroviruses and its possible role in regulating type 1 diabetes. Clin. Exp. Immunol. 2012, 168, 30–38. [Google Scholar] [CrossRef]
- Sin, J.; Mangale, V.; Thienphrapa, W.; Gottlieb, R.A.; Feuer, R. Recent progress in understanding coxsackievirus replication, dissemination, and pathogenesis. Virology 2015, 484, 288–304. [Google Scholar] [CrossRef]
- Bozym, R.A.; Morosky, S.A.; Kim, K.S.; Cherry, S.; Coyne, C.B. Release of intracellular calcium stores facilitates coxsackievirus entry into polarized endothelial cells. PLoS Pathog. 2010, 6, e1001135. [Google Scholar] [CrossRef] [PubMed]
- Coyne, C.B.; Bergelson, J.M. Virus-induced Abl and Fyn kinase signals permit coxsackievirus entry through epithelial tight junctions. Cell 2006, 124, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Harrath, R.; Bourlet, T.; Delezay, O.; Douche-Aourik, F.; Omar, S.; Aouni, M.; Pozzetto, B. Coxsackievirus B3 replication and persistence in intestinal cells from mice infected orally and in the human CaCo-2 cell line. J. Med. Virol. 2004, 74, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Riabi, S.; Gaaloul, I.; Harrath, R.; Aouni, M. Persistent infection of human intestinal Caco-2 cell line by Coxsackieviruses B. Pathol. Biol. 2012, 60, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Drummond, C.G.; Bolock, A.M.; Ma, C.; Luke, C.J.; Good, M.; Coyne, C.B. Enteroviruses infect human enteroids and induce antiviral signaling in a cell lineage-specific manner. Proc. Natl. Acad. Sci. USA 2017, 114, 1672–1677. [Google Scholar] [CrossRef]
- Drummond, C.G.; Nickerson, C.A.; Coyne, C.B. A Three-Dimensional Cell Culture Model To Study Enterovirus Infection of Polarized Intestinal Epithelial Cells. mSphere 2016, 1. [Google Scholar] [CrossRef] [PubMed]
- Villenave, R.; Wales, S.Q.; Hamkins-Indik, T.; Papafragkou, E.; Weaver, J.C.; Ferrante, T.C.; Bahinski, A.; Elkins, C.A.; Kulka, M.; Ingber, D.E. Human Gut-On-A-Chip Supports Polarized Infection of Coxsackie B1 Virus In Vitro. PLoS ONE 2017, 12, e0169412. [Google Scholar] [CrossRef]
- Oikarinen, M.; Tauriainen, S.; Oikarinen, S.; Honkanen, T.; Collin, P.; Rantala, I.; Maki, M.; Kaukinen, K.; Hyoty, H. Type 1 diabetes is associated with enterovirus infection in gut mucosa. Diabetes 2012, 61, 687–691. [Google Scholar] [CrossRef]
- Flodstrom, M.; Maday, A.; Balakrishna, D.; Cleary, M.M.; Yoshimura, A.; Sarvetnick, N. Target cell defense prevents the development of diabetes after viral infection. Nat. Immunol. 2002, 3, 373–382. [Google Scholar] [CrossRef]
- Lutton, C.W.; Gauntt, C.J. Ameliorating effect of IFN-beta and anti-IFN-beta on coxsackievirus B3-induced myocarditis in mice. J. Interferon Res. 1985, 5, 137–146. [Google Scholar] [CrossRef]
- Koestner, W.; Spanier, J.; Klause, T.; Tegtmeyer, P.K.; Becker, J.; Herder, V.; Borst, K.; Todt, D.; Lienenklaus, S.; Gerhauser, I.; et al. Interferon-beta expression and type I interferon receptor signaling of hepatocytes prevent hepatic necrosis and virus dissemination in Coxsackievirus B3-infected mice. PLoS Pathog. 2018, 14, e1007235. [Google Scholar] [CrossRef] [PubMed]
- Hultcrantz, M.; Huhn, M.H.; Wolf, M.; Olsson, A.; Jacobson, S.; Williams, B.R.; Korsgren, O.; Flodstrom-Tullberg, M. Interferons induce an antiviral state in human pancreatic islet cells. Virology 2007, 367, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Lind, K.; Richardson, S.J.; Leete, P.; Morgan, N.G.; Korsgren, O.; Flodstrom-Tullberg, M. Induction of an antiviral state and attenuated coxsackievirus replication in type III interferon-treated primary human pancreatic islets. J. Virol. 2013, 87, 7646–7654. [Google Scholar] [CrossRef] [PubMed]
