Structural Insight into CVB3-VLP Non-Adjuvanted Vaccine

 ,
, 
 , , , ,
, , , ,  ,
,  and
and 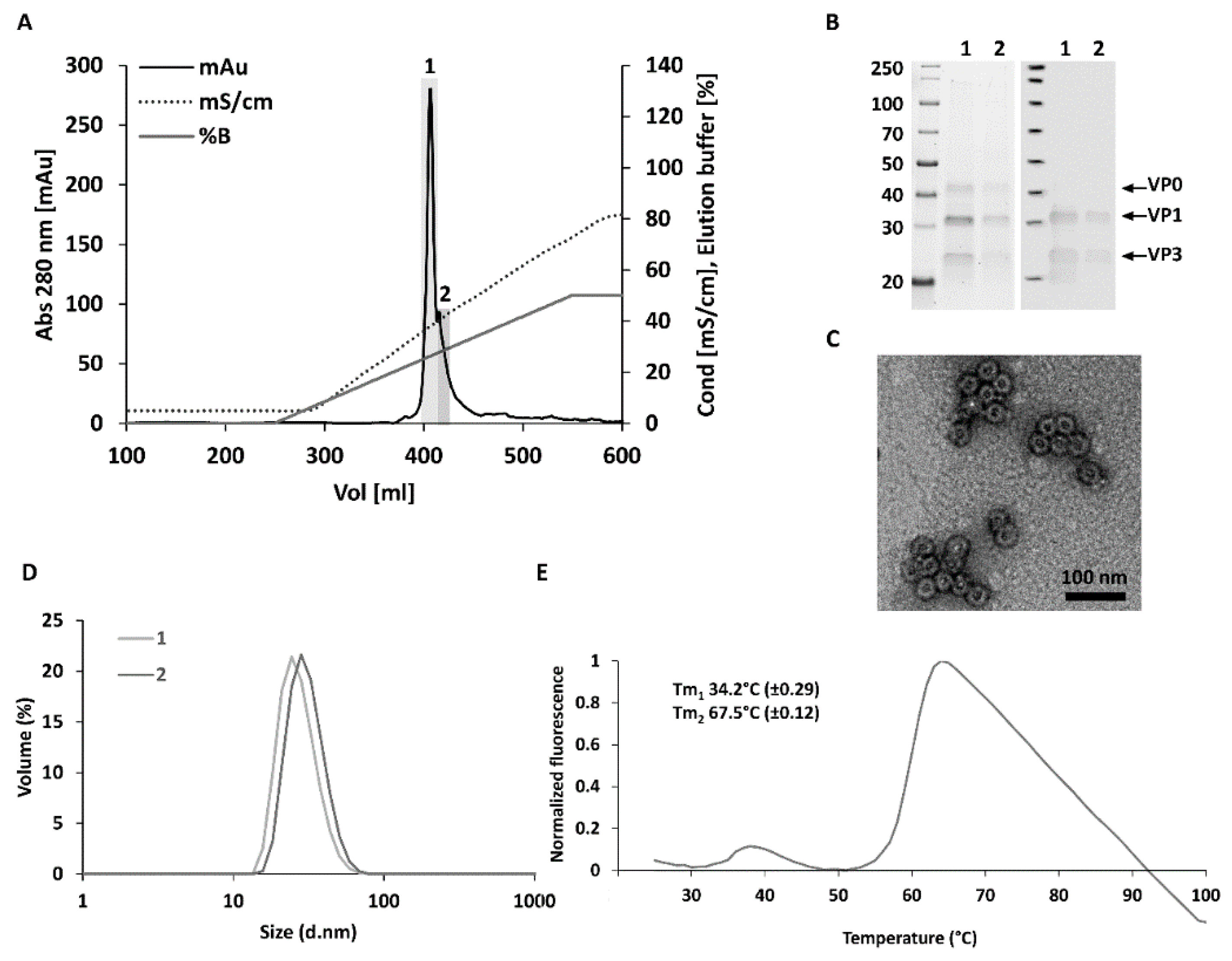{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Generation of the CVB3-VLP-Encoding Baculoviruses
2.2. Production, Purification and Chracterization of CVB3-VLPs
2.3. Cryo-Electron Microscopy Structural Determination of CVB3-VLP
2.4. Mouse Immunizations
2.5. CVB3-VLP Specific ELISA and Neutralization Assay
3. Results
3.1. Optimized Construct Design, Production Yields and Purity for CVB3-VLP
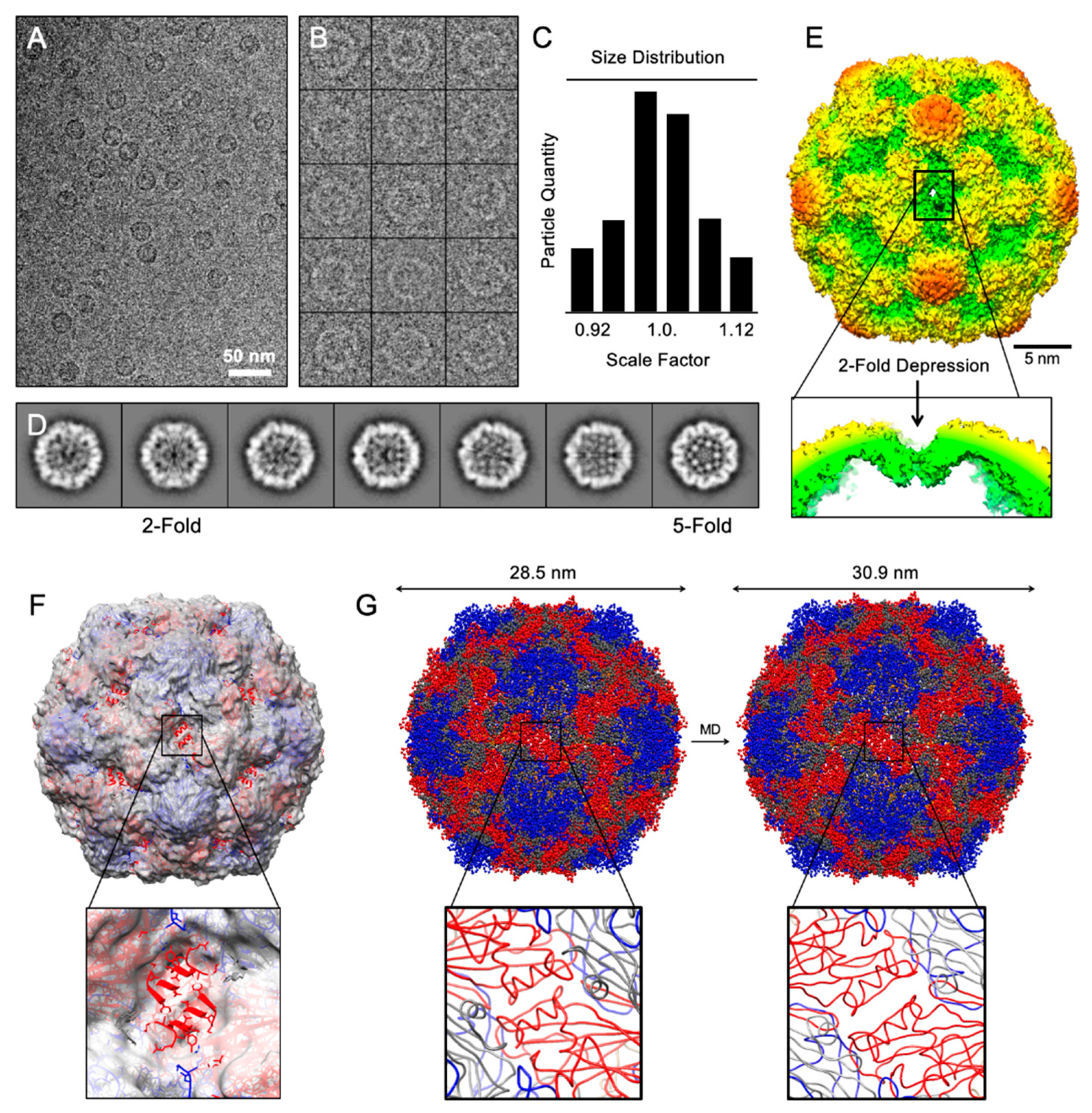
3.2. Structural Analysis of CVB3-VLP
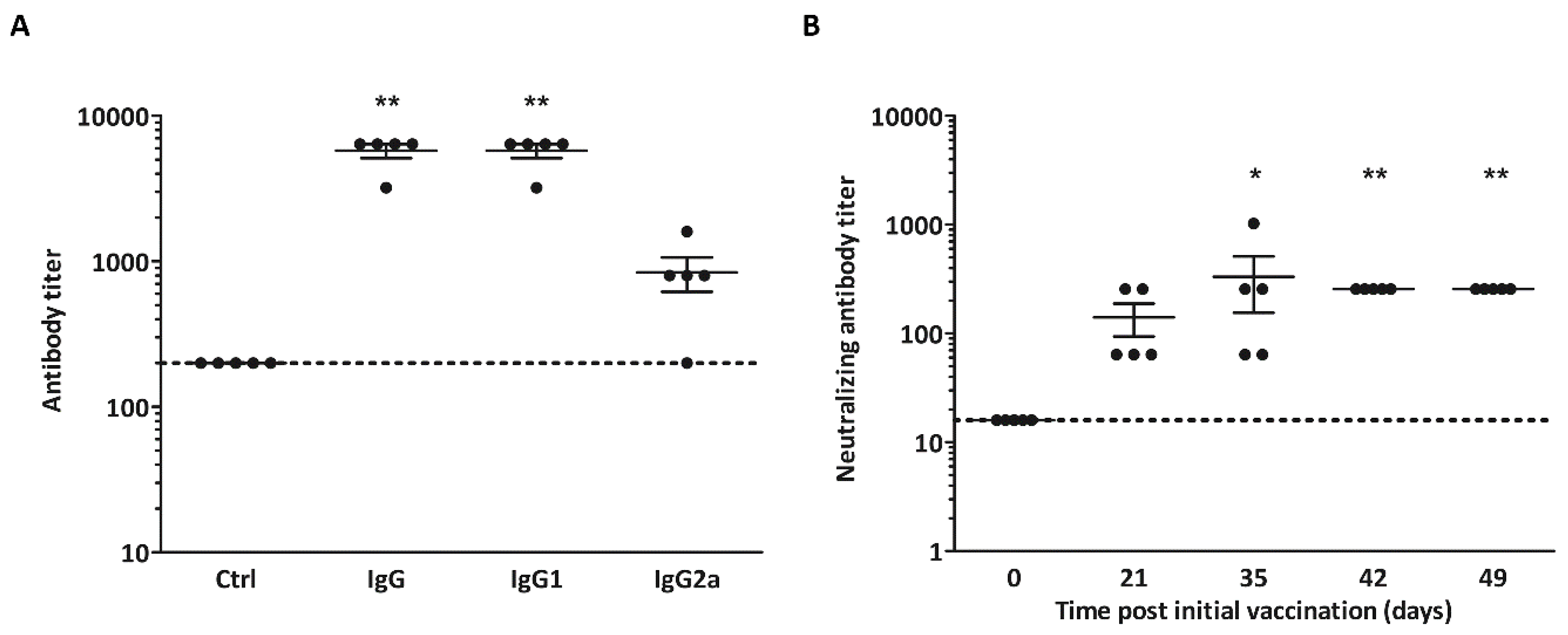
3.3. Non-Adjuvanted CVB3-VLPs Induce CVB3-Specific IgG, IgG1-, IgG2a and Neutralizing Antibody Responses in C57BL/6J Mice
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sin, J.; Mangale, V.; Thienphrapa, W.; Gottlieb, R.A.; Feuer, R. Recent progress in understanding coxsackievirus replication, dissemination, and pathogenesis. Virology 2015, 484, 288–304. [Google Scholar] [CrossRef]
- Peischard, S.; Ho, H.T.; Theiss, C.; Strutz-Seebohm, N.; Seebohm, G. A kidnapping story: How Coxsackievirus B3 and its host cell interact. Cell. Physiol. Biochem. 2019, 53, 121–140. [Google Scholar] [CrossRef]
- Han, Z.; Zhang, Y.; Huang, K.; Wang, J.; Tian, H.; Song, Y.; Yang, Q.; Yan, D.; Zhu, S.; Yao, M.; et al. Two COXSACKIEVIRUS B3 outbreaks associated with hand, foot, and mouth disease in China and the evolutionary history worldwide. BMC Infect. Dis. 2019, 19, 466. [Google Scholar] [CrossRef] [PubMed]
- Laitinen, O.H.; Honkanen, H.; Pakkanen, O.; Oikarinen, S.; Hankaniemi, M.M.; Huhtala, H.; Ruokoranta, T.; Lecouturier, V.; Andre, P.; Harju, R.; et al. Coxsackievirus B1 is associated with induction of beta-cell autoimmunity that portends type 1 diabetes. Diabetes 2014, 63, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Sioofy-Khojine, A.; Lehtonen, J.; Nurminen, N.; Laitinen, O.H.; Oikarinen, S.; Huhtala, H.; Pakkanen, O.; Ruokoranta, T.; Hankaniemi, M.M.; Toppari, J.; et al. Coxsackievirus B1 infections are associated with the initiation of insulin-driven autoimmunity that progresses to type 1 diabetes. Diabetologia 2018, 61, 1193–1202. [Google Scholar] [CrossRef] [PubMed]
