Helicobacter pylori-Derived Outer Membrane Vesicles (OMVs): Role in Bacterial Pathogenesis?



Abstract
1. Background
2. Physicochemical Characteristics of H. pylori-Derived OMVs
3. H. pylori OMVs Promote Biofilm Formation
4. The Content of H. pylori OMVs
4.1. Lipids, Peptidoglycan, and Nucleic Acids in H. pylori OMVs
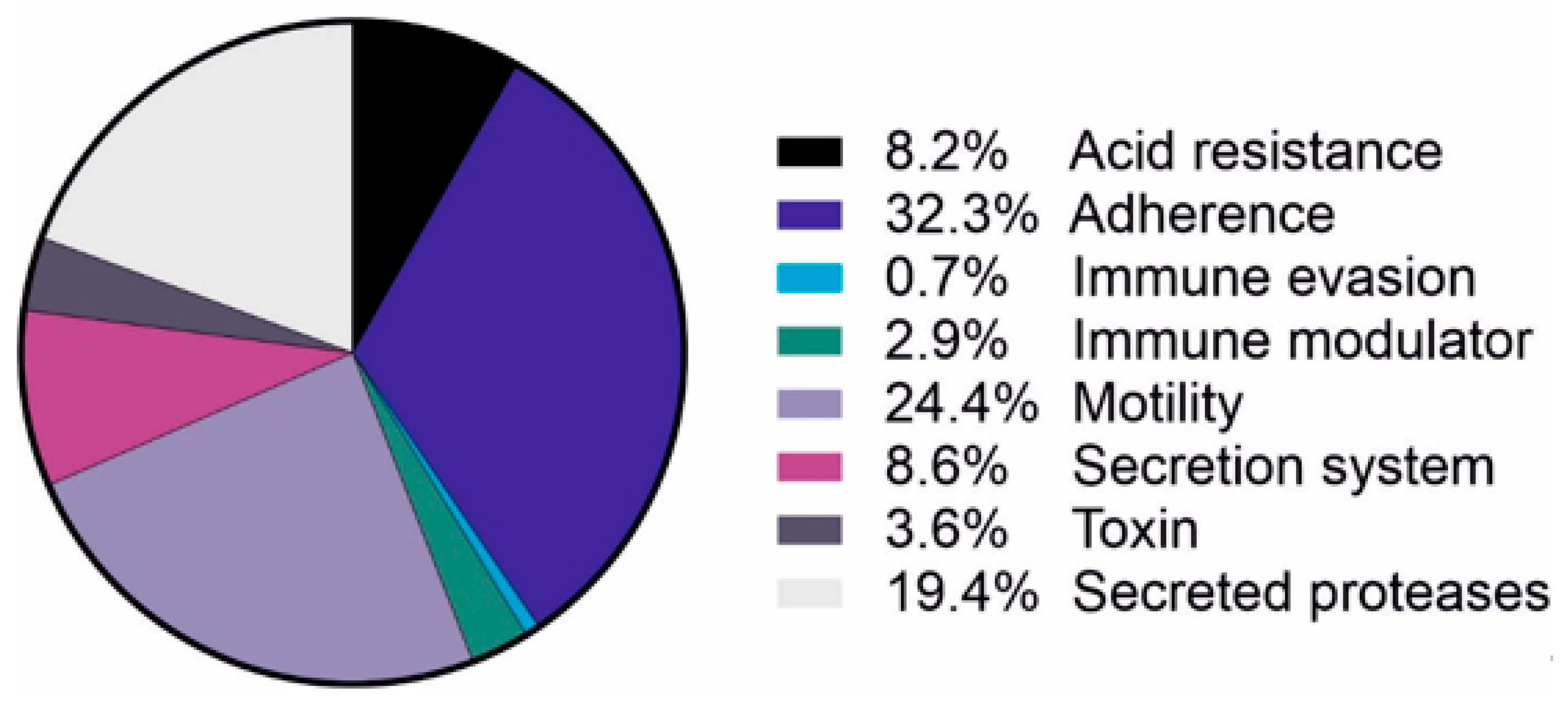
4.2. The Protein Content of H. pylori OMVs
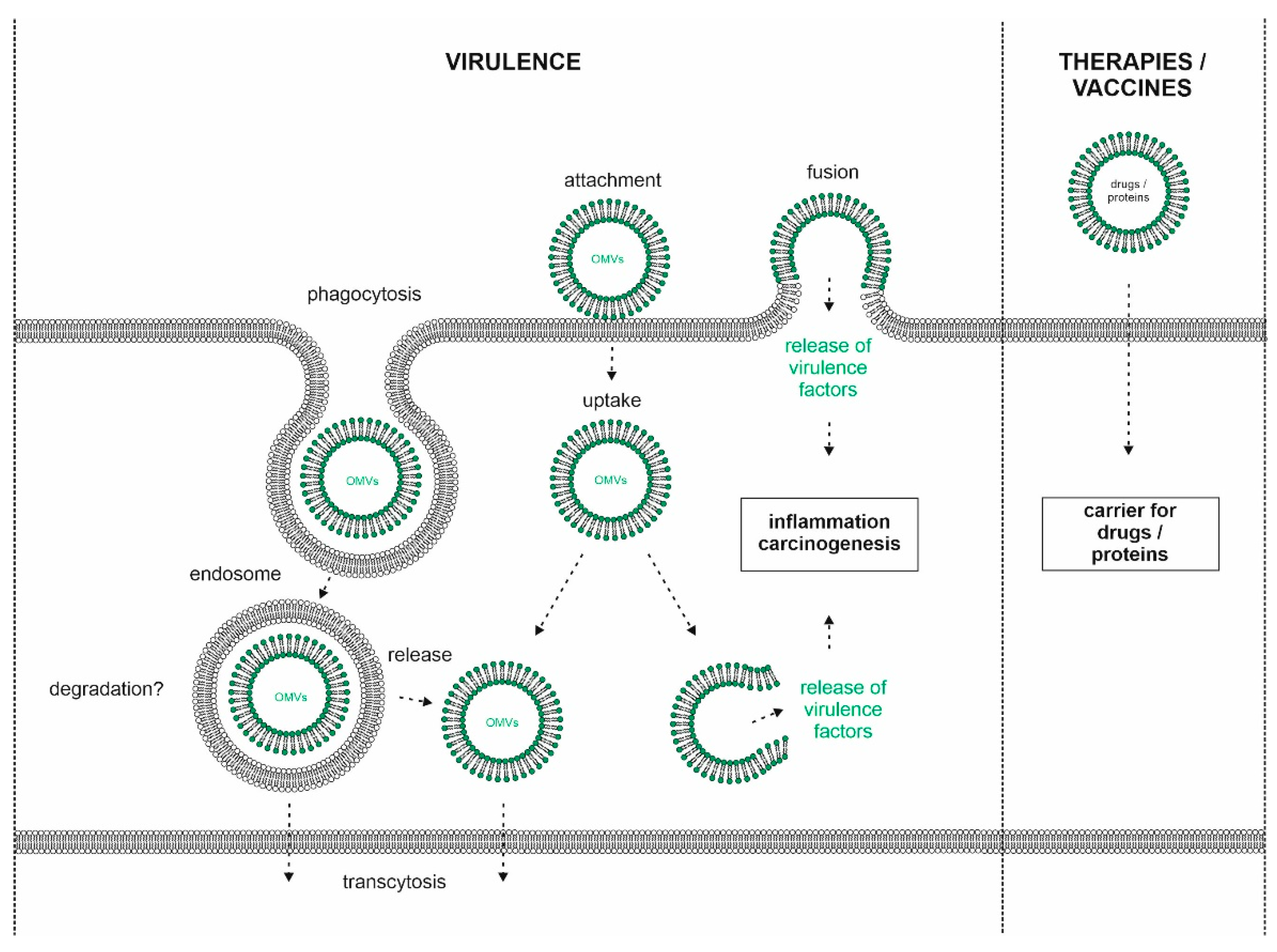
Uptake of H. pylori OMVs: Delivery of Virulence Factors into Host Cells?

