1. Introduction
Functional diversity includes all biological processes performed within an ecosystem and it has been suggested that the more functionally diverse an ecosystem is, the more resilient it is to environmental change [
1]. However, functional diversity studies based on ecological networks have suggested that most ecosystems include a relatively small core set of functions (whose depletion could lead to ecological collapse), and a large set of peripheral functions (that can be depleted without an observable impact on the ecosystem)—such a pattern has been termed a scale-free network [
2]. Following on from this, an increase in overall functional diversity should also lead to a proportional increase in these peripheral functions (whose depletion would not impact the core function of the ecosystem) hence conferring resilience. Such a mechanistic link has not been tested previously.
Microbial communities are ideal systems in which to test ecological theories, for their relative simplicity and the direct link between genetic composition and ecological function [
3]. Understanding functional resilience and the potential impact of changes in microbial community structure and ecology remains a major challenge, as microbes tend to show high levels of both metabolic flexibility and functional redundancy [
4]. Despite this, microbial community composition changes rapidly with environmental perturbation and they appear to show little tendency to return to the original structure following such a disturbance [
5,
6]. This could be due to changes in the competitive advantage in co-occurring microorganisms (e.g., [
7]). Conversely, recent studies have also found unexpected levels of ecological resilience, such as within sub-Arctic phytoplankton communities [
8] and the reason behind such conflicting results could partly be because traditionally microbial resilience tends to be measured in terms of their ability to recover back to an original state after a disturbance based on taxonomy, i.e., community composition [
6] or population dynamics [
9,
10] alone. To complicate matters, functionality is also still commonly assessed using environmental conditions as proxy [
11] or by overly relying on taxonomic markers [
12,
13,
14]. In any case, the potential impact that changes at the microbial level could have on upper trophic levels would strongly depend on whether the remaining taxa are able to maintain the same level of ecological function (ecosystem service) as the original community.
Polar ecosystems represent an ideal platform to test such hypotheses about ecological resilience, as they are often considered fragile or close to ecological tipping points [
15,
16,
17,
18]. For example, functional redundancy is predicted to be low in Antarctic soils [
17], while polar marine ecosystems are thought to be potentially close to ecological collapse as a consequence of rapid sea ice loss and temperature fluctuations [
18,
19]. Furthermore, each microbial species is predicted to fulfil distinct ecological functions in these comparatively species-poor environments [
17,
18], representing a system which, in theory, is particularly vulnerable to environmental change and biodiversity loss.
In this study, we analyzed the structure of functional groupings (as defined in [
20,
21] and summarized in
Supplementary Table S1) arising from common methods to measure ecological resilience, such as network analysis and cluster analysis, to identify whether;
(1) there is a link between network structure and the distribution of functional genes;
(2) the level of functional redundancy in this system is indicative of either resilience or a close to ‘collapse’ scenario;
(3) there are any signs of deterioration in the investigated microbial system at the functional level.
Thus, we analyzed the network structure and distribution of microbial functions based on the co-occurrence of functional genes (as defined in [
20]) across a broad geographical range. This analysis allowed the identification of specific characteristics of the functional network that could explain the microbial ability to survive and potentially thrive even in the most extreme environments on Earth.