- Hyöty, H.; Leon, F.; Knip, M. Developing a vaccine for type 1 diabetes by targeting coxsackievirus B. Expert Rev. Vaccines 2018, 17, 1071–1083. [Google Scholar] [CrossRef] [PubMed]
- Dunne, J.L.; Richardson, S.J.; Atkinson, M.A.; Craig, M.E.; Dahl-Jørgensen, K.; Flodström-Tullberg, M.; Hyöty, H.; Insel, R.A.; Lernmark, Å.; Lloyd, R.E.; et al. Rationale for enteroviral vaccination and antiviral therapies in human type 1 diabetes. Diabetologia 2019, 62, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Stone, V.M.; Hankaniemi, M.M.; Laitinen, O.H.; Sioofy-Khojine, A.B.; Lin, A.; Lozano, I.M.D.; Mazur, M.A.; Marjomäki, V.; Loré, K.; Hyöty, H.; et al. A hexavalent Coxsackievirus B vaccine is highly immunogenic and has a strong protective capacity in mice and nonhuman primates. Sci. Adv. 2020, 6, eaaz2433. [Google Scholar] [CrossRef]
- Lien, G.; Heymann, D.L. The problems with polio: Toward eradication. Infect. Dis. Ther. 2013, 2, 167–174. [Google Scholar] [CrossRef]
- Chang, P.-C.; Chen, S.-C.; Chen, K.-T. The current status of the disease caused by enterovirus 71 infections: Epidemiology, pathogenesis, molecular epidemiology, and vaccine development. Int. J. Environ. Res. Public Health 2016, 13, 890. [Google Scholar] [CrossRef]
- Lukashev, A.N. Recombination among picornaviruses. Rev. Med. Virol. 2010, 20, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Parham, N.; Zhang, F.; Aasa-Chapman, M.M.I.; Gould, E.; Zhang, H. Vaccination with coxsackievirus B3 virus-like particles elicits humoral immune response and protects mice against myocarditis. Vaccine 2012, 30, 2301–2308. [Google Scholar] [CrossRef] [PubMed]