- Domsgen, E.; Lind, K.; Kong, L.; Huhn, M.H.; Rasool, O.; van Kuppeveld, F.; Korsgren, O.; Lahesmaa, R.; Flodstrom-Tullberg, M. An IFIH1 gene polymorphism associated with risk for autoimmunity regulates canonical antiviral defence pathways in Coxsackievirus infected human pancreatic islets. Sci. Rep. 2016, 6, 39378. [Google Scholar] [CrossRef]
- Lind, K.; Svedin, E.; Utorova, R.; Stone, V.M.; Flodstrom-Tullberg, M. Type III interferons are expressed by Coxsackievirus-infected human primary hepatocytes and regulate hepatocyte permissiveness to infection. Clin. Exp. Immunol. 2014, 177, 687–695. [Google Scholar] [CrossRef]
- Laitinen, O.H.; Svedin, E.; Kapell, S.; Nurminen, A.; Hytonen, V.P.; Flodstrom-Tullberg, M. Enteroviral proteases: Structure, host interactions and pathogenicity. Rev. Med. Virol. 2016, 26, 251–267. [Google Scholar] [CrossRef]
- Wells, A.I.; Coyne, C.B. Enteroviruses: A Gut-Wrenching Game of Entry, Detection, and Evasion. Viruses 2019, 11, 460. [Google Scholar] [CrossRef]
- Visser, L.J.; Langereis, M.A.; Rabouw, H.H.; Wahedi, M.; Muntjewerff, E.M.; de Groot, R.J.; van Kuppeveld, F.J.M. Essential Role of Enterovirus 2A Protease in Counteracting Stress Granule Formation and the Induction of Type I Interferon. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Saeed, M.; Kapell, S.; Hertz, N.T.; Wu, X.; Bell, K.; Ashbrook, A.W.; Mark, M.T.; Zebroski, H.A.; Neal, M.L.; Flodstrom-Tullberg, M.; et al. Defining the proteolytic landscape during enterovirus infection. PLoS Pathog. 2020, 16, e1008927. [Google Scholar] [CrossRef]
- Feng, Q.; Langereis, M.A.; Lork, M.; Nguyen, M.; Hato, S.V.; Lanke, K.; Emdad, L.; Bhoopathi, P.; Fisher, P.B.; Lloyd, R.E.; et al. Enterovirus 2Apro targets MDA5 and MAVS in infected cells. J. Virol. 2014, 88, 3369–3378. [Google Scholar] [CrossRef]
- Harris, K.G.; Coyne, C.B. Death waits for no man--does it wait for a virus? How enteroviruses induce and control cell death. Cytokine Growth Factor Rev. 2014, 25, 587–596. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jagdeo, J.M.; Dufour, A.; Klein, T.; Solis, N.; Kleifeld, O.; Kizhakkedathu, J.; Luo, H.; Overall, C.M.; Jan, E. N-Terminomics TAILS Identifies Host Cell Substrates of Poliovirus and Coxsackievirus B3 3C Proteinases That Modulate Virus Infection. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Lind, K.; Svedin, E.; Domsgen, E.; Kapell, S.; Laitinen, O.H.; Moll, M.; Flodstrom-Tullberg, M. Coxsackievirus counters the host innate immune response by blocking type III interferon expression. J. Gen. Virol. 2016, 97, 1368–1380. [Google Scholar] [CrossRef] [PubMed]
- Laitinen, O.H.; Honkanen, H.; Pakkanen, O.; Oikarinen, S.; Hankaniemi, M.M.; Huhtala, H.; Ruokoranta, T.; Lecouturier, V.; Andre, P.; Harju, R.; et al. Coxsackievirus B1 is associated with induction of beta-cell autoimmunity that portends type 1 diabetes. Diabetes 2014, 63, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Brand, S.; Beigel, F.; Olszak, T.; Zitzmann, K.; Eichhorst, S.T.; Otte, J.M.; Diebold, J.; Diepolder, H.; Adler, B.; Auernhammer, C.J.; et al. IL-28A and IL-29 mediate antiproliferative and antiviral signals in intestinal epithelial cells and murine CMV infection increases colonic IL-28A expression. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G960–G968. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Mikamo-Satoh, E.; Hirai, R.; Kawai, T.; Matsushita, K.; Hiiragi, A.; Dermody, T.S.; Fujita, T.; Akira, S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 2008, 205, 1601–1610. [Google Scholar] [CrossRef]