2. Materials and Methods
2.1. Sampling
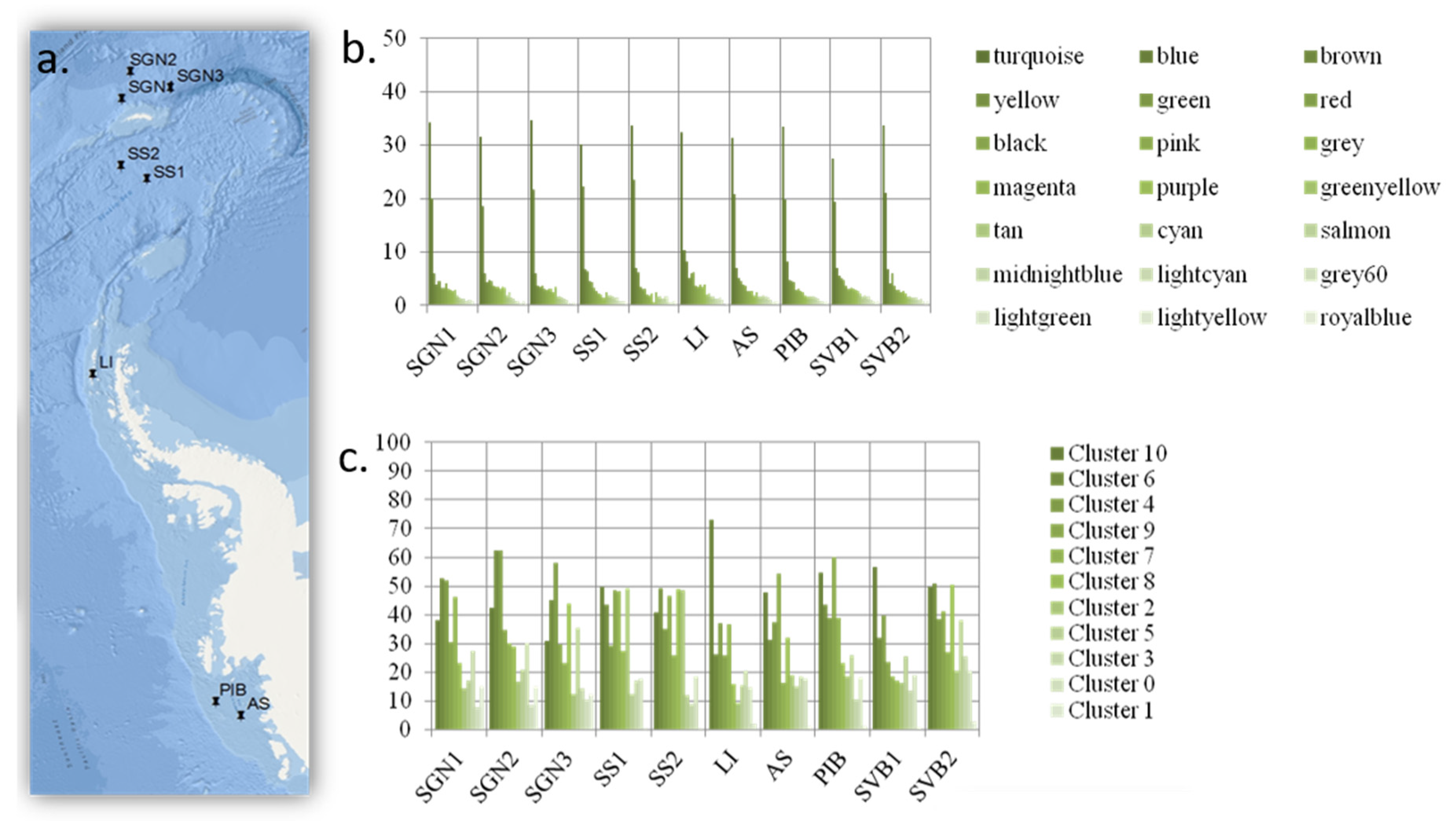
A quantity of 300 L of seawater was collected 30 m below the surface (at the chlorophyll maximum) using a CTD (Conductivity, Temperature, and Depth) sensor, from 10 high latitude locations (the Antarctic locations are shown in Figure 3; the Arctic location was at the water confluence between Kongsfjord and Krossfjord in the West Coast of Svalbard, Norway).
The cruise data were deposited and are available at the BODC (British Oceanographic data Centre), including cruise reports, sampling station locations and cruise tracks for the following relevant oceanographic cruises: JR144 (26/2/06–17/4/06) to the Scotia Sea and the South Sandwich Islands; IPY Kinnvika RV Horyzont II to Magdalenefjord, Krossfjord and Kongsfjord, Svalbard (9/7/08–13/7/08); JR179 (21/2/08–11/4/08) to the Amundsen Sea, Bellingshausen Sea and Pine Island Bay; JR262 (24/9/11–20/11/11) to South Georgia and the South Orkney Islands; and JR230 (02/12/09–11/12/09) to the Bellingshausen Sea and Adelaide Island. This represents a sampling acquisition effort of 6 cruises and 50 days of sampling covering areas above and below the Southern Antarctic Circumpolar Current and above the Arctic Front.