- Koho, T.; Koivunen, M.R.; Oikarinen, S.; Kummola, L.; Mäkinen, S.; Mähönen, A.J.; Sioofy-Khojine, A.-B.; Marjomäki, V.S.; Kazmertsuk, A.; Junttila, I.S.; et al. Coxsackievirus B3 VLPs purified by ion exchange chromatography elicit strong immune responses in mice. Antivir. Res. 2014, 104, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Salmons, B.; Lim, P.-Y.; Djurup, R.; Cardosa, J. Non-clinical safety assessment of repeated intramuscular administration of an EV-A71 VLP vaccine in rabbits. Vaccine 2018, 36, 6623–6630. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Ku, Z.; Zhou, Y.; Li, D.; Wang, L.; Lan, K.; Liu, Q.; Huang, Z. Virus-like particle-based vaccine against coxsackievirus A6 protects mice against lethal infections. Vaccine 2016, 34, 4025–4031. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Yan, K.; Feng, Y.; Huang, X.; Ku, Z.; Cai, Y.; Liu, F.; Shi, J.; Huang, Z. A virus-like particle vaccine for coxsackievirus A16 potently elicits neutralizing antibodies that protect mice against lethal challenge. Vaccine 2012, 30, 6642–6648. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Zhang, C.; Zhang, X.; Xiong, P.; Liu, Q.; Gong, S.; Geng, L.; Zhou, D.; Huang, Z. A virus-like particle vaccine confers protection against enterovirus D68 lethal challenge in mice. Vaccine 2018, 36, 653–659. [Google Scholar] [CrossRef]
- Zhang, W.; Dai, W.; Zhang, C.; Zhou, Y.; Xiong, P.; Wang, S.; Ye, X.; Liu, Q.; Zhou, D.; Huang, Z. A virus-like particle-based tetravalent vaccine for hand, foot, and mouth disease elicits broad and balanced protective immunity. Emerg. Microbes Infect. 2018, 7. [Google Scholar] [CrossRef]
- Hankaniemi, M.M.; Stone, V.M.; Andrejeff, T.; Heinimäki, S.; Sioofy-Khojine, A.-B.; Marjomäki, V.; Hyöty, H.; Blazevic, V.; Flodström-Tullberg, M.; Hytönen, V.P.; et al. Formalin treatment increases the stability and immunogenicity of coxsackievirus B1 VLP vaccine. Antivir. Res. 2019, 171, 104595. [Google Scholar] [CrossRef]
- Laitinen, O.H.; Svedin, E.; Kapell, S.; Nurminen, A.; Hytönen, V.P.; Flodström-Tullberg, M. Enteroviral proteases: Structure, host interactions and pathogenicity. Rev. Med. Virol. 2016, 26, 251–267. [Google Scholar] [CrossRef]