- Feng, Q.; Langereis, M.A.; van Kuppeveld, F.J. Induction and suppression of innate antiviral responses by picornaviruses. Cytokine Growth Factor Rev. 2014, 25, 577–585. [Google Scholar] [CrossRef]
- Chau, D.H.; Yuan, J.; Zhang, H.; Cheung, P.; Lim, T.; Liu, Z.; Sall, A.; Yang, D. Coxsackievirus B3 proteases 2A and 3C induce apoptotic cell death through mitochondrial injury and cleavage of eIF4GI but not DAP5/p97/NAT1. Apoptosis 2007, 12, 513–524. [Google Scholar] [CrossRef]
- Wu, S.; Wang, Y.; Lin, L.; Si, X.; Wang, T.; Zhong, X.; Tong, L.; Luan, Y.; Chen, Y.; Li, X.; et al. Protease 2A induces stress granule formation during coxsackievirus B3 and enterovirus 71 infections. Virol. J. 2014, 11, 192. [Google Scholar] [CrossRef]
- Zhang, Q.M.; Song, W.Q.; Li, Y.J.; Qian, J.; Zhai, A.X.; Wu, J.; Li, A.M.; He, J.M.; Zhao, J.Y.; Yu, X.; et al. Over-expression of mitochondrial antiviral signaling protein inhibits coxsackievirus B3 infection by enhancing type-I interferons production. Virol. J. 2012, 9, 312. [Google Scholar] [CrossRef]
- Mukherjee, A.; Morosky, S.A.; Delorme-Axford, E.; Dybdahl-Sissoko, N.; Oberste, M.S.; Wang, T.; Coyne, C.B. The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog. 2011, 7, e1001311. [Google Scholar] [CrossRef] [PubMed]
- Haller, O.; Kochs, G. Human MxA protein: An interferon-induced dynamin-like GTPase with broad antiviral activity. J. Interferon Cytokine Res. 2011, 31, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Rahnefeld, A.; Klingel, K.; Schuermann, A.; Diny, N.L.; Althof, N.; Lindner, A.; Bleienheuft, P.; Savvatis, K.; Respondek, D.; Opitz, E.; et al. Ubiquitin-like protein ISG15 (interferon-stimulated gene of 15 kDa) in host defense against heart failure in a mouse model of virus-induced cardiomyopathy. Circulation 2014, 130, 1589–1600. [Google Scholar] [CrossRef] [PubMed]
- Flodstrom-Tullberg, M.; Hultcrantz, M.; Stotland, A.; Maday, A.; Tsai, D.; Fine, C.; Williams, B.; Silverman, R.; Sarvetnick, N. RNase L and double-stranded RNA-dependent protein kinase exert complementary roles in islet cell defense during coxsackievirus infection. J. Immunol. 2005, 174, 1171–1177. [Google Scholar] [CrossRef]
- Flodstrom, M.; Horwitz, M.S.; Maday, A.; Balakrishna, D.; Rodriguez, E.; Sarvetnick, N. A critical role for inducible nitric oxide synthase in host survival following coxsackievirus B4 infection. Virology 2001, 281, 205–215. [Google Scholar] [CrossRef]
- Lowenstein, C.J.; Hill, S.L.; Lafond-Walker, A.; Wu, J.; Allen, G.; Landavere, M.; Rose, N.R.; Herskowitz, A. Nitric oxide inhibits viral replication in murine myocarditis. J. Clin. Investig. 1996, 97, 1837–1843. [Google Scholar] [CrossRef]
- Broquet, A.H.; Hirata, Y.; McAllister, C.S.; Kagnoff, M.F. RIG-I/MDA5/MAVS are required to signal a protective IFN response in rotavirus-infected intestinal epithelium. J. Immunol. 2011, 186, 1618–1626. [Google Scholar] [CrossRef]
- Pott, J.; Mahlakoiv, T.; Mordstein, M.; Duerr, C.U.; Michiels, T.; Stockinger, S.; Staeheli, P.; Hornef, M.W. IFN-lambda determines the intestinal epithelial antiviral host defense. Proc. Natl. Acad. Sci. USA 2011, 108, 7944–7949. [Google Scholar] [CrossRef]
- Baldridge, M.T.; Nice, T.J.; McCune, B.T.; Yokoyama, C.C.; Kambal, A.; Wheadon, M.; Diamond, M.S.; Ivanova, Y.; Artyomov, M.; Virgin, H.W. Commensal microbes and interferon-lambda determine persistence of enteric murine norovirus infection. Science 2015, 347, 266–269. [Google Scholar] [CrossRef]