{kind=link}
{kind=link}
{kind=link}
| Virulence Factors—Acid Resistance | |||||
|---|---|---|---|---|---|
| Gene/Chromosome (NC_000915) | Function/Description | Virulence Factors | Confirmed by Mass Spectroscopy (Reference) | Confirmed by Western Blotting (Reference) | |
| ureH/HP0067 | urease accessory proteins, form a complex that acts as a GTP-hydrolysis-dependent molecular chaperone, activating the urease | Urease | [41] | ||
| ureG/HP0068 | [39] | ||||
| ureF/HP0069 | [39,41] | ||||
| ureB/HP0072 | urease subunit beta | neutralizes the gastric acidity, NH3 damages the gastric epithelium | [38,39,40,41,42,45] | [20] | |
| ureA/HP0073 | urease subunit alpha | [38,39,40,41,42] | [20] | ||
| Virulence factors—Adherence | |||||
| hopZ/HP0009 | Outer membrane protein | HopZ | [38,40,42,45] | ||
| horB/HP0127 | Outer membrane protein 4 | HorB | [38,39,40,41,45] | ||
| babA/hopS/HP0317/HP1243 | Outer membrane protein 9/28, binds with the epithelial cell receptor Leb, mediates bacterial attachment and colonization | Blood group antigen binding adhesins | [39,40,41,42] | [41] | |
| babB/hopT/HP0896 | Outer membrane protein 19, babA paralog | [38,39,40,41,42] | |||
| sabB/hopO/HP0722 | sabA homologue | Sialic acid binding adhesins | [40,41,42] | ||
| sabA/hopP/HP0725 | binds with sialyl-Lex antigen, mediates bacterial attachment and colonization | [38,40,41,42] | [41] | ||
| hpaA/HP0797 | neuraminyllactose-binding hemagglutinin | H. pylori adhesin A | [38,39,40,41,42] | [38] | |
| alpA/hopC/HP0912 | Outer membrane protein 20 | adherence-associated lipoprotein | [38,39,40,41,42] | ||
| alpB/hopB/HP0913 | Outer membrane protein 21 | [39,40,41,42] | [34,41] | ||
| Virulence factors—Immune evasion | |||||
| futA/HP0379 | alpha-(1,3)-fucosyltransferases, LPS oligosaccharide biosynthesis | Lipopoly-saccharide Lewis antigens | [39] | ||
| futB/HP0651 | [41] | ||||
| Virulence factors—Immune modulator | |||||
| napA/HP0243 | DNA protection during starvation, activates neutrophils, mast cells and monocytes | Neutrophil-activating protein (HP-NAP) | [39,40,42] | [38] | |
| oipA/hopH/HP0638 | Outer membrane protein 13, bacterial adherence to the gastric epithelium, damages mucosal layer, induces apoptosis and IL-8 expression | Outer inflammatory protein | [38,39,41,42] | [38,41] | |
| Virulence factors—Motility | |||||
| flaB/HP0115 | minor flagellin subunit, polymerizes to form the filaments of bacterial flagella | helps bacteria to minimize contact to acidic environment | Flagella | [39,41,42] | |
| flgI/HP0246 | P-ring protein, assembles around the rod | [39,41,42] | |||
| flgL/HP0295 | Hook-associated protein 3 | [39,41,42] | |||
| flgH/HP0325 | L-ring protein, assembles around the rod | [39,45] | |||
| flaA/HP0601 | predominant flagellin subunit, polymerizes to form the filaments of bacterial flagella | [38,39,40,41] | |||
| flaG/HP0751 | polar flagellin, rotor/switch protein | [39,42] | |||
| fliD/HP0752 | filament-capping protein, flagellin folding chaperone, required for the morphogenesis and for the elongation of the flagellar filament | [39,41,42] | |||
| flgE_1/HP0870 | hook protein, links the flagellar filament to the drive apparatus in the basal body | [39,40,41] | |||
| flgD/HP0907 | basal body rod modification protein, required for flagellar hook formation, scaffolding protein | [39,40,41,42] | |||
| flgE_2/HP0908 | flagellar basal body protein | [39,41] | |||
| flgG_1/HP1092 | flagellar basal body protein | [39,42] | |||
| flgK/HP1119 | first hook-filament junction protein | [39,41,42] | |||
| flgA/HP1477 | involved in the assembly process of the P-ring formation | [40,41] | |||
| fliE/HP1557 | MS-ring rod junction protein, | [39,40,42] | |||
| flgC/HP1558 | rod protein | [39] | |||
| flgB/HP1559 | rod protein of flagellar basal body | [39,42] | |||
| flgG_2/HP1585 | distal rod protein | [39,42] | |||
| Virulence factors—Secretion system (Proteins Required for Cag T4SS Activity) | |||||
| cag1/HP0520 | membrane protein in T4SS, associated with IL-8 expression induction and CagA delivery to host cells | Cag PAI type IV secretion system | [41] | ||
| cag3/HP0522 | defined localization in T4SS apparatus | [39] | |||
| cagX/HP0528 | defined localization in T4SS apparatus | [39] | [41] | ||
| cagT/HP532 | defined localization in T4SS apparatus, core complex protein in T4SS, helps in the translocation of CagA | [39,41] | [41] | ||
| cagM/HP0537 | defined localization in T4SS apparatus | [39,41] | [41] | ||
| cagN/HP0538 | localization in T4SS apparatus is not yet defined | [39] | [41] | ||
| cagF/HP0543 | localization in T4SS apparatus is not yet defined, CagA chaperone | [41] | |||