- Muckelbauer, J.K.; Kremer, M.; Minor, I.; Diana, G.; Dutko, F.J.; Groarke, J.; Pevear, D.C.; Rossmann, M.G. The structure of coxsackievirus B3 at 3.5 å resolution. Structure 1995, 3, 653–667. [Google Scholar] [CrossRef]
- Lyu, K.; Ding, J.; Han, J.-F.; Zhang, Y.; Wu, X.-Y.; He, Y.-L.; Qin, C.-F.; Chen, R. Human enterovirus 71 uncoating captured at atomic resolution. J. Virol. 2013, 88, 3114–3126. [Google Scholar] [CrossRef] [PubMed]
- Fricks, C.E.; Hogle, J.M. Cell-induced conformational change in poliovirus: Externalization of the amino terminus of VP1 is responsible for liposome binding. J. Virol. 1990, 64, 1934–1945. [Google Scholar] [CrossRef] [PubMed]
- De Sena, J.; Mandel, B. Studies on the in vitro uncoating of poliovirus. Virology 1976, 70, 470–483. [Google Scholar] [CrossRef]
- Lin, S.-Y.; Yeh, C.-T.; Li, W.-H.; Yu, C.-P.; Lin, W.-C.; Yang, J.-Y.; Wu, H.-L.; Hu, Y.-C. Enhanced enterovirus 71 virus-like particle yield from a new baculovirus design. Biotechnol. Bioeng. 2015, 112, 2005–2015. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Hankaniemi, M.M.; Laitinen, O.H.; Stone, V.M.; Sioofy-Khojine, A.-B.; Määttä, J.; Larsson, P.G.; Marjomäki, V.S.; Hyöty, H.; Flodström-Tullberg, M.; Hytönen, V.P. Optimized production and purification of Coxsackievirus B1 vaccine and its preclinical evaluation in a mouse model. Vaccine 2017, 35, 3718–3725. [Google Scholar] [CrossRef]
- Tang, G.; Peng, L.; Baldwin, P.R.; Mann, D.S.; Jiang, W.; Rees, I.; Ludtke, S.J. EMAN2: An extensible image processing suite for electron microscopy. J. Struct. Biol. 2007, 157, 38–46. [Google Scholar] [CrossRef]
- Baker, T.S.; Cheng, R. A model-based approach for determining orientations of biological macromolecules imaged by cryoelectron microscopy. J. Struct. Biol. 1996, 116, 120–130. [Google Scholar] [CrossRef]