The sample water was passed through a sonication bath to disrupt suspended particles and through a 2.7 µm prefilter using a sterile stainless-steel housing. The water was then subjected to ultrafiltration in a Pellicon 2 ultrafiltration system (EMD Millipore, Billerica, MA, USA) with 1 × 10,000 Da molecular weight cut-off filters. The permeate was excluded and the retentate recycled through the system until the total seawater volume reached <300 mL, and frozen at −20 °C for further analysis. The 300 mL retentate was defrosted slowly and centrifuged at 38,000 revolutions per minute (r.p.m.) to recover whole cells in a centrifugal concentrator. The DNA from cells concentrated in this way was subjected to DNA extraction using the MoBio water DNA extraction kit (MoBio, Carlsbad, CA, USA) according to the manufacturer’s instructions.
2.2. Microarray
To characterize the functional composition of the microbial communities in the marine samples investigated, we used a functional gene array (GeoChip 3.0, [
20]), a high throughput metagenomic tool to detect and characterize microbial communities. We chose this microarray for its high specificity, low false-positive rates, inherent signal quantification and to eliminate potential observer bias. The full design pipeline for the functional chip array is previously described in [
20], whereby CommOligo 2.0 [
22] was used to design both gene- and group-specific probes, which were subsequently synthesized by Invitrogen (Carlsbad, CA, USA) and arrayed onto Corning UltraGAPS (Corning, NY, USA) slides using a Microgrid II Arrayer.
GeoChip 3.0 targets a variety of functions involved in carbon, nitrogen, phosphorus and sulphur cycles, energy metabolism, antibiotic resistance, metal resistance and organic contaminant degradation [
20], which are considered relevant to those selected ecosystems, especially for biogeochemical cycling genes. Therefore, this selection of functions also covers major environmental processes. Even if we could expect the polar oceans to hold a proportion of genes, microorganisms and pathways not present elsewhere, and consequently not detectible by the GeoChip, we can still conclude that this pattern of modular resilience holds for all the main biogeochemical processes identified for microbial communities worldwide. This also opens the question about whether the pattern would hold for community-specific genes and pathways. More interestingly, if future studies show that this pattern does not hold for community-specific functions, this could further show why this set of functions and not others have evolutionarily become biogeochemically important worldwide. As we used DNA samples instead of mRNA for GeoChip analyses, these data are interpreted as being the functional potential instead of functional activity, drawing in the pool of functional pre-adaptations available within the community studied for these essential functions. In addition, the level of accuracy of the GeoChip 3.0 is to be considered, as it can detect down to 1 or 2 cells [
21].The genes in the microarray were selected to cover a large proportion of common functional genes involved in major known ecological and environmental processes across the world.
2.3. Taxonomic and Species Richness Analysis
As a taxonomic marker, we used the gene
gyrB encoding DNA gyrase b-subunit as the phylogenetic marker on GeoChip 3.0. Sequences of 16S rRNA genes are widely used as phylogenetic markers for bacterial/archaeal systematics and ecology [
23]; however, it is difficult to use them to obtain fine-scale resolution at species and/or strain level as is required here. A phylogenetic tree based on
gyrB results in a magnitude higher resolution than a tree based on 16S rRNA genes [
24,
25].