| cagD/HP0545 | localization in T4SS apparatus is not yet defined | [39,42] | |||
| cagA/HP0547 | Scaffold/hub protein, oncoprotein, becomes phosphorylated in the host cells, causes cellular proliferation and elongation, induces IL-8 expression | Secreted T4SS effector cytotoxin-associated gene A | [38,39,41,42,45] | [29,38,41] | |
| Virulence factors—Toxin | |||||
| vacA/HP0887 | induces vacuolization of epithelial cells and endoplasmic reticulum stress, causes cell vacuolization, necrosis and apoptosis, enhances activation of autophagy and increased cellular death | Vacuolating cytotoxin | [39,40,41,42,45] | [27,29,32,38,41,65,66] | |
| Other | |||||
| katA/HP0875 | antioxidant enzyme, neutralization of H2O2 and NaClO | [36,38,39,40,41] | enzyme activity determined in [62] | ||
| hcpD/HP0160 | beta-lactamase | Putative solenoid proteins | [39,40,41] | ||
| hcpA/HP0211 | beta-lactamase, cysteine-rich 28 kD protein | [39,40,42] | |||
| hcpE/HP0235 | beta-lactamase | [39,40,41,42] | |||
| hcpC/HP1098 | beta-lactamase, cysteine-rich protein C | [38,39,40,42] | |||
| tolB/HP1126 | periplasmic protein interacting with outer membrane proteins (OMPs) | [38,39,40,41,42] | |||
| csd3/HP0506 | conserved hypothetical secreted protein | Secreted proteases | [39,41] | ||
| ymxG/HP0657 | processing protease | [38,39,41,42] | |||
| pqqE/HP1012 | metalloendopeptidase | [38,39,40,41,42,45] | |||
| htrA/HP1018-9 | acts as protease, degrades misfolded proteins and tight junction protein enabling delivery of CagA | [38,39,40,41,42] | |||
| HP1037 | metal ion binding aminopeptidase | [39,41] | |||
| ggt/HP1118 | transpeptidation and amino acid synthesis, enhances cell apoptosis, inhibits cellular proliferation | [38,39,40,41,42] | |||
| ctpB/HP1350 | serine-type endopeptidase | [38,39,40,41] | |||
4.3. Do H. pylori OMVs Modulate Host Immune Regulation?
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Piazuelo, M.B.; Epplein, M.; Correa, P. Gastric Cancer: An Infectious Disease. Infect. Dis. Clin. N. Am. 2010, 24, 853–869. [Google Scholar] [CrossRef]
- Waskito, L.A.; Salama, N.R.; Yamaoka, Y. Pathogenesis of Helicobacter pylori infection. Helicobacter 2018, 23, e12516. [Google Scholar] [CrossRef]
- Venerito, M.; Vasapolli, R.; Rokkas, T.; Malfertheiner, P. Gastric cancer: Epidemiology, prevention, and therapy. Helicobacter 2018, 23, e12518. [Google Scholar] [CrossRef]
- Schistosomes, liver flukes and Helicobacter pylori. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon, 7–14 June 1994. IARC Monogr. Eval. Carcinog. Risks Hum. 1994, 61, 1–241.
- Parkin, D.M. The global health burden of infection-associated cancers in the year 2002. Int. J. Cancer 2006, 118, 3030–3044. [Google Scholar] [CrossRef]
- Moss, S.F. The Clinical Evidence Linking Helicobacter pylori to Gastric Cancer. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 183–191. [Google Scholar] [CrossRef]
- Correa, P.; Piazuelo, M.B. The gastric precancerous cascade. J. Dig. Dis. 2011, 13, 2–9. [Google Scholar] [CrossRef]
- Correa, P. Human gastric carcinogenesis: A multistep and multifactorial process—First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 1992, 52, 6735–6740. [Google Scholar]
- Choi, I.J.; Kook, M.-C.; Kim, Y.-I.; Cho, S.-J.; Lee, J.Y.; Kim, C.G.; Park, B.; Nam, B.H. Helicobacter pylori Therapy for the Prevention of Metachronous Gastric Cancer. N. Engl. J. Med. 2018, 378, 1085–1095. [Google Scholar] [CrossRef]
- Ford, A.C.; Forman, D.; Hunt, R.H.; Yuan, Y.; Moayyedi, P. Helicobacter pylori eradication therapy to prevent gastric cancer in healthy asymptomatic infected individuals: Systematic review and meta-analysis of randomised controlled trials. BMJ 2014, 348, g3174. [Google Scholar] [CrossRef]
- Šterbenc, A.; Jarc, E.; Poljak, M.; Homan, M. Helicobacter pylori virulence genes. World J. Gastroenterol. 2019, 25, 4870–4884. [Google Scholar] [CrossRef]
- Posselt, G.; Backert, S.; Wessler, S. The functional interplay of Helicobacter pylori factors with gastric epithelial cells induces a multi-step process in pathogenesis. Cell Commun. Signal. 2013, 11, 77. [Google Scholar] [CrossRef]
- Backert, S.; Clyne, M. Pathogenesis of Helicobacter pylori Infection. Helicobacter 2011, 16, 19–25. [Google Scholar] [CrossRef]
- Zhang, X.-Y.; Zhang, P.-Y.; Aboul-Soud, M.A. From inflammation to gastric cancer: Role of Helicobacter pylori. Oncol. Lett. 2016, 13, 543–548. [Google Scholar] [CrossRef]
- Deatherage, B.L.; Cookson, B.T. Membrane Vesicle Release in Bacteria, Eukaryotes, and Archaea: A Conserved yet Underappreciated Aspect of Microbial Life. Infect. Immun. 2012, 80, 1948–1957. [Google Scholar] [CrossRef]