- Guo, F.; Jiang, W. Single particle cryo-electron microscopy and 3-D reconstruction of viruses. Breast Cancer 2014, 1117, 401–443. [Google Scholar] [CrossRef]
- Cheng, R.H.; Olson, N.H.; Baker, T.S. Cauliflower mosaic virus: A 420 subunit (T = 7), multilayer structure. Virology 1992, 186, 655–668. [Google Scholar] [CrossRef]
- Henderson, R.; Sali, A.; Baker, M.L.; Carragher, B.; Devkota, B.; Downing, K.H.; Egelman, E.H.; Feng, Z.; Frank, J.; Grigorieff, N.; et al. Outcome of the first electron microscopy validation task force meeting. Structure 2012, 20, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Stone, V.M.; Hankaniemi, M.M.; Svedin, E.; Sioofy-Khojine, A.-B.; Oikarinen, S.; Hyöty, H.; Laitinen, O.H.; Hytönen, V.P.; Flodström-Tullberg, M. A Coxsackievirus B vaccine protects against virus-induced diabetes in an experimental mouse model of type 1 diabetes. Diabetologia 2017, 61, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Hitchman, R.B.; Possee, R.D.; Crombie, A.T.; Chambers, A.; Ho, K.; Siaterli, E.; Lissina, O.; Sternard, H.; Novy, R.; Loomis, K.; et al. Genetic modification of a baculovirus vector for increased expression in insect cells. Cell Biol. Toxicol. 2009, 26, 57–68. [Google Scholar] [CrossRef]
- Krammer, F.; Schinko, T.; Palmberger, D.; Tauer, C.; Messner, P.; Grabherr, R. Trichoplusia ni cells (High Five) are highly efficient for the production of influenza a virus-like particles: A comparison of two insect cell lines as production platforms for influenza vaccines. Mol. Biotechnol. 2010, 45, 226–234. [Google Scholar] [CrossRef]
- Niesen, F.H.; Berglund, H.; Vedadi, M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007, 2, 2212–2221. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, C.; Zhou, Y.; Zhang, X.; Shen, C.; Ye, X.; Jiang, W.; Huang, Z.; Cong, Y. A 3.0-angstrom resolution cryo-electron microscopy structure and antigenic sites of coxsackievirus A6-like particles. J. Virol. 2018, 92, e01257-17. [Google Scholar] [CrossRef]