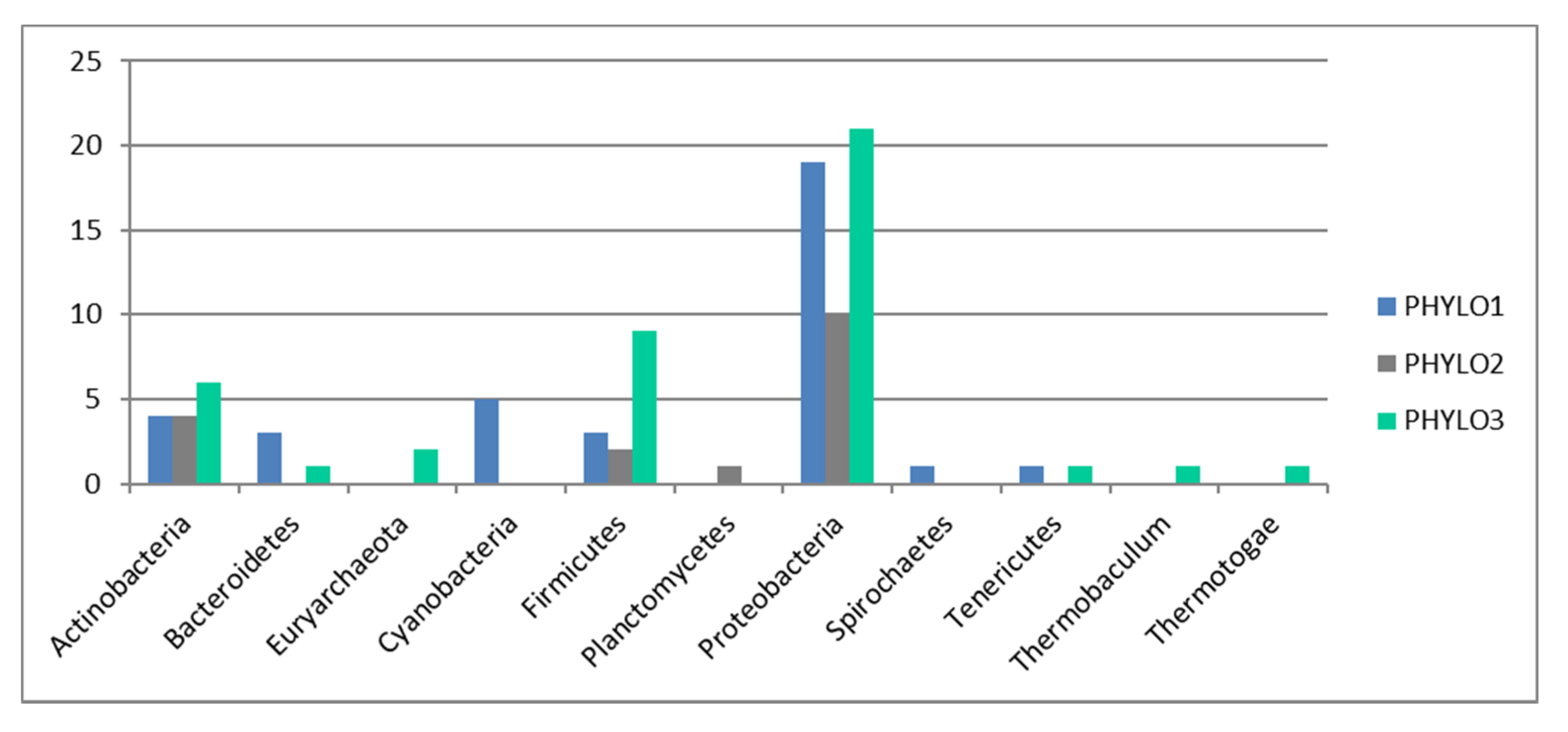
GyrB is used here as an indicator of taxonomic diversity. This way the phylogenetic markers could be used to conduct a similar analysis to the functional genes and test whether the pattern for functional genes can be explained by taxonomical distribution alone.
The pattern of presence/absence of taxa based on this marker was used to assess whether the sites differed in relation to species distribution and richness. Thus, species richness index was estimated for each site. A pairwise t-test with Holm adjustment (significance level p < 0.05) was performed to assess the distribution of taxonomic species and individual functional genes across sites.