- Malloci, M.; Perdomo, L.; Veerasamy, M.; Andriantsitohaina, R.; Simard, G.; Martínez, M.C. Extracellular Vesicles: Mechanisms in Human Health and Disease. Antioxid. Redox Signal. 2019, 30, 813–856. [Google Scholar] [CrossRef]
- Bitto, N.J.; Kaparakis-Liaskos, M. The Therapeutic Benefit of Bacterial Membrane Vesicles. Int. J. Mol. Sci. 2017, 18, 1287. [Google Scholar] [CrossRef]
- Schwechheimer, C.; Kuehn, M.J. Outer-membrane vesicles from Gram-negative bacteria: Biogenesis and functions. Nat. Rev. Genet. 2015, 13, 605–619. [Google Scholar] [CrossRef]
- Fiocca, R.; Necchi, V.; Sommi, P.; Ricci, V.; Telford, J.; Cover, T.L.; Solcia, E. Release of Helicobacter pylori vacuolating cytotoxin by both a specific secretion pathway and budding of outer membrane vesicles. Uptake of released toxin and vesicles by gastric epithelium. J. Pathol. 1999, 188, 220–226. [Google Scholar] [CrossRef]
- Kaparakis, M.; Turnbull, L.; Carneiro, L.; Firth, S.; Coleman, H.A.; Parkington, H.C.; Le Bourhis, L.; Karrar, A.; Viala, J.; Mak, J.; et al. Bacterial membrane vesicles deliver peptidoglycan to NOD1 in epithelial cells. Cell. Microbiol. 2010, 12, 372–385. [Google Scholar] [CrossRef]
- Shen, Y.; Torchia, M.L.G.; Lawson, G.W.; Karp, C.L.; Ashwell, J.D.; Mazmanian, S.K. Outer membrane vesicles of a human commensal mediate immune regulation and disease protection. Cell Host Microbe 2012, 12, 509–520. [Google Scholar] [CrossRef]
- Ismail, S.; Hampton, M.B.; Keenan, J. Helicobacter pylori Outer Membrane Vesicles Modulate Proliferation and Interleukin-8 Production by Gastric Epithelial Cells. Infect. Immun. 2003, 71, 5670–5675. [Google Scholar] [CrossRef]
- Parker, H.; Keenan, J.I. Composition and function of Helicobacter pylori outer membrane vesicles. Microbes Infect. 2012, 14, 9–16. [Google Scholar] [CrossRef]
- Sgouras, D.; Tegtmeyer, N.; Wessler, S. Activity and Functional Importance of Helicobacter pylori Virulence Factors. Adv. Exp. Med. Biol. 2019, 35–56. [Google Scholar] [CrossRef]
- Wessler, S.; Backert, S. Molecular mechanisms of epithelial-barrier disruption by Helicobacter pylori. Trends Microbiol. 2008, 16, 397–405. [Google Scholar] [CrossRef]
- Keenan, J.I.; Allardyce, R.A.; Bagshaw, P.F. Dual silver staining to characterise Helicobacter spp. outer membrane components. J. Immunol. Methods 1997, 209, 17–24. [Google Scholar] [CrossRef]
- Keenan, J.; Day, T.; Neal, S.; Cook, B.; Perez-Perez, G.; Allardyce, R.; Bagshaw, P. A role for the bacterial outer membrane in the pathogenesis of Helicobacter pylori infection. FEMS Microbiol. Lett. 2000, 182, 259–264. [Google Scholar] [CrossRef]
- Hynes, S.O.; Keenan, J.I.; Ferris, J.A.; Annuk, H.; Moran, A.P. Lewis Epitopes on Outer Membrane Vesicles of Relevance to Helicobacter pylori Pathogenesis. Helicobacter 2005, 10, 146–156. [Google Scholar] [CrossRef]
- Choi, H.-I.; Choi, J.-P.; Seo, J.; Kim, B.J.; Rho, M.; Han, J.K.; Kim, J.G. Helicobacter pylori-derived extracellular vesicles increased in the gastric juices of gastric adenocarcinoma patients and induced inflammation mainly via specific targeting of gastric epithelial cells. Exp. Mol. Med. 2017, 49, e330. [Google Scholar] [CrossRef]
- Grande, R.; Di Marcantonio, M.C.; Robuffo, I.; Pompilio, A.; Celia, C.; Di Marzio, L.; Paolino, D.; Codagnone, M.; Muraro, R.; Stoodley, P.; et al. Helicobacter pylori ATCC 43629/NCTC 11639 Outer Membrane Vesicles (OMVs) from Biofilm and Planktonic Phase Associated with Extracellular DNA (eDNA). Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef]
- Heczko, U.; Smith, V.C.; Meloche, R.M.; Buchan, A.M.J.; Finlay, B.B. Characteristics of Helicobacter pylori attachment to human primary antral epithelial cells. Microbes Infect. 2000, 2, 1669–1676. [Google Scholar] [CrossRef]
- Sommi, P.; Ricci, V.; Fiocca, R.; Necchi, V.; Romano, M.; Telford, J.L.; Solcia, E.; Ventura, U. Persistence of Helicobacter pylori VacA toxin and vacuolating potential in cultured gastric epithelial cells. Am. J. Physiol. Content 1998, 275, G681–G688. [Google Scholar] [CrossRef]
- Yonezawa, H.; Osaki, T.; Woo, T.; Kurata, S.; Zaman, C.; Hojo, F.; Hanawa, T.; Kato, S.; Kamiya, S. Analysis of outer membrane vesicle protein involved in biofilm formation of Helicobacter pylori. Anaerobe 2011, 17, 388–390. [Google Scholar] [CrossRef]
- Yonezawa, H.; Osaki, T.; Fukutomi, T.; Hanawa, T.; Kurata, S.; Zaman, C.; Hojo, F.; Kamiya, S. Diversification of the AlpB Outer Membrane Protein of Helicobacter pylori Affects Biofilm Formation and Cellular Adhesion. J. Bacteriol. 2016, 199, e00729-16. [Google Scholar] [CrossRef]
- Yonezawa, H.; Osaki, T.; Kurata, S.; Fukuda, M.; Kawakami, H.; Ochiai, K.; Hanawa, T.; Kamiya, S. Outer Membrane Vesicles of Helicobacter pylori TK1402 are Involved in Biofilm Formation. BMC Microbiol. 2009, 9, 197. [Google Scholar] [CrossRef]