- Nice, T.J.; Baldridge, M.T.; McCune, B.T.; Norman, J.M.; Lazear, H.M.; Artyomov, M.; Diamond, M.S.; Virgin, H.W. Interferon-lambda cures persistent murine norovirus infection in the absence of adaptive immunity. Science 2015, 347, 269–273. [Google Scholar] [CrossRef]
- Stanifer, M.L.; Kee, C.; Cortese, M.; Zumaran, C.M.; Triana, S.; Mukenhirn, M.; Kraeusslich, H.G.; Alexandrov, T.; Bartenschlager, R.; Boulant, S. Critical Role of Type III Interferon in Controlling SARS-CoV-2 Infection in Human Intestinal Epithelial Cells. Cell Rep. 2020, 32, 107863. [Google Scholar] [CrossRef] [PubMed]
- Mahlakoiv, T.; Hernandez, P.; Gronke, K.; Diefenbach, A.; Staeheli, P. Leukocyte-derived IFN-alpha/beta and epithelial IFN-lambda constitute a compartmentalized mucosal defense system that restricts enteric virus infections. PLoS Pathog. 2015, 11, e1004782. [Google Scholar] [CrossRef] [PubMed]
- Alidjinou, E.K.; Sane, F.; Engelmann, I.; Geenen, V.; Hober, D. Enterovirus persistence as a mechanism in the pathogenesis of type 1 diabetes. Discov. Med. 2014, 18, 273–282. [Google Scholar] [PubMed]
- Vehik, K.; Lynch, K.F.; Wong, M.C.; Tian, X.; Ross, M.C.; Gibbs, R.A.; Ajami, N.J.; Petrosino, J.F.; Rewers, M.; Toppari, J.; et al. Prospective virome analyses in young children at increased genetic risk for type 1 diabetes. Nat. Med. 2019, 25, 1865–1872. [Google Scholar] [CrossRef]
- Hirata, Y.; Broquet, A.H.; Menchen, L.; Kagnoff, M.F. Activation of innate immune defense mechanisms by signaling through RIG-I/IPS-1 in intestinal epithelial cells. J. Immunol. 2007, 179, 5425–5432. [Google Scholar] [CrossRef]
- Su, R.; Shereen, M.A.; Zeng, X.; Liang, Y.; Li, W.; Ruan, Z.; Li, Y.; Liu, W.; Liu, Y.; Wu, K.; et al. The TLR3/IRF1/Type III IFN Axis Facilitates Antiviral Responses against Enterovirus Infections in the Intestine. mBio 2020, 11. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stone, V.M.; Ringqvist, E.E.; Larsson, P.G.; Domsgen, E.; Holmlund, U.; Sverremark-Ekström, E.; Flodström-Tullberg, M. Inhibition of Type III Interferon Expression in Intestinal Epithelial Cells—A Strategy Used by Coxsackie B Virus to Evade the Host’s Innate Immune Response at the Primary Site of Infection? Microorganisms 2021, 9, 105. https://doi.org/10.3390/microorganisms9010105
Stone VM, Ringqvist EE, Larsson PG, Domsgen E, Holmlund U, Sverremark-Ekström E, Flodström-Tullberg M. Inhibition of Type III Interferon Expression in Intestinal Epithelial Cells—A Strategy Used by Coxsackie B Virus to Evade the Host’s Innate Immune Response at the Primary Site of Infection? Microorganisms. 2021; 9(1):105. https://doi.org/10.3390/microorganisms9010105
Chicago/Turabian StyleStone, Virginia M., Emma E. Ringqvist, Pär G. Larsson, Erna Domsgen, Ulrika Holmlund, Eva Sverremark-Ekström, and Malin Flodström-Tullberg. 2021. "Inhibition of Type III Interferon Expression in Intestinal Epithelial Cells—A Strategy Used by Coxsackie B Virus to Evade the Host’s Innate Immune Response at the Primary Site of Infection?" Microorganisms 9, no. 1: 105. https://doi.org/10.3390/microorganisms9010105
APA StyleStone, V. M., Ringqvist, E. E., Larsson, P. G., Domsgen, E., Holmlund, U., Sverremark-Ekström, E., & Flodström-Tullberg, M. (2021). Inhibition of Type III Interferon Expression in Intestinal Epithelial Cells—A Strategy Used by Coxsackie B Virus to Evade the Host’s Innate Immune Response at the Primary Site of Infection? Microorganisms, 9(1), 105. https://doi.org/10.3390/microorganisms9010105