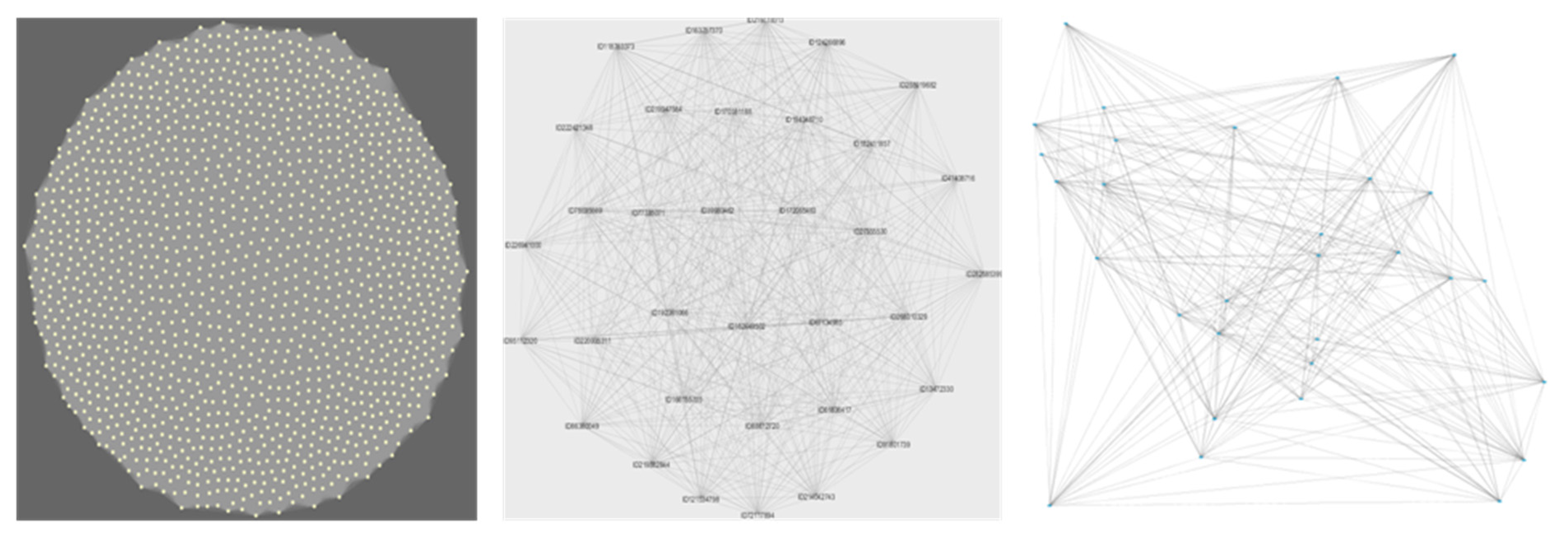
2.4. Network Construction and Cluster Analysis
We used weighted gene co-expression network analysis (WGCNA) and KMeans++ clustering to identify whether, at the functional level, microbial communities are arranged in distinguishable interactive units (modules or clusters). WGCNA is a widely used data mining method, especially for studying biological networks based on pairwise correlations between variables. Network analyses are particularly useful to represent the level of interconnectivity within interactive units. We used WGCNA as it was tailored specifically for microarray data [
26].
The groups formed using WGCNA are known as modules, while the groups identified by KMeans++ are defined as clusters. In both cases, they will correspond to a set of bacteria, or a bacterial network, whose genes are highly likely to interact within the environment as they tend to co-occur.
We used the specifically designed WGCNA R package for network detection, gene selection and network construction [
26,
27,
28]. A total of 4915 functional genes were included in the network analysis. These were the genes present in at least six of the sites, and hence useful to establish meaningful multiple correlations. After assuming an initial scale-free topology, we identified a soft threshold power of 10 as the highest power showing a significant increase in mean connectivity among module items. We used the module eigengenes to plot the network heatmap.
WGCNA allocates to a module all genes that tend to co-occur. If a specific gene is found more than a threshold number of times (80%) in the same sample than another gene, these two genes are linked together and grouped in the same module. Each gene included in the analysis also fulfils a specific function (i.e., antibiotic resistance, carbon sequestration, etc.) for being an essential part of a metabolic path related to the allocated function. Note that the GeoChip was specifically designed to include genes unambiguously assigned to a unique function.
To ensure that the pattern found was not an artefact of the clustering method used, we re-analyzed the data using a completely different clustering method, known as the K++ mean. K++, as distinct from WGCNA, can allocate a gene to more than one cluster instead of allocating the gene to the first group where the threshold is met. Therefore, the order in which the genes are included in the analysis has a lower influence in the result than for WGCNA.