- Ronci, M.; Del Prete, S.; Puca, V.; Carradori, S.; Carginale, V.; Muraro, R.; Mincione, G.; Aceto, A.; Sisto, F.; Supuran, C.T.; et al. Identification and characterization of the α-CA in the outer membrane vesicles produced by Helicobacter pylori. J. Enzym. Inhib. Med. Chem. 2019, 34, 189–195. [Google Scholar] [CrossRef]
- Rozo, A.J.; Cox, M.H.; Devitt, A.; Rothnie, A.J.; Goddard, A.D. Biophysical analysis of lipidic nanoparticles. Methods 2020. [Google Scholar] [CrossRef]
- Mullaney, E.; Brown, P.; Smith, S.M.; Botting, C.H.; Yamaoka, Y.; Terres, A.M.; Kelleher, D.P.; Windle, H.J. Proteomic and functional characterization of the outer membrane vesicles from the gastric pathogen Helicobacter pylori. Proteom. Clin. Appl. 2009, 3, 785–796. [Google Scholar] [CrossRef]
- Zavan, L.; Bitto, N.J.; Johnston, E.L.; Greening, D.W.; Kaparakis-Liaskos, M. Helicobacter pylori Growth Stage Determines the Size, Protein Composition, and Preferential Cargo Packaging of Outer Membrane Vesicles. Proteomics 2018, 19, e1800209. [Google Scholar] [CrossRef]
- Turner, L.; Bitto, N.J.; Steer, D.L.; Lo, C.; D’Costa, K.; Ramm, G.; Shambrook, M.; Hill, A.F.; Ferrero, R.L.; Kaparakis-Liaskos, M. Helicobacter pylori Outer Membrane Vesicle Size Determines Their Mechanisms of Host Cell Entry and Protein Content. Front. Immunol. 2018, 9, 9. [Google Scholar] [CrossRef]
- Olofsson, A.; Vallström, A.; Petzold, K.; Tegtmeyer, N.; Schleucher, J.; Carlsson, S.; Haas, R.; Backert, S.; Wai, S.N.; Gröbner, G.; et al. Biochemical and functional characterization of Helicobacter pylori vesicles. Mol. Microbiol. 2010, 77, 1539–1555. [Google Scholar] [CrossRef]
- Turner, L.; Praszkier, J.; Hutton, M.L.; Steer, D.; Ramm, G.; Kaparakis-Liaskos, M.; Ferrero, R.L. Increased Outer Membrane Vesicle Formation in a Helicobacter pylori tolB Mutant. Helicobacter 2015, 20, 269–283. [Google Scholar] [CrossRef]
- Penfold, C.N.; Li, C.; Zhang, Y.; Vankemmelbeke, M.; James, R. Colicin A binds to a novel binding site of TolA in the Escherichia coli periplasm. Biochem. Soc. Trans. 2012, 40, 1469–1474. [Google Scholar] [CrossRef]
- Godlewska, R.; Wiśniewska, K.; Pietras, Z.; Jagusztyn-Krynicka, E.K. Peptidoglycan-associated lipoprotein (Pal) of Gram-negative bacteria: Function, structure, role in pathogenesis and potential application in immunoprophylaxis. FEMS Microbiol. Lett. 2009, 298, 1–11. [Google Scholar] [CrossRef]
- Liu, Q.; Li, X.; Zhang, Y.; Song, Z.; Li, R.; Ruan, H.; Huang, X. Orally-administered outer-membrane vesicles from Helicobacter pylori reduce H. pylori infection via Th2-biased immune responses in mice. Pathog. Dis. 2019, 77. [Google Scholar] [CrossRef]
- Yonezawa, H.; Osaki, T.; Kamiya, S. Biofilm Formation by Helicobacter pylori and Its Involvement for Antibiotic Resistance. BioMed. Res. Int. 2015, 2015, 1–9. [Google Scholar] [CrossRef]
- Carron, M.A.; Tran, V.R.; Sugawa, C.; Coticchia, J.M. Identification of Helicobacter pylori Biofilms in Human Gastric Mucosa. J. Gastrointest. Surg. 2006, 10, 712–717. [Google Scholar] [CrossRef]
- Coticchia, J.; Sugawa, C.; Tran, V.; Gurrola, J.; Kowalski, E.; Carron, M. Presence and Density of Helicobacter pylori Biofilms in Human Gastric Mucosa in Patients with Peptic Ulcer Disease. J. Gastrointest. Surg. 2006, 10, 883–889. [Google Scholar] [CrossRef]
- Skotland, T.; Sandvig, K.; Llorente, A. Lipids in exosomes: Current knowledge and the way forward. Prog. Lipid Res. 2017, 66, 30–41. [Google Scholar] [CrossRef]
- Keenan, J.I.; Davis, K.A.; Beaugie, C.R.; McGovern, J.J.; Moran, A.P. Alterations in Helicobacter pylori outer membrane and outer membrane vesicle-associated lipopolysaccharides under iron-limiting growth conditions. Innate Immun. 2008, 14, 279–290. [Google Scholar] [CrossRef]
- Koeppen, K.; Hampton, T.H.; Jarek, M.; Scharfe, M.; Gerber, S.A.; Mielcarz, D.W.; Demers, E.G.; Dolben, E.L.; Hammond, J.H.; Hogan, D.A.; et al. A Novel Mechanism of Host-Pathogen Interaction through sRNA in Bacterial Outer Membrane Vesicles. PLoS Pathog. 2016, 12, e1005672. [Google Scholar] [CrossRef]
- Polakovicova, I.; Jerez, S.; Wichmann, I.A.; Sandoval-Bórquez, A.; Carrasco-Véliz, N.; Corvalán, A.L. Role of microRNAs and Exosomes in Helicobacter pylori and Epstein-Barr Virus Associated Gastric Cancers. Front. Microbiol. 2018, 9, 636. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, Y.; Song, Z.; Li, R.; Ruan, H.; Liu, Q.; Huang, X. sncRNAs packaged by Helicobacter pylori outer membrane vesicles attenuate IL-8 secretion in human cells. Int. J. Med. Microbiol. 2020, 310, 151356. [Google Scholar] [CrossRef]