- Shingler, K.L.; Yoder, J.L.; Carnegie, M.S.; Ashley, R.E.; Makhov, A.M.; Conway, J.F.; Hafenstein, S. The enterovirus 71 A-particle forms a gateway to allow genome release: A cryoEM study of picornavirus uncoating. PLoS Pathog. 2013, 9, e1003240. [Google Scholar] [CrossRef]
- Wang, X.; Peng, W.; Ren, J.; Hu, Z.; Xu, J.; Lou, Z.; Li, X.; Yin, W.; Shen, X.; Porta, C.; et al. A sensor-adaptor mechanism for enterovirus uncoating from structures of EV71. Nat. Struct. Mol. Biol. 2012, 19, 424–429. [Google Scholar] [CrossRef]
- Ren, J.; Wang, X.; Zhu, L.; Hu, Z.; Gao, Q.; Yang, P.; Li, X.; Wang, J.; Shen, X.; Fry, E.; et al. Structures of Coxsackievirus A16 capsids with native antigenicity: Implications for particle expansion, receptor binding, and immunogenicity. J. Virol. 2015, 89, 10500–10511. [Google Scholar] [CrossRef]
- Ren, J.; Wang, X.; Hu, Z.; Gao, Q.; Sun, Y.; Li, X.; Porta, C.; Walter, T.S.; Gilbert, R.J.C.; Zhao, Y.; et al. Picornavirus uncoating intermediate captured in atomic detail. Nat. Commun. 2013, 4, 1929. [Google Scholar] [CrossRef] [PubMed]
- López-Blanco, J.R.; Chacón, P. iMODFIT: Efficient and robust flexible fitting based on vibrational analysis in internal coordinates. J. Struct. Biol. 2013, 184, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ku, Z.; Zhang, X.; Ye, X.; Chen, J.; Liu, Q.; Zhang, W.; Zhang, C.; Fu, Z.; Jin, X.; et al. Structure, immunogenicity, and protective mechanism of an engineered enterovirus 71-like particle vaccine mimicking 80S empty capsid. J. Virol. 2017, 92, e01330-17. [Google Scholar] [CrossRef] [PubMed]
- Ruokolainen, V.; Domanska, A.; Laajala, M.; Pelliccia, M.; Butcher, S.J.; Marjomäki, V. Extracellular albumin and endosomal ions prime enterovirus particles for uncoating that can be prevented by fatty acid saturation. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Ward, T.; Powell, R.M.; Chaudhry, Y.; Meredith, J.; Almond, J.W.; Kraus, W.; Nelsen-Salz, B.; Eggers, H.J.; Evans, D.J. Fatty acid-depleted albumin induces the formation of echovirus a particles. J. Virol. 2000, 74, 3410–3412. [Google Scholar] [CrossRef]
- Adeyemi, O.O.; Nicol, C.; Stonehouse, N.; Rowlands, D.J. Increasing type 1 poliovirus capsid stability by thermal selection. J. Virol. 2016, 91, e01586-16. [Google Scholar] [CrossRef]
- Fox, H.; Knowlson, S.; Minor, P.D.; Macadam, A. Genetically Thermo-Stabilised, Immunogenic Poliovirus Empty Capsids; a Strategy for Non-replicating Vaccines. PLoS Pathog. 2017, 13, e1006117. [Google Scholar] [CrossRef]
- Cimica, V.; Galarza, J.M. Adjuvant formulations for virus-like particle (VLP) based vaccines. Clin. Immunol. 2017, 183, 99–108. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, X.; Zhang, W.; Dai, W.; Xie, J.; Ye, L.; Wang, H.; Chen, H.; Liu, Q.; Gong, S.; et al. Enterovirus D68 virus-like particles expressed in Pichia pastoris potently induce neutralizing antibody responses and confer protection against lethal viral infection in mice. Emerg. Microbes Infect. 2018, 7. [Google Scholar] [CrossRef]
- Wang, C.; Zheng, X.; Gai, W.; Wong, G.; Wang, H.; Jin, H.; Feng, N.; Zhao, Y.; Zhang, W.; Li, N.; et al. Novel chimeric virus-like particles vaccine displaying MERS-CoV receptor-binding domain induce specific humoral and cellular immune response in mice. Antivir. Res. 2017, 140, 55–61. [Google Scholar] [CrossRef]
- Ku, Z.; Liu, Q.; Ye, X.; Cai, Y.; Wang, X.; Shi, J.; Li, D.; Jin, X.; An, W.; Huang, Z. A virus-like particle based bivalent vaccine confers dual protection against enterovirus 71 and coxsackievirus A16 infections in mice. Vaccine 2014, 32, 4296–4303. [Google Scholar] [CrossRef] [PubMed]
- Braun, M.; Jandus, C.; Maurer, P.; Hammann-Haenni, A.; Schwarz, K.; Bachmann, M.F.; Speiser, D.E.; Romero, P. Virus-like particles induce robust human T-helper cell responses. Eur. J. Immunol. 2011, 42, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Svedin, E.; Utorova, R.; Hühn, M.H.; Larsson, P.G.; Stone, V.M.; Garimella, M.G.; Lind, K.; Hägglöf, T.; Pincikova, T.; Laitinen, O.H.; et al. A link between a common mutation in cftr and impaired innate and adaptive viral defense. J. Infect. Dis. 2017, 216, 1308–1317. [Google Scholar] [CrossRef] [PubMed]
- Torfason, E.G.; Reimer, C.B.; Keyserling, H.L. Subclass restriction of human enterovirus antibodies. J. Clin. Microbiol. 1987, 25, 1376–1379. [Google Scholar] [CrossRef]
- Hankaniemi, M.M.; Stone, V.M.; Sioofy-Khojine, A.-B.; Heinimäki, S.; Marjomäki, V.; Hyöty, H.; Blazevic, V.; Laitinen, O.H.; Flodström-Tullberg, M.; Hytönen, V.P. A comparative study of the effect of UV and formalin inactivation on the stability and immunogenicity of a Coxsackievirus B1 vaccine. Vaccine 2019, 37, 5962–5971. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hankaniemi, M.M.; Baikoghli, M.A.; Stone, V.M.; Xing, L.; Väätäinen, O.; Soppela, S.; Sioofy-Khojine, A.; Saarinen, N.V.V.; Ou, T.; Anson, B.; et al. Structural Insight into CVB3-VLP Non-Adjuvanted Vaccine. Microorganisms 2020, 8, 1287. https://doi.org/10.3390/microorganisms8091287
Hankaniemi MM, Baikoghli MA, Stone VM, Xing L, Väätäinen O, Soppela S, Sioofy-Khojine A, Saarinen NVV, Ou T, Anson B, et al. Structural Insight into CVB3-VLP Non-Adjuvanted Vaccine. Microorganisms. 2020; 8(9):1287. https://doi.org/10.3390/microorganisms8091287
Chicago/Turabian StyleHankaniemi, Minna M., Mo A. Baikoghli, Virginia M. Stone, Li Xing, Outi Väätäinen, Saana Soppela, Amirbabak Sioofy-Khojine, Niila V. V. Saarinen, Tingwei Ou, Brandon Anson, and et al. 2020. "Structural Insight into CVB3-VLP Non-Adjuvanted Vaccine" Microorganisms 8, no. 9: 1287. https://doi.org/10.3390/microorganisms8091287
APA StyleHankaniemi, M. M., Baikoghli, M. A., Stone, V. M., Xing, L., Väätäinen, O., Soppela, S., Sioofy-Khojine, A., Saarinen, N. V. V., Ou, T., Anson, B., Hyöty, H., Marjomäki, V., Flodström-Tullberg, M., Cheng, R. H., Hytönen, V. P., & Laitinen, O. H. (2020). Structural Insight into CVB3-VLP Non-Adjuvanted Vaccine. Microorganisms, 8(9), 1287. https://doi.org/10.3390/microorganisms8091287