Thus, for the second clustering analysis (results in Figure 3), the KMeans++ clustering method with seeding [
29] was implemented in the data mining software WEKA [
30]. WEKA compiles a collection of machine learning algorithms tailored for data mining, whilst the KMeans++ method applied implemented these algorithms to find the centroid for each cluster with iterations (repeated until the process provides stable results). The centroids are originally chosen at random from the data points, but the algorithm weighs the data points according to their squared distance from the closest center already chosen, which has been proven to provide accurate meaningful clusters. Once the centroids are identified, the clusters are built around them based on a distance formula estimated using the number of matches/no matches among vectors, indicating the presence/absence of each functional gene per location. Points with equal distances to more than one centroid were allocated to more than one cluster, assuming each cluster is an open interactive unit, whereby each function can interact with more than one functional cluster. We used the Apriori algorithm [
31] to find specific associations (association algorithms find association of the type, e.g., whenever a specific function occurs in A and B, a specific associated function will also occur in A and B; or, if a function occurs in A and B, it will always occur/be absent in C). We also used WEKA to implement the association algorithms. In particular, Apriori seeks simultaneous occurrences of subgroups of attributes, allowing the identification of co-occurrences among subgroups of characteristics (in this case, functions) within specific geographic locations. The levels of significance of the associations thus found are given as confidence levels.
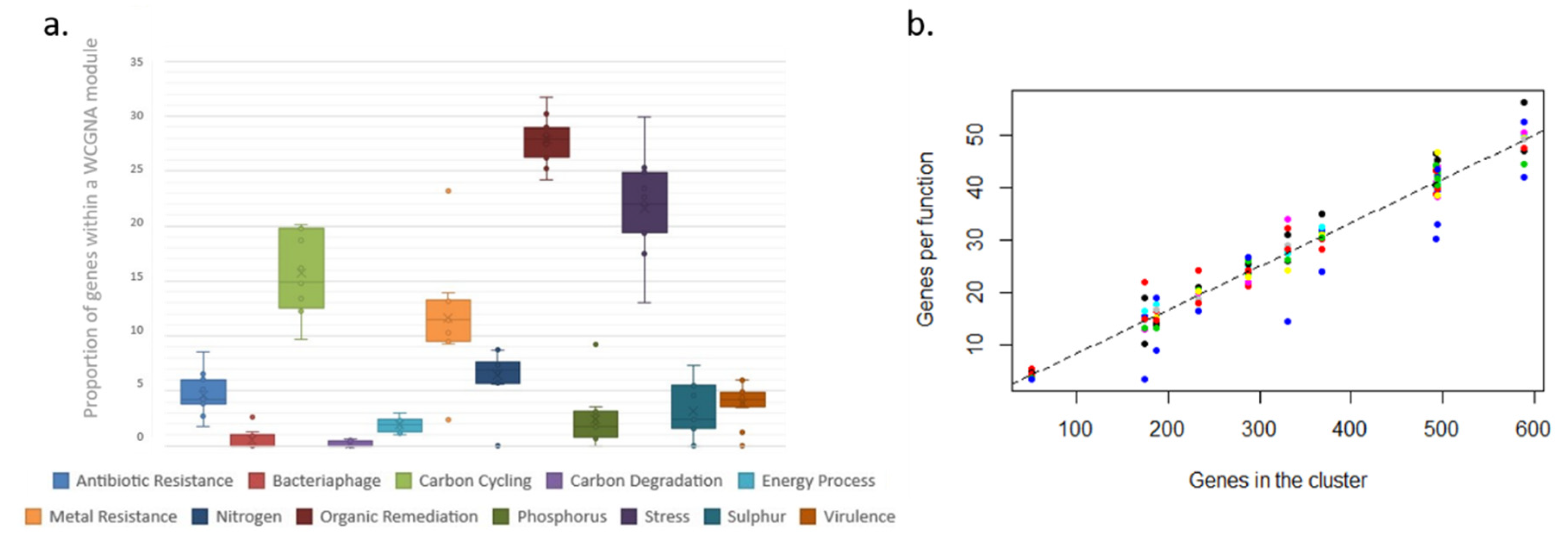
For comparative purposes, the number of genes in a cluster for each functional group was divided by the number of genes in the microarray for that function and presented as the proportional representation of each function within each site, WGCNA module or K++ means cluster. Thus, per each gene functional grouping, the results are shown as the proportion of genes from the category included in the analyses that appear in the cluster/module/site. The full list of functional groupings included in the microarray is shown in
Supplementary Table S1.