- Lee, H.-J. Microbe-Host Communication by Small RNAs in Extracellular Vesicles: Vehicles for Transkingdom RNA Transportation. Int. J. Mol. Sci. 2019, 20, 1487. [Google Scholar] [CrossRef]
- Patton, J.G.; Franklin, J.L.; Weaver, A.M.; Vickers, K.; Zhang, B.; Coffey, R.J.; Ansel, K.M.; Blelloch, R.; Goga, A.; Huang, B.; et al. Biogenesis, delivery, and function of extracellular RNA. J. Extracell. Vesicles 2015, 4, 27494. [Google Scholar] [CrossRef]
- Parker, H.; Chitcholtan, K.; Hampton, M.B.; Keenan, J.I. Uptake of Helicobacter pylori Outer Membrane Vesicles by Gastric Epithelial Cells. Infect. Immun. 2010, 78, 5054–5061. [Google Scholar] [CrossRef]
- Olofsson, A.; Skalman, L.N.; Obi, I.; Lundmark, R.; Arnqvist, A. Uptake of Helicobacter pylori Vesicles Is Facilitated by Clathrin-Dependent and Clathrin-Independent Endocytic Pathways. mBio 2014, 5, e00979-14. [Google Scholar] [CrossRef]
- Irving, A.T.; Mimuro, H.; Kufer, T.A.; Lo, C.Y.; Wheeler, R.; Turner, L.; Thomas, B.J.; Malosse, C.; Gantier, M.P.; Casillas, L.N.; et al. The Immune Receptor NOD1 and Kinase RIP2 Interact with Bacterial Peptidoglycan on Early Endosomes to Promote Autophagy and Inflammatory Signaling. Cell Host Microbe 2014, 15, 623–635. [Google Scholar] [CrossRef]
- Heusermann, W.; Hean, J.; Trojer, D.; Steib, E.; Von Bueren, S.; Graff-Meyer, A.; Genoud, C.; Martin, K.; Pizzato, N.; Voshol, J.; et al. Exosomes surf on filopodia to enter cells at endocytic hot spots, traffic within endosomes, and are targeted to the ER. J. Cell Biol. 2016, 213, 173–184. [Google Scholar] [CrossRef]
- Chitcholtan, K.; Hampton, M.B.; Keenan, J.I. Outer membrane vesicles enhance the carcinogenic potential of Helicobacter pylori. Carcinogenesis 2008, 29, 2400–2405. [Google Scholar] [CrossRef]
- Van Der Pol, L.; Stork, M.; Van Der Ley, P. Outer membrane vesicles as platform vaccine technology. Biotechnol. J. 2015, 10, 1689–1706. [Google Scholar] [CrossRef]
- Lekmeechai, S.; Su, Y.-C.; Brant, M.; Alvarado-Kristensson, M.; Vallström, A.; Obi, I.; Arnqvist, A.; Riesbeck, K. Helicobacter pylori Outer Membrane Vesicles Protect the Pathogen From Reactive Oxygen Species of the Respiratory Burst. Front. Microbiol. 2018, 9, 9. [Google Scholar] [CrossRef]
- Eaton, K.A.; Brooks, C.L.; Morgan, D.R.; Krakowka, S. Essential role of urease in pathogenesis of gastritis induced by Helicobacter pylori in gnotobiotic piglets. Infect. Immun. 1991, 59, 2470–2475. [Google Scholar] [CrossRef]
- Hathroubi, S.; Servetas, S.L.; Windham, I.; Merrell, D.S.; Ottemann, K.M. Helicobacter pylori Biofilm Formation and Its Potential Role in Pathogenesis. Microbiol. Mol. Biol. Rev. 2018, 82, e00001-18. [Google Scholar] [CrossRef]
- Winter, J.; Letley, D.; Rhead, J.; Atherton, J.; Robinson, K. Helicobacter pylori Membrane Vesicles Stimulate Innate Pro- and Anti-Inflammatory Responses and Induce Apoptosis in Jurkat T Cells. Infect. Immun. 2014, 82, 1372–1381. [Google Scholar] [CrossRef]
- Ayala, G.; Torres, L.; Espinosa, M.; Fierros-Zarate, G.; Maldonado, V.; Melendez-Zajgla, J. External membrane vesicles from Helicobacter pylori induce apoptosis in gastric epithelial cells. FEMS Microbiol. Lett. 2006, 260, 178–185. [Google Scholar] [CrossRef]
- Ilver, D.; Barone, S.; Mercati, D.; Lupetti, P.; Telford, J.L. Helicobacter pylori toxin VacA is transferred to host cells via a novel contact-dependent mechanism. Cell. Microbiol. 2004, 6, 167–174. [Google Scholar] [CrossRef]
- Molinari, M.; Galli, C.; Norais, N.; Telford, J.L.; Rappuoli, R.; Luzio, J.P.; Montecucco, C. Vacuoles Induced by Helicobacter pylori Toxin Contain Both Late Endosomal and Lysosomal Markers. J. Biol. Chem. 1997, 272, 25339–25344. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Wu, J.-J.; Lei, H.-Y. The Autophagic Induction in Helicobacter pylori-Infected Macrophage. Exp. Biol. Med. 2009, 234, 171–180. [Google Scholar] [CrossRef]
- Galmiche, A.; Rassow, J.; Doye, A.; Cagnol, S.; Chambard, J.C.; Contamin, S.; De Thillot, V.; Just, I.; Ricci, V.; Solcia, E.; et al. The N-terminal 34 kDa fragment of Helicobacter pylori vacuolating cytotoxin targets mitochondria and induces cytochrome c release. EMBO J. 2000, 19, 6361–6370. [Google Scholar] [CrossRef]
- Papini, E.; Satin, B.; Norais, N.; De Bernard, M.; Telford, J.L.; Rappuoli, R.; Montecucco, C. Selective increase of the permeability of polarized epithelial cell monolayers by Helicobacter pylori vacuolating toxin. J. Clin. Investig. 1998, 102, 813–820. [Google Scholar] [CrossRef]
- Keenan, J.; Allardyce, R.A. Iron influences the expression of Helicobacter pylori outer membrane vesicle-associated virulence factors. Eur. J. Gastroenterol. Hepatol. 2000, 12, 1267–1273. [Google Scholar] [CrossRef]
- Ricci, V.; Chiozzi, V.; Necchi, V.; Oldani, A.; Romano, M.; Solcia, E.; Ventura, U. Free-soluble and outer membrane vesicle-associated VacA from Helicobacter pylori: Two forms of release, a different activity. Biochem. Biophys. Res. Commun. 2005, 337, 173–178. [Google Scholar] [CrossRef]
- Yamasaki, E.; Wada, A.; Kumatori, A.; Nakagawa, I.; Funao, J.; Nakayama, M.; Hisatsune, J.; Kimura, M.; Moss, J.; Hirayama, T. Helicobacter pylori Vacuolating Cytotoxin Induces Activation of the Proapoptotic Proteins Bax and Bak, Leading to Cytochrome c Release and Cell Death, Independent of Vacuolation. J. Biol. Chem. 2006, 281, 11250–11259. [Google Scholar] [CrossRef]
- Hoy, B.; Löwer, M.; Weydig, C.; Carra, G.; Tegtmeyer, N.; Geppert, T.; Schröder, P.; Sewald, N.; Backert, S.; Schneider, G.; et al. Helicobacter pylori HtrA is a new secreted virulence factor that cleaves E-cadherin to disrupt intercellular adhesion. EMBO Rep. 2010, 11, 798–804. [Google Scholar] [CrossRef]
- Tegtmeyer, N.; Wessler, S.; Necchi, V.; Rohde, M.; Harrer, A.; Rau, T.T.; Asche, C.I.; Boehm, M.; Loessner, H.; Figueiredo, C.; et al. Helicobacter pylori Employs a Unique Basolateral Type IV Secretion Mechanism for CagA Delivery. Cell Host Microbe 2017, 22, 552.e5–560.e5. [Google Scholar] [CrossRef]
- Zhang, G.; Ducatelle, R.; Pasmans, F.; D’Herde, K.; Huang, L.; Smet, A.; Haesebrouck, F.; Flahou, B. Effects of Helicobacter suis γ-Glutamyl Transpeptidase on Lymphocytes: Modulation by Glutamine and Glutathione Supplementation and Outer Membrane Vesicles as a Putative Delivery Route of the Enzyme. PLoS ONE 2013, 8, e77966. [Google Scholar] [CrossRef]
- Hatakeyama, M. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat. Rev. Cancer 2004, 4, 688–694. [Google Scholar] [CrossRef]
- Poppe, M.; Feller, S.M.; Romer, G.; Wessler, S. Phosphorylation of Helicobacter pylori CagA by c-Abl leads to cell motility. Oncogene 2006, 26, 3462–3472. [Google Scholar] [CrossRef]
- Mueller, D.; Tegtmeyer, N.; Brandt, S.; Yamaoka, Y.; De Poire, E.; Sgouras, D.; Wessler, S.; Torres, J.; Smolka, A.; Backert, S. c-Src and c-Abl kinases control hierarchic phosphorylation and function of the CagA effector protein in Western and East Asian Helicobacter pylori strains. J. Clin. Investig. 2012, 122, 1553–1566. [Google Scholar] [CrossRef]
- Krisch, L.M.; Posselt, G.; Hammerl, P.; Wessler, S. CagA Phosphorylation in Helicobacter pylori-Infected B Cells Is Mediated by the Nonreceptor Tyrosine Kinases of the Src and Abl Families. Infect. Immun. 2016, 84, 2671–2680. [Google Scholar] [CrossRef]
- Selbach, M.; Moese, S.; Hauck, C.R.; Meyer, T.F.; Backert, S. Src Is the Kinase of the Helicobacter pylori CagA Protein in Vitro and in Vivo. J. Biol. Chem. 2002, 277, 6775–6778. [Google Scholar] [CrossRef]
- Stein, M.; Bagnoli, F.; Halenbeck, R.; Rappuoli, R.; Fantl, W.J.; Covacci, A. c-Src/Lyn kinases activate Helicobacter pylori CagA through tyrosine phosphorylation of the EPIYA motifs. Mol. Microbiol. 2002, 43, 971–980. [Google Scholar] [CrossRef]
- Selbach, M.; Paul, F.E.; Brandt, S.; Guye, P.; Daumke, O.; Backert, S.; Dehio, C.; Mann, M. Host Cell Interactome of Tyrosine-Phosphorylated Bacterial Proteins. Cell Host Microbe 2009, 5, 397–403. [Google Scholar] [CrossRef]
- Shimoda, A.; Ueda, K.; Nishiumi, S.; Murata-Kamiya, N.; Mukai, S.-A.; Sawada, S.-I.; Azuma, T.; Hatakeyama, M.; Akiyoshi, K. Exosomes as nanocarriers for systemic delivery of the Helicobacter pylori virulence factor CagA. Sci. Rep. 2016, 6, 18346. [Google Scholar] [CrossRef]
- Ko, S.H.; Rho, D.J.; Jeon, J.I.; Kim, Y.-J.; Woo, H.A.; Kim, N.; Kim, J.M. Crude Preparations of Helicobacter pylori Outer Membrane Vesicles Induce Upregulation of Heme Oxygenase-1 via Activating Akt-Nrf2 and mTOR–IκB Kinase–NF-κB Pathways in Dendritic Cells. Infect. Immun. 2016, 84, 2162–2174. [Google Scholar] [CrossRef]
- Turkina, M.V.; Olofsson, A.; Magnusson, K.-E.; Arnqvist, A.; Vikström, E. Helicobacter pylori vesicles carrying CagA localize in the vicinity of cell–cell contacts and induce histone H1 binding to ATP in epithelial cells. FEMS Microbiol. Lett. 2015, 362, 362. [Google Scholar] [CrossRef]
- Fischer, W.; Püls, J.; Buhrdorf, R.; Gebert, B.; Odenbreit, S.; Haas, R. Systematic mutagenesis of the Helicobacter pylori cag pathogenicity island: Essential genes for CagA translocation in host cells and induction of interleukin-8. Mol. Microbiol. 2002, 42, 1337–1348. [Google Scholar] [CrossRef]
- Viala, J.; Chaput, C.; Boneca, I.G.; Cardona, A.; Girardin, S.E.; Moran, A.P.; Athman, R.; Mémet, S.; Huerre, M.R.; Coyle, A.J.; et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat. Immunol. 2004, 5, 1166–1174. [Google Scholar] [CrossRef]
- Pfannkuch, L.; Hurwitz, R.; Traulsen, J.; Sigulla, J.; Poeschke, M.; Matzner, L.; Kosma, P.; Schmid, M.; Meyer, T.F. ADP heptose, a novel pathogen-associated molecular pattern identified in Helicobacter pylori. FASEB J. 2019, 33, 9087–9099. [Google Scholar] [CrossRef]
- Zimmermann, S.; Pfannkuch, L.; Al-Zeer, M.A.; Bartfeld, S.; Koch, M.; Liu, J.; Rechner, C.; Soerensen, M.; Sokolova, O.; Zamyatina, A.; et al. ALPK1- and TIFA-Dependent Innate Immune Response Triggered by the Helicobacter pylori Type IV Secretion System. Cell Rep. 2017, 20, 2384–2395. [Google Scholar] [CrossRef]
- Ko, S.H.; Jeon, J.I.; Kim, Y.-J.; Yoon, H.J.; Kim, H.; Kim, N.; Kim, J.S.; Kim, J.M. Helicobacter pylori Outer Membrane Vesicle Proteins Induce Human Eosinophil Degranulation via a β2 Integrin CD11/CD18- and ICAM-1-Dependent Mechanism. Mediat. Inflamm. 2015, 2015, 301716. [Google Scholar] [CrossRef]
- Hock, B.D.; McKenzie, J.L.; Keenan, J.I. Helicobacter pylori outer membrane vesicles inhibit human T cell responses via induction of monocyte COX-2 expression. Pathog. Dis. 2017, 75. [Google Scholar] [CrossRef]
- Keenan, J.I.; Allardyce, R.; Bagshaw, P.F. Lack of protection following immunisation with H. pylori outer membrane vesicles highlights antigenic differences between H. felis and H. pylori. FEMS Microbiol. Lett. 1998, 161, 21–27. [Google Scholar] [CrossRef][Green Version]
- Keenan, J.; Oliaro, J.; Domigan, N.; Potter, H.; Aitken, G.; Allardyce, R.; Roake, J. Immune Response to an 18-Kilodalton Outer Membrane Antigen Identifies Lipoprotein 20 as a Helicobacter pylori Vaccine Candidate. Infect. Immun. 2000, 68, 3337–3343. [Google Scholar] [CrossRef][Green Version]
- Keenan, J.; Rijpkema, S.; Durrani, Z.; Roake, J.A. Differences in immunogenicity and protection in mice and guinea pigs following intranasal immunization with Helicobacter pylori outer membrane antigens. FEMS Immunol. Med. Microbiol. 2003, 36, 199–205. [Google Scholar] [CrossRef]
- Jain, S.; Pillai, J. Bacterial membrane vesicles as novel nanosystems for drug delivery. Int. J. Nanomed. 2017, 12, 6329–6341. [Google Scholar] [CrossRef]
- Pathirana, R.D.; Kaparakis-Liaskos, M. Bacterial membrane vesicles: Biogenesis, immune regulation and pathogenesis. Cell. Microbiol. 2016, 18, 1518–1524. [Google Scholar] [CrossRef]
- Tan, K.; Li, R.; Huang, X.; Liu, Q. Outer Membrane Vesicles: Current Status and Future Direction of These Novel Vaccine Adjuvants. Front. Microbiol. 2018, 9, 783. [Google Scholar] [CrossRef]
- Irene, C.; Fantappiè, L.; Caproni, E.; Zerbini, F.; Anesi, A.; Tomasi, M.; Zanella, I.; Stupia, S.; Prete, S.; Valensin, S.; et al. Bacterial outer membrane vesicles engineered with lipidated antigens as a platform for Staphylococcus aureus vaccine. Proc. Natl. Acad. Sci. USA 2019, 116, 21780–21788. [Google Scholar] [CrossRef]
- Toledo, A.; Coleman, J.L.; Kuhlow, C.J.; Crowley, J.T.; Benach, J.L. The Enolase of Borrelia burgdorferi Is a Plasminogen Receptor Released in Outer Membrane Vesicles. Infect. Immun. 2011, 80, 359–368. [Google Scholar] [CrossRef]
- Gankema, H.; Wensink, J.; Guinée, P.A.M.; Jansen, W.H.; Witholt, B. Some characteristics of the outer membrane material released by growing enterotoxigenic Escherichia coli. Infect. Immun. 1980, 29, 704–713. [Google Scholar]
- Horstman, A.L.; Kuehn, M.J. Enterotoxigenic Escherichia coli Secretes Active Heat-labile Enterotoxin via Outer Membrane Vesicles. J. Biol. Chem. 2000, 275, 12489–12496. [Google Scholar] [CrossRef]
- Kato, S.; Kowashi, Y.; DeMuth, D.R. Outer membrane-like vesicles secreted by Actinobacillus actinomycetemcomitans are enriched in leukotoxin. Microb. Pathog. 2002, 32, 1–13. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jarzab, M.; Posselt, G.; Meisner-Kober, N.; Wessler, S. Helicobacter pylori-Derived Outer Membrane Vesicles (OMVs): Role in Bacterial Pathogenesis? Microorganisms 2020, 8, 1328. https://doi.org/10.3390/microorganisms8091328
Jarzab M, Posselt G, Meisner-Kober N, Wessler S. Helicobacter pylori-Derived Outer Membrane Vesicles (OMVs): Role in Bacterial Pathogenesis? Microorganisms. 2020; 8(9):1328. https://doi.org/10.3390/microorganisms8091328
Chicago/Turabian StyleJarzab, Miroslaw, Gernot Posselt, Nicole Meisner-Kober, and Silja Wessler. 2020. "Helicobacter pylori-Derived Outer Membrane Vesicles (OMVs): Role in Bacterial Pathogenesis?" Microorganisms 8, no. 9: 1328. https://doi.org/10.3390/microorganisms8091328
APA StyleJarzab, M., Posselt, G., Meisner-Kober, N., & Wessler, S. (2020). Helicobacter pylori-Derived Outer Membrane Vesicles (OMVs): Role in Bacterial Pathogenesis? Microorganisms, 8(9), 1328. https://doi.org/10.3390/microorganisms8091328