Evaluation of Extraction Methods for Clinical Metagenomic Assay

 ,
, 
Abstract
:1. Introduction
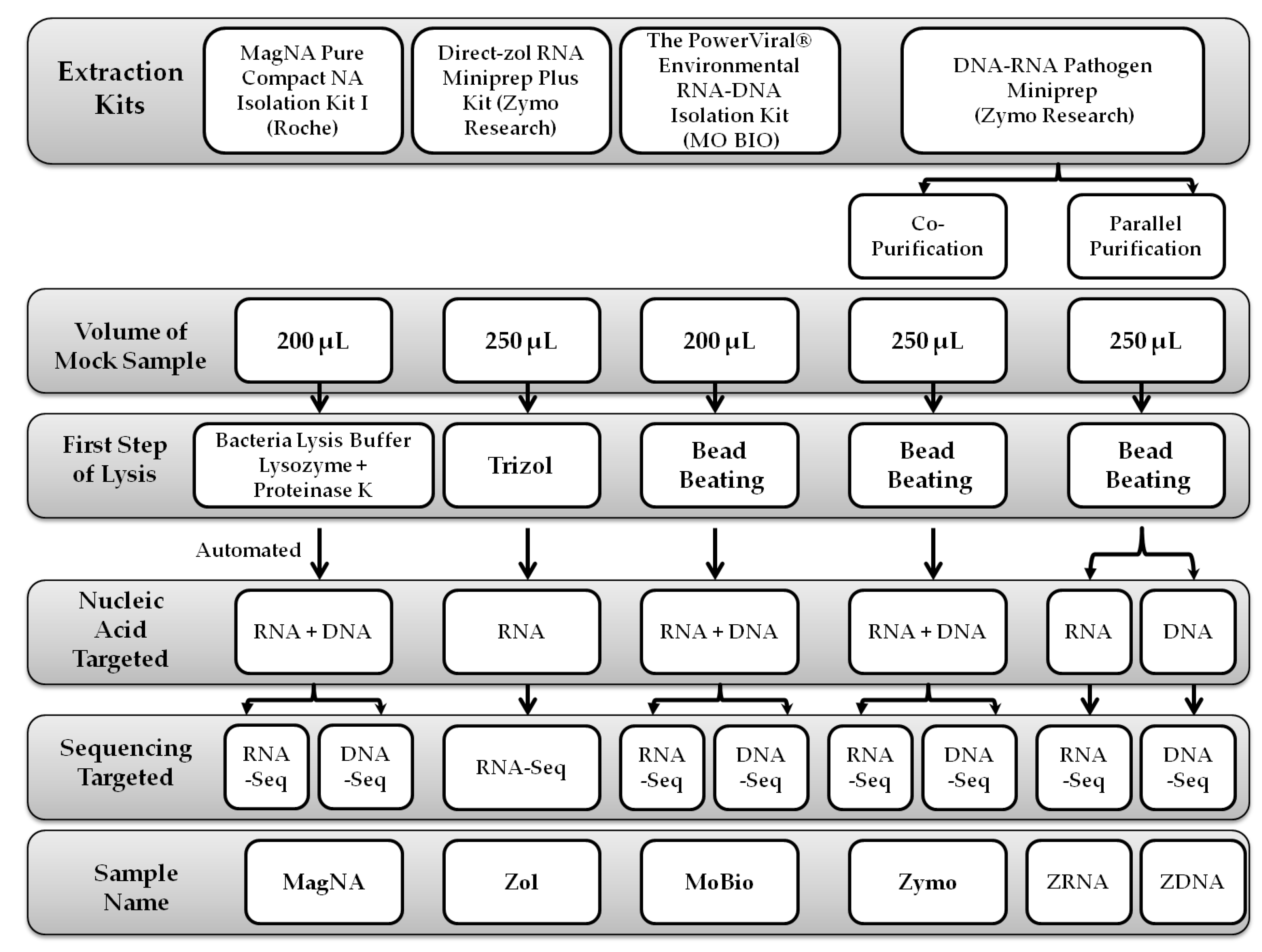
2. Materials and Methods
2.1. Mock Sample Preparation
2.2. Extraction Protocols
2.2.1. MagNA Pure Compact NA Isolation Protocol
2.2.2. Direct-zol™ RNA Miniprep Plus Protocol
2.2.3. PowerViral® Environmental RNA/DNA Isolation Protocol
2.2.4. ZymoBIOMICS™ DNA/RNA Miniprep Protocols
2.3. Nucleic Acid Quantification
2.4. Library Preparation
2.4.1. Preparation of the RNA Library
2.4.2. Preparation of the DNA Library
2.5. MiSeq Sequencing
2.6. Analysis of NGS Data
2.7. Bead Beating Optimization
2.8. Statistical Analysis
2.9. Clinical Samples
2.10. Analysis of Clinical Metagenomics
3. Results
3.1. Sequencing RNA Targets
3.2. Sequencing DNA Targets
3.3. Bead Beating Optimization
3.4. Clinical Samples Analysis
3.4.1. Sample No. 1
3.4.2. Sample No. 2
3.4.3. Sample No. 3
3.4.4. Sample No. 4
3.4.5. Sample No. 5
3.4.6. Sample No. 6
3.4.7. Negative Control
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Bead Beating Optimization

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Name | Sequence | Ref |
|---|---|---|---|
| C. neoformans | CneoFwd | 5′-GCCGCGACCTGCAAAG-3′ | [37] |
| CneoRev | 5′-GGTAATCACCTTCCCACTAACACAT-3′ | ||
| CneoProbe | 5′-FAM-ACGTCGGCTCGCC-BHQ1-3′ | ||
| ALKV | KFDVF | 5′-GAGGCTGCGTCATGGACAT-3′ | [38] |
| KFDVR | 5′-CCTTGATGTTCGTGAGGGTGTT-3′ | ||
| KFDVP | 5′-HEX-CAACGTGGTTCAGGY1CAGGTGGT-BHQ1-3′ |
| Name | Bead Beating Cycles | Total Beating | C. neoformans Ct | ALKV Ct |
|---|---|---|---|---|
| Sample 1 | 25 s.5 s × 2 | 50 s | 26.74 | 20.87 |
| Sample 2 | 25 s.5 s × 3 | 1 min + 15 s | 26.28 | 21.25 |
| Sample 3 | 25 s.5 s × 4 | 1 min + 40 s | 25.69 | 21.46 |
| Sample 4 | 25 s.5 s × 5 | 2 min + 5 s | 24.95 | 21.66 |
| Sample 5 | 25 s.5 s × 6 | 2 min + 30 s | 23.71 | 21.37 |
| Sample 6 | 25 s.5 s × 7 | 2 min + 55 s | 25.12 | 21.93 |
| Sample 7 | 25 s.5 s × 8 | 3 min + 20 s | 25.29 | 21.68 |
| Sample 8 | 25 s.5 s × 9 | 3 min + 45 s | 25.24 | 20.65 |
| Sample 9 | 25 s.5 s ×10 | 4 min + 10 s | 23.95 | 21.61 |
| Sample 10 | 25 s.5 s × 11 | 4 min + 35 s | 24.6 | 22.68 |
| Sample 11 | 25 s.5 s × 12 | 5 min | 23.82 | 20.72 |
| Sample 12 | 5 min | 5 min | 22.7 | 21.2 |
Appendix B
Metagenomics Protocol
Appendix C

Appendix D
| Sample Number | 1 | 2 | 3 | 4 | 5 | 6 | |
| Fungi | Candida albicans | 387 | 0 | 2965 | 0 | 0 | 0 |
| Candida glabrata | 799 | 0 | 0 | 0 | 0 | 0 | |
| Kluyveromyces marxianus | 352 | 0 | 0 | 0 | 0 | 0 | |
| Bacteria | Abiotrophia defectiva | 0 | 0 | 0 | 0 | 0 | 5591 |
| Achromobacter | 1690 | 0 | 0 | 0 | 0 | 0 | |
| Actinomyces graevenitzii | 0 | 12,585 | 0 | 0 | 0 | 2546 | |
| Actinomyces odontolyticus | 0 | 0 | 0 | 0 | 0 | 1647 | |
| Aggregatibacter | 116 | 0 | 0 | 0 | 0 | 0 | |
| Atopobium parvulum | 0 | 0 | 0 | 0 | 0 | 8819 | |
| Atopobium sp ICM42b | 0 | 0 | 0 | 0 | 0 | 9724 | |
| Atopobium sp ICM58 | 0 | 0 | 0 | 0 | 0 | 94,149 | |
| Bacteroidales Order 27 | 0 | 0 | 0 | 0 | 0 | 869 | |
| Bifidobacterium breve | 0 | 141 | 0 | 0 | 0 | 0 | |
| Campylobacter concisus | 0 | 0 | 744 | 0 | 0 | 0 | |
| Campylobacter showae | 0 | 0 | 286 | 0 | 0 | 0 | |
| Candidate division TM7 | 0 | 0 | 3017 | 0 | 0 | 0 | |
| Capnocytophaga | 0 | 0 | 378 | 0 | 0 | 0 | |
| Capnocytophaga gingivalis | 0 | 0 | 0 | 0 | 0 | 26,919 | |
| Capnocytophaga ochracea | 0 | 0 | 613 | 0 | 0 | 0 | |
| Capnocytophaga sp 123 Branch | 0 | 0 | 0 | 0 | 0 | 383 | |
| Capnocytophaga sp. Oral | 0 | 0 | 661 | 0 | 0 | 0 | |
| Christensenella Family Node 1 | 0 | 0 | 0 | 0 | 0 | 1381 | |
| Corynebacterium matruchotii | 0 | 0 | 1739 | 0 | 0 | 0 | |
| Corynebacterium pseudodiphtheriticum | 0 | 2429 | 0 | 306 | 1396 | 0 | |
| Dermabacter | 0 | 1562 | 0 | 0 | 0 | 0 | |
| Dolosigranulum pigrum | 0 | 85,751 | 0 | 12,087 | 33,972 | 0 | |
| Eikenella corrodens | 0 | 0 | 0 | 0 | 0 | 7923 | |
| Escherichia coli | 0 | 23 | 588 | 0 | 0 | 4226 | |
| Fusobacterium nucleatum | 0 | 0 | 445 | 0 | 0 | 0 | |
| Fusobacterium periodonticum | 0 | 0 | 122 | 0 | 0 | 1283 | |
| Gemella | 0 | 8 | 0 | 19 | 18 | 0 | |
| Gemella haemolysans | 0 | 0 | 31,095 | 0 | 0 | 0 | |
| Gemella morbillorum | 0 | 0 | 2887 | 0 | 0 | 0 | |
| Gemella sanguinis | 0 | 0 | 4067 | 0 | 0 | 0 | |
| Granulicatella | 0 | 78 | 44 | 0 | 0 | 0 | |
| Granulicatella adiacens | 0 | 0 | 0 | 0 | 0 | 45,700 | |
| Haemophilus | 0 | 10 | 0 | 26 | 0 | 0 | |
| Haemophilus haemolyticus | 0 | 0 | 0 | 25 | 0 | 0 | |
| Haemophilus influenzae | 398 | 0 | 0 | 0 | 0 | 0 | |
| Haemophilus parainfluenzae | 0 | 0 | 16,865 | 0 | 0 | 2469 | |
| Kingella denitrificans | 0 | 0 | 3037 | 0 | 0 | 0 | |
| Lachnoanaerobaculum saburreum | 0 | 0 | 1223 | 0 | 0 | 906 | |
| Lachnoanaerobaculum sp ICM7 | 0 | 0 | 0 | 0 | 0 | 111,698 | |
| Lachnospiraceae | 0 | 0 | 30 | 0 | 0 | 0 | |
| Lachnospiraceae bacterium oral | 0 | 0 | 0 | 0 | 0 | 8288 | |
| Lachnospiraceae oral | 0 | 0 | 0 | 0 | 0 | 3738 | |
| Lactobacillus | 0 | 50 | 0 | 0 | 0 | 0 | |
| Lactobacillus fermentum | 0 | 8 | 0 | 0 | 0 | 0 | |
| Lactobacillus gasseri | 0 | 123 | 0 | 0 | 0 | 0 | |
| Lactobacillus oris | 0 | 0 | 0 | 0 | 1888 | 0 | |
| Lactobacillus salivarius | 0 | 0 | 0 | 0 | 24,443 | 0 | |
| Lactobacillus vaginalis | 0 | 0 | 0 | 0 | 56,698 | 0 | |
| Lautropia mirabilis | 0 | 0 | 0 | 0 | 0 | 74,316 | |
| Leptotrichia | 0 | 80 | 0 | 0 | 0 | 0 | |
| Leptotrichia buccalis | 0 | 0 | 3077 | 0 | 0 | 0 | |
| Leptotrichia hofstadii | 0 | 0 | 1694 | 0 | 0 | 0 | |
| Leptotrichia shahii | 0 | 0 | 988 | 0 | 0 | 0 | |
| Leptotrichia sp. Oral | 0 | 0 | 5321 | 0 | 0 | 56,298 | |
| Leptotrichia trevisanii | 0 | 0 | 1530 | 0 | 0 | 0 | |
| Leptotrichia wadei | 0 | 0 | 1497 | 0 | 0 | 0 | |
| Moraxella catarrhalis | 867 | 535 | 0 | 309 | 36 | 0 | |
| Negativicoccus succinicivorans | 0 | 0 | 0 | 0 | 0 | 2945 | |
| Neisseria cinerea | 0 | 0 | 0 | 1518 | 0 | 0 | |
| Neisseria flavescens | 0 | 0 | 0 | 0 | 0 | 1617 | |
| Neisseria lactamica | 0 | 0 | 0 | 1260 | 0 | 0 | |
| Neisseria meningitidis | 0 | 0 | 0 | 10 | 0 | 0 | |
| Neisseria mucosa | 0 | 0 | 0 | 0 | 0 | 1856 | |
| Neisseria polysaccharea | 0 | 0 | 0 | 817 | 0 | 0 | |
| Neisseria subflava | 0 | 0 | 0 | 0 | 0 | 3363 | |
| Oribacterium | 0 | 0 | 49 | 0 | 0 | 0 | |
| Oribacterium 27619 Branch | 0 | 0 | 0 | 0 | 0 | 835 | |
| Oribacterium parvum | 0 | 0 | 0 | 0 | 0 | 690 | |
| Oribacterium sinus | 0 | 0 | 5749 | 0 | 0 | 8051 | |
| Peptoniphilus lacrimalis | 0 | 0 | 0 | 0 | 0 | 3520 | |
| Peptostreptococcus 26809 Branch | 0 | 0 | 0 | 0 | 0 | 25 | |
| Porphyromonas | 11 | 110 | 0 | 0 | 0 | 0 | |
| Porphyromonas sp COT 1360 Branch | 0 | 0 | 0 | 0 | 0 | 88 | |
| Porphyromonas sp oral | 0 | 0 | 0 | 0 | 0 | 133,736 | |
| Prevotella | 170 | 0 | 8 | 0 | 54 | 0 | |
| Prevotella denticola | 0 | 0 | 26 | 0 | 0 | 0 | |
| Prevotella histicola | 0 | 2166 | 1799 | 0 | 0 | 184,392 | |
| Prevotella intermedia | 0 | 0 | 476 | 0 | 0 | 0 | |
| Prevotella melaninogenica | 0 | 0 | 11,664 | 0 | 0 | 113,376 | |
| Prevotella nanceiensis | 0 | 0 | 0 | 0 | 0 | 131,564 | |
| Prevotella nigrescens | 0 | 0 | 820 | 0 | 0 | 174 | |
| Prevotella oris 764 Branch | 0 | 0 | 0 | 0 | 0 | 381 | |
| Prevotella pallens | 0 | 0 | 4867 | 0 | 0 | 87,205 | |
| Prevotella salivae | 0 | 0 | 1358 | 0 | 0 | 61,527 | |
| Prevotella scopos | 0 | 0 | 0 | 0 | 0 | 18,536 | |
| Prevotella shahii ] | 0 | 0 | 0 | 0 | 0 | 120,417 | |
| Prevotella sp. ICM33 | 0 | 0 | 14,193 | 0 | 0 | 112,154 | |
| Prevotella veroralis | 0 | 0 | 1078 | 0 | 0 | 0 | |
| Propionibacteriaceae | 0 | 0 | 0 | 0 | 12 | 0 | |
| Propionibacterium acnes | 0 | 0 | 0 | 0 | 0 | 420 | |
| Propionibacterium sp DORA 15 | 0 | 0 | 0 | 0 | 0 | 1892 | |
| Rothia dentocariosa | 0 | 0 | 1728 | 0 | 0 | 0 | |
| Rothia mucilaginosa | 0 | 14,625 | 790 | 0 | 0 | 3622 | |
| Solobacterium moorei | 0 | 0 | 0 | 0 | 0 | 731 | |
| Staphylococcus aureus | 0 | 242 | 0 | 0 | 0 | 0 | |
| Staphylococcus epidermidis | 0 | 0 | 0 | 0 | 0 | 48 | |
| Staphylococcus sp DORA 6 22 | 0 | 0 | 0 | 0 | 0 | 247 | |
| Stomatobaculum longum | 0 | 0 | 0 | 0 | 0 | 27,025 | |
| Streptococcus | 47 | 15 | 241 | 0 | 2368 | 0 | |
| Streptococcus agalactiae | 0 | 81 | 282 | 0 | 140 | 0 | |
| Streptococcus anginosus | 0 | 0 | 0 | 0 | 5011 | 0 | |
| Streptococcus australis | 0 | 0 | 0 | 0 | 0 | 32,772 | |
| Streptococcus infantis | 0 | 0 | 0 | 0 | 0 | 9143 | |
| Streptococcus mitis | 0 | 758 | 1257 | 1218 | 9294 | 0 | |
| Streptococcus oralis | 0 | 0 | 1671 | 0 | 0 | 0 | |
| Streptococcus parasanguinis | 0 | 0 | 0 | 0 | 0 | 3898 | |
| Streptococcus peroris | 0 | 0 | 0 | 0 | 0 | 1559 | |
| Streptococcus pneumoniae | 0 | 28 | 230 | 11 | 101 | 0 | |
| Streptococcus pseudopneumoniae | 0 | 37 | 0 | 16 | 388 | 0 | |
| Streptococcus salivarius | 0 | 1966 | 0 | 0 | 9599 | 0 | |
| Streptococcus salivarius | 0 | 0 | 0 | 0 | 0 | 0 | |
| Streptococcus sp I P16 | 0 | 0 | 0 | 0 | 0 | 3220 | |
| Streptococcus sp. C150 | 0 | 2308 | 0 | 0 | 20,769 | 0 | |
| Streptococcus sp. C150 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Streptococcus sp. DBCMS | 0 | 0 | 2063 | 0 | 0 | 0 | |
| Streptococcus thermophilus | 0 | 0 | 0 | 0 | 10 | 0 | |
| Streptococcus vestibularis | 0 | 0 | 0 | 0 | 3396 | 0 | |
| Tannerella sp oral | 0 | 0 | 0 | 0 | 0 | 84 | |
| Veillonella | 0 | 101 | 49 | 0 | 144 | 0 | |
| Veillonella atypica | 0 | 520 | 0 | 0 | 0 | 2196 | |
| Veillonella dispar | 0 | 0 | 6874 | 0 | 0 | 138,748 | |
| Veillonella parvula | 0 | 0 | 543 | 0 | 0 | 3730 | |
| Veillonella sp oral | 0 | 0 | 0 | 0 | 0 | 327,939 | |
| Veillonella sp. 3_1_44 | 0 | 0 | 355 | 0 | 0 | 0 | |
| Veillonella sp. HPA0037 | 0 | 837 | 0 | 0 | 0 | 0 | |
| Viruses | Dill cryptic virus | 158 | 158 | 64 | 22 | 203 | 211 |
| Dulcamara mottle virus | 0 | 0 | 0 | 0 | 0 | 12 | |
| Groundnut ringspot and Tomato chlorotic spot virus reassortant | 0 | 0 | 20 | 0 | 0 | 0 | |
| Haemophilus virus | 0 | 0 | 0 | 0 | 0 | 68 | |
| Hp1virus | 0 | 13 | 115 | 24 | 0 | 0 | |
| Human adenovirus 41 | 0 | 0 | 0 | 0 | 45 | 0 | |
| Human betaherpesvirus 5 (CMV) | 0 | 0 | 0 | 809 | 0 | 0 | |
| Human gammaherpesvirus 4 (EBV) | 0 | 0 | 173 | 0 | 0 | 0 | |
| Human orthopneumovirus (RSV) | 0 | 0 | 0 | 845 | 2398 | 0 | |
| Human parainfluenza virus 2 | 2363 | 0 | 0 | 0 | 0 | 0 | |
| Influenza A virus (H1N1) | 0 | 1891 | 0 | 0 | 0 | 0 | |
| Influenza A virus (H3N2) | 0 | 0 | 0 | 0 | 0 | 1365 | |
| Piscine myocarditis-like virus | 0 | 31 | 0 | 7 | 52 | 106 | |
| Pseudomonas virus Pf1 | 65 | 0 | 0 | 0 | 0 | 0 | |
| Red clover cryptic virus | 137 | 104 | 45 | 26 | 146 | 220 | |
| Rosellinia necatrix partitivirus | 82 | 95 | 85 | 66 | 261 | 311 | |
| Sclerotinia sclerotiorum partitivirus | 0 | 0 | 0 | 0 | 0 | 13 | |
| Staphylococcus virus | 21 | 67 | 0 | 0 | 0 | 27 | |
| Streptococcus virus | 0 | 0 | 0 | 0 | 720 | 0 | |
| Tobacco mosaic virus | 0 | 0 | 0 | 0 | 27 | 0 | |
| Torque teno mini virus | 0 | 0 | 0 | 10 | 0 | 0 | |
| Vicia cryptic virus | 46 | 31 | 0 | 0 | 82 | 0 | |
| White clover cryptic virus | 18 | 0 | 16 | 6 | 0 | 193 |
References
- Bullman, S.; Meyerson, M.; Kostic, A.D. Emerging Concepts and Technologies for the Discovery of Microorganisms Involved in Human Disease. Annu. Rev. Pathol. 2017, 12, 217–244. [Google Scholar] [CrossRef] [PubMed]
- Bousbia, S.; Raoult, D.; La Scola, B. Pneumonia pathogen detection and microbial interactions in polymicrobial episodes. Future Microbiol. 2013, 8, 633–660. [Google Scholar] [CrossRef] [PubMed]
- The Top 10 Causes of Death. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 4 November 2018).
- Caliendo, A.M.; Gilbert, D.N.; Ginocchio, C.C.; Hanson, K.E.; May, L.; Quinn, T.C.; Tenover, F.C.; Alland, D.; Blaschke, A.J.; Bonomo, R.A.; et al. Better tests, better care: Improved diagnostics for infectious diseases. Clin. Infect. Dis. 2013, 57, S139–S170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, R.R.; Montoya, V.; Gardy, J.L.; Patrick, D.M.; Tang, P. Metagenomics for pathogen detection in public health. Genome Med. 2013, 5, 81. [Google Scholar] [CrossRef] [Green Version]
- Polage, C.R.; Cohen, S.H. State-of-the-Art Microbiologic Testing for Community-Acquired Meningitis and Encephalitis. J. Clin. Microbiol. 2016, 54, 1197–1202. [Google Scholar] [CrossRef] [Green Version]
- Susilawati, T.N.; Jex, A.R.; Cantacessi, C.; Pearson, M.; Navarro, S.; Susianto, A.; Loukas, A.C.; McBride, W.J. Deep sequencing approach for investigating infectious agents causing fever. Eur. J. Clin. Microbiol. Infect. Dis. Off. Publ. Eur. Soc. Clin. Microbiol. 2016, 35, 1137–1149. [Google Scholar] [CrossRef] [Green Version]
- Datta, S.; Budhauliya, R.; Das, B.; Chatterjee, S.; Vanlalhmuaka; Veer, V. Next-generation sequencing in clinical virology: Discovery of new viruses. World J. Virol. 2015, 4, 265–276. [Google Scholar] [CrossRef]
- Weinstock, G.M.; Goldberg, B.; Ledeboer, N.; Rubin, E.; Sichtig, H.; Geyer, C. Applications of clinical microbial next-generation sequencing. Am. Acad. Microbiol. 2016. [Google Scholar] [CrossRef]
- Forbes, J.D.; Knox, N.C.; Peterson, C.L.; Reimer, A.R. Highlighting Clinical Metagenomics for Enhanced Diagnostic Decision-making: A Step towards Wider Implementation. Comput. Struct. Biotechnol. J. 2018, 16, 108–120. [Google Scholar] [CrossRef]
- Bag, S.; Saha, B.; Mehta, O.; Anbumani, D.; Kumar, N.; Dayal, M.; Pant, A.; Kumar, P.; Saxena, S.; Allin, K.H.; et al. An Improved Method for High Quality Metagenomics DNA Extraction from Human and Environmental Samples. Sci. Rep. 2016, 6, 26775. [Google Scholar] [CrossRef]
- Mulcahy-O’Grady, H.; Workentine, M.L. The Challenge and Potential of Metagenomics in the Clinic. Front. Immunol. 2016, 7, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madani, T.A.; Azhar, E.I.; el Abuelzein, T.M.; Kao, M.; Al-Bar, H.M.; Farraj, S.A.; Masri, B.E.; Al-Kaiedi, N.A.; Shakil, S.; Sohrab, S.S.; et al. Complete genome sequencing and genetic characterization of Alkhumra hemorrhagic fever virus isolated from Najran, Saudi Arabia. Intervirology 2014, 57, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, K.; Izumikawa, K.; Yamamoto, Y.; Yanagihara, K.; Ohkusu, K.; Kohno, S. Multiple lung abscesses caused by Actinomyces graevenitzii mimicking acute pulmonary coccidioidomycosis. J. Clin. Microbiol. 2012, 50, 3125–3128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maraki, S.; Papadakis, I.S. Rothia mucilaginosa pneumonia: A literature review. Infect. Dis. 2015, 47, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Burkovski, A. Corynebacterium pseudodiphtheriticum: Putative probiotic, opportunistic infector, emerging pathogen. Virulence 2015, 6, 673–674. [Google Scholar] [CrossRef]
- Raabe, V.N.; Shane, A.L. Group B Streptococcus (Streptococcus agalactiae). Microbiol. Spectr. 2019, 7, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Haddar, C.H.; Joly, J.; Carricajo, A.; Verhoeven, P.O.; Grattard, F.; Mory, O.; Begaud, E.; Germani, Y.; Cantais, A.; Pozzetto, B. Strategy using a new antigenic test for rapid diagnosis of Streptococcus pneumoniae infection in respiratory samples from children consulting at hospital. BMC Microbiol. 2020, 20, 79. [Google Scholar] [CrossRef] [Green Version]
- Kilian, M.; Tettelin, H. Identification of Virulence-Associated Properties by Comparative Genome Analysis of Streptococcus pneumoniae, S. pseudopneumoniae, S. mitis, Three S. oralis Subspecies, and S. infantis. mBio 2019, 10, 1–32. [Google Scholar] [CrossRef] [Green Version]
- Emmanouilidou, G.; Voukelatou, P.; Vrettos, I.; Aftzi, V.; Dodos, K.; Koumpouli, D.; Avgeropoulos, G.; Kalliakmanis, A. A Case Report of Successful Conservative Treatment for Infective Endocarditis Caused by Gemella sanguinis. Case Rep. Infect. Dis. 2019, 2019, 9382395. [Google Scholar] [CrossRef]
- Garcia Lopez, E.; Martin-Galiano, A.J. The Versatility of Opportunistic Infections Caused by Gemella Isolates Is Supported by the Carriage of Virulence Factors from Multiple Origins. Front. Microbiol. 2020, 11, 524. [Google Scholar] [CrossRef]
- Feldman, C.; Anderson, R. Meningococcal pneumonia: A review. Pneumonia 2019, 11, 3. [Google Scholar] [CrossRef] [Green Version]
- Reis-Melo, A.; Soares, D.; Magalhaes, M.F.; Ferraz, C.; Vaz, L. Complicated Pneumonia with Empyema Caused by Streptococcus Anginosus in a Child. Rev. Paul. Pediatr. 2020, 38, e2018258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Garg, M.; Misra, S.; Singhal, S. Granulicatella adiacens abscess: Two rare cases and review. J. Lab. Physicians 2018, 10, 121–123. [Google Scholar] [CrossRef] [PubMed]
- Ehrmann, E.; Jolivet-Gougeon, A.; Bonnaure-Mallet, M.; Fosse, T. Multidrug-resistant oral Capnocytophaga gingivalis responsible for an acute exacerbation of chronic obstructive pulmonary disease: Case report and literature review. Anaerobe 2016, 42, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Rossmann, S.N.; Wilson, P.H.; Hicks, J.; Carter, B.; Cron, S.G.; Simon, C.; Flaitz, C.M.; Demmler, G.J.; Shearer, W.T.; Kline, M.W. Isolation of Lautropia mirabilis from oral cavities of human immunodeficiency virus-infected children. J. Clin. Microbiol. 1998, 36, 1756–1760. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Nie, H. Severe purulent pericarditis caused by invasive Eikenella corrodens: Case report and literature review. BMC Infect. Dis. 2019, 19, 657. [Google Scholar] [CrossRef]
- Murphy, E.C.; Frick, I.M. Gram-positive anaerobic cocci—Commensals and opportunistic pathogens. FEMS Microbiol. Rev. 2013, 37, 520–553. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Ma, L.; Fan, K.; Li, Y.; Xie, L.; Xia, W.; Gu, B.; Liu, G. Necrotizing pneumonia and empyema caused by Neisseria flavescens infection. J. Thorac. Dis. 2014, 6, 553–557. [Google Scholar] [CrossRef]
- Leite, G.M.; Magan, N.; Medina, A. Comparison of different bead-beating RNA extraction strategies: An optimized method for filamentous fungi. J. Microbiol. Methods 2012, 88, 413–418. [Google Scholar] [CrossRef] [Green Version]
- Kajiura, L.N.; Stewart, S.D.; Dresios, J.; Uyehara, C.F.T. Simultaneous Extraction of Viral and Bacterial Nucleic Acids for Molecular. J. Biomol. Tech. 2015, 26, 118–124. [Google Scholar] [CrossRef] [Green Version]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017, 35, 833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kresse, S.H.; Namlos, H.M.; Lorenz, S.; Berner, J.M.; Myklebost, O.; Bjerkehagen, B.; Meza-Zepeda, L.A. Evaluation of commercial DNA and RNA extraction methods for high-throughput sequencing of FFPE samples. PLoS ONE 2018, 13, e0197456. [Google Scholar] [CrossRef] [PubMed]
- Langelier, C.; Zinter, M.S.; Kalantar, K.; Yanik, G.A.; Christenson, S.; O’Donovan, B.; White, C.; Wilson, M.; Sapru, A.; Dvorak, C.C.; et al. Metagenomic Sequencing Detects Respiratory Pathogens in Hematopoietic Cellular Transplant Patients. Am. J. Respir. Crit. Care Med. 2018, 197, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szczesniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef] [Green Version]
- Simner, P.J.; Miller, H.B.; Breitwieser, F.P.; Pinilla Monsalve, G.; Pardo, C.A.; Salzberg, S.L.; Sears, C.L.; Thomas, D.L.; Eberhart, C.G.; Carroll, K.C. Development and Optimization of Metagenomic Next-Generation Sequencing Methods for Cerebrospinal Fluid Diagnostics. J. Clin. Microbiol. 2018, 56. [Google Scholar] [CrossRef] [Green Version]
- Veron, V.; Simon, S.; Blanchet, D.; Aznar, C. Real-time polymerase chain reaction detection of Cryptococcus neoformans and Cryptococcus gattii in human samples. Diagn. Microbiol. Infect. Dis. 2009, 65, 69–72. [Google Scholar] [CrossRef]
- Pang, Z.; Li, A.; Li, J.; Qu, J.; He, C.; Zhang, S.; Li, C.; Zhang, Q.; Liang, M.; Li, D. Comprehensive multiplex one-step real-time TaqMan qRT-PCR assays for detection and quantification of hemorrhagic fever viruses. PLoS ONE 2014, 9, e95635. [Google Scholar] [CrossRef]
- Endoh, D.; Mizutani, T.; Kirisawa, R.; Maki, Y.; Saito, H.; Kon, Y.; Morikawa, S.; Hayashi, M. Species-independent detection of RNA virus by representational difference analysis using non-ribosomal hexanucleotides for reverse transcription. Nucleic Acids Res. 2005, 33, e65. [Google Scholar] [CrossRef]


| Microorganism | Reference Name | Accession |
|---|---|---|
| C. neoformans | Cryptococcus neoformans var. neoformans JEC21 | AE017341-356 |
| K. pneumoniae | Klebsiella pneumoniae subsp. pneumoniae HS11286 | CP003223-228 + CP003200 |
| S. aureus | Staphylococcus aureus subsp. aureus NCTC 8325 | CP000253 |
| AdV | Human mastadenovirus E strain HAdVE/USA_ New York/38813/2014/P4H4F4 | KY996444 |
| ALKV | Alkhumra hemorrhagic fever virus strain SCVHF001 | JN860200 |
| Average RPM | MagNA | Zol | MoBio | Zymo | ZRNA |
|---|---|---|---|---|---|
| C. neoformans | 19,257 | 82,441 | 30,427 | 90,684 | 75,359 |
| K. pneumoniae | 784,302 | 117,122 | 255,302 | 184,597 | 177,745 |
| S. aureus | 46,263 | 182,489 | 210,541 | 143,569 | 216,660 |
| AdV | 11,193 | 21,834 | 54,898 | 53,243 | 7593 |
| ALKV | 2866 | 8182 | 2247 | 12,830 | 12,945 |
| Average of Coverage % | MagNA | Zol | MoBio | Zymo | ZRNA |
|---|---|---|---|---|---|
| C. neoformans | 0.04 | 0.06 | 0.03 | 0.09 | 0.08 |
| K. pneumoniae | 5.98 | 7.96 | 7.20 | 5.29 | 4.64 |
| S. aureus | 13.40 | 36.35 | 64.10 | 45.90 | 24.95 |
| Adv | 100.00 | 100.00 | 100.00 | 100.00 | 99.80 |
| ALKV | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 |
| Average of RPM | MagNA | MoBio | Zymo | ZDNA |
|---|---|---|---|---|
| C. neoformans | 34 | 601 | 56 | 163 |
| K. pneumoniae | 152,023 | 192,812 | 100,536 | 91,928 |
| S. aureus | 1590 | 40,813 | 37,205 | 34,785 |
| Adv | 119,851 | 53,233 | 40,054 | 72,556 |
| Average of Coverage % | MagNA | MoBio | Zymo | ZDNA |
|---|---|---|---|---|
| C. neoformans | 0.02 | 0.03 | 0.11 | 0.48 |
| K. pneumoniae | 13.51 | 12.41 | 13.51 | 13.51 |
| S. aureus | 19.90 | 23.20 | 99.85 | 99.90 |
| Adv | 100.00 | 100.00 | 100.00 | 100.00 |
| Average of RPM | Average of Coverage % | |||
|---|---|---|---|---|
| Sample Name | MoBio | Zymo | MoBio | Zymo |
| C. neoformans | 1379 | 410 | 3.46 | 0.54 |
| K. pneumoniae | 232,653 | 240,061 | 18.57 | 18.45 |
| S. aureus | 51,102 | 42,645 | 99.90 | 99.80 |
| Adv | 145,277 | 80,502 | 100.00 | 100.00 |
| Name | MoBio 5-min | Zymo 5-min |
|---|---|---|
| Naegleria fowleri | ||
| Staphylococcus aureus subsp. Aureus | ||
| Staphylococcus lugdunensis | ||
| Streptococcus | ||
| Actinomyces sp. HPA0247 | ||
| Rothia | ||
| Eikenella corrodens ATCC 23834 | ||
| Neisseria | ||
| Kingella denitrificans ATCC 33394 | ||
| Pseudomonas aeruginosa | ||
| Aggregatibacter segnis ATCC 33393 | ||
| Stenotrophomonas maltophilia | ||
| Campylobacter | ||
| Fusobacterium | ||
| Porphyromonas | ||
| Prevotella nanceiensis DSM 19126 | ||
| Human metapneumovirus | ||
| Pseudomonas phage | ||
| Staphylococcus phage | ||
| Staphylococcus prophage phiPV83 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farraj, S.A.; El-Kafrawy, S.A.; Kumosani, T.A.; Yousef, J.M.; Azhar, E.I. Evaluation of Extraction Methods for Clinical Metagenomic Assay. Microorganisms 2020, 8, 1128. https://doi.org/10.3390/microorganisms8081128
Farraj SA, El-Kafrawy SA, Kumosani TA, Yousef JM, Azhar EI. Evaluation of Extraction Methods for Clinical Metagenomic Assay. Microorganisms. 2020; 8(8):1128. https://doi.org/10.3390/microorganisms8081128
Chicago/Turabian StyleFarraj, Suha A., Shreif A. El-Kafrawy, Taha A. Kumosani, Jehad M. Yousef, and Esam I. Azhar. 2020. "Evaluation of Extraction Methods for Clinical Metagenomic Assay" Microorganisms 8, no. 8: 1128. https://doi.org/10.3390/microorganisms8081128
APA StyleFarraj, S. A., El-Kafrawy, S. A., Kumosani, T. A., Yousef, J. M., & Azhar, E. I. (2020). Evaluation of Extraction Methods for Clinical Metagenomic Assay. Microorganisms, 8(8), 1128. https://doi.org/10.3390/microorganisms8081128