2.5. Comparative Analysis between Function and Taxonomic Distributions
To assess whether the pattern observed in functional distribution was a direct result of phylogenetic distribution, we repeated the WGCNA analysis as above based only on the phylogenetic markers. Subsequently, we used Pearson’s correlation to check whether the geographical distribution of the WGCNA phylogenetic modules followed the distribution of the WGCNA functional modules. A Student’s t-test was used to assess whether the correlation was significant at a 5% level of significance.
4. Discussion
Initially, we set out to test whether an increase in functional diversity should lead to a proportional increase in the peripheral functions of the expected scale-free network conferring resilience. What we found, however, was a very different picture. The network did not organize as a scale-free network but as a set of random networks (each network represented as a WGCNA module, represented by colours in
Figure 3, panel b). In such a structure, each co-occurring module is in itself weak, as every node (gene in this case) has the equivalent effect on the overall module stability (and is proportional to the number of nodes in the network). In other words, every node (gene) removed from the module, e.g., through extinction, influences the overall functioning of the module (which in turns represents an interacting community).
In contrast, the modules and clusters found using two different methods organize in an even manner, based on how frequently or infrequently functional genes linked to each functional group appear in the system. Thus, all functional groups are equally represented within each WCGMA module or K++ means cluster. We find that, in such systems, frequent functions associate together in big clusters, while infrequent ones associate together in smaller clusters. The size of the cluster thus represents the number of genes present per functional group in the cluster. In this case, each functional group includes an equal share of 8% of the genes in the cluster. This way, each module always includes a proportional representation of all the functional groups and there are no functional groups missing on any module or cluster.
Thus, the overall ecosystem is organized in such a way that a different co-occurring module is prepared to take the place of any collapsing module. We do not have an ecological mechanism for this organization, but we hypothesize that an ecosystem organized in such a way would be ready to take over any novel niche it opens in the environment (even if the new niche comes from extinction of the existing community) without the immediate need of genetic adaptation, through reproduction rate alone before evolutionary adaptation for optimal growth kicks in. The mechanism we describe here will not ensure colonization of any environment on its own unless the pre-adapted genetic pool is present but would allow colonizing a new environment as a fully functioning community, potentially speeding up colonization.
Indeed, our results also show a very striking pattern—that functions do not associate following a biological pattern (e.g., we do not find a stress function module or a virulence function, nor do we find functions linked to specific metabolic pathways, e.g., functions linked to carbon degradation do not associate in a particular module). The pattern also does not follow a phylogenetic pattern, as the phylogenetic module grouping (three distinct modules only) does not relate to the 21 functional modules found. A correlation analysis also show that the majority of the patterns found for functional distribution cannot be explained by phylogenetic distribution.
Association algorithms [
31] used to test whether there is any specific relationship between functional groups and the K++ means clusters did not provide any significant results either (not even random noise, as would be expected if the functional groups were distributed randomly among the clusters). This further supports the hypothesis that the functional associations (i.e., patterns seen for both WCNGA and K++ means algorithms) we find here are indeed evolutionarily homogeneous among clusters and that this is not the result of a random distribution.
We also found that this homogeneous distribution of functional groups within the WGCNA modules or the K++ means clusters is apparent regardless of the clustering method used. Or, in other words, the results obtained here are not an artifact of the method. Our analysis includes two independent methodologies using different algorithms (WGCNA network analysis and K++ clustering [
26,
29]) and both return the same proportional association. Furthermore, this association is significant after correcting for the number of functional genes per functional group included in the microarray. The main difference between the results from these two methods (WGCNA module detection and K++ clustering) is that, even if the overall pattern is similar (i.e., even module distribution across sites and more abundant modules dominating the functional landscape), the main module identified by WGCNA includes many more functions than the main cluster in the K++ cluster analysis, and it is also more predominant (i.e., in relation to the predominance of all other modules/clusters) in each site than the main cluster in the cluster analysis. This difference potentially relates to the fact that WGCNA only included those functions occurring in six sites or more (dominant functions), while the K++ means cluster analyses included all genes nested by functional category under the ecological assumption that similar functions are subjected to similar selective pressures (i.e., antibiotic resistance, carbon sequestration, metal resistance, etc.).
This pattern of functional group distribution would result in a resilient system based on a structural redundancy, composed of a series of independent functionally-sound clusters, and therefore for the system to ‘collapse’ or become non-functional all the clusters would need to collapse simultaneously. Furthermore, the pattern found at the functional level does not follow the pattern found for taxonomic diversity. Therefore, the pattern found for functional groups does not represent taxonomic units. This pattern is consistent with the scale-free network patterns found for taxonomic associations, as it would allow for a large number of taxonomic units to be depleted without altering the proportional distribution of functional genes with a minimum level of functional gene redundancy in the system (as each taxonomic unit would need to impair the functionality of all functional clusters before the system became dysfunctional).
If this pattern holds for other polar environments, our analyses would explain why we have not found strong evidence of recent environmental impact on polar marine microbial communities at the functional level (unless all communities analyzed have changed exactly in the same direction and intensity simultaneously, which is unlikely given we are comparing areas changing at different rates [
32]). Whilst arguably the temperature threshold might have not been reached for bacterial communities [
33], strong changes have been reported for phytoplankton communities from the maritime Antarctic and widespread evidence ecological changes at the taxonomic level is available from both the Arctic and the Antarctic [
34,
35]. Therefore, even if microbial community composition is changing rapidly in these regions due to climate warming, the change is not yet obvious at the functional level. The pattern we see here is not what we would expect from a genetic erosion scenario either, as all sites include tightly packed complex networks of essential genes and a series of smaller, but well-structured and homogeneously present, clusters of less frequent genes. Genetic erosion will not only be expected to homogenize the landscape, but to reduce functional diversity throughout. The scenario we see here is that of diverse functionally redundant clusters homogeneously distributed, which is a pattern more likely to reflect the very nature of functional gene distribution in microbial communities than a consequence of genetic erosion. Moreover, this pattern holds and is significant despite the large geographical range of the sampling covered, including areas above and below the Antarctic Convergence and an Arctic site. This pattern is also significant and holds despite the temporal variability in the sampling, suggesting again that it is characteristic of the system under study, and not an artefact of seasonal or geographical variation.
Overall, our results show a fairly even functional structure within microbial communities in high latitude oceans based on the presence/absence of specific functional genes and their relative co-occurrence. The sampling sites have repeatedly been acknowledged as some of the fastest warming environments on Earth [
17,
32]. Such changes, however, do not seem to influence the functional structure of the microbial communities present. Our results also show that 8% of the genes within a cluster are devoted to any particular functional group (
Figure 4), suggesting that, in order for a new function to become established within a functional cluster, the proportion of genes involved in the function in relation to the cluster gene pool must surpass a minimum threshold; less frequent functions might not succeed in integrating themselves within functional clusters. This mechanism seems to ensure that microbial networks do not rely on less frequent functions (the extreme of this situation would be ensuring that the ecosystem function does not rely on rare functions). In addition, no function becomes dominant over the others, also reducing the risk of communities relying on a small set of keystone functions. This also raises the question of the importance of rarity for overall ecosystem function. At higher taxonomic levels, rarity seems to be essential to maintain ecosystem resilience in highly diverse ecosystems [
36]; however, our results show that a high level of rarity does not necessarily translate into higher levels of redundancy, as there is no link between the distribution of less frequent functions and the distribution of taxonomic diversity. Consequently, less frequent genes do not contribute to the overall ecosystem function in the polar oceans at the ecological time scales considered here, at least for microbial communities.
Here, we have used two different clustering approaches, based on co-occurrence and presence-absence data, to support our conclusions. Whilst the network analysis is based on the strength of the signal in the microarray, the K++ clustering method is based on presence/absence data. These two methods were specifically chosen to reduce the probability of a mathematical bias or a methodological bias based on the strength of the molecular signal. The algorithms used in the network analysis and in the K++ cluster analysis indeed differ considerably, yet the conclusions are significantly similar (a homogeneous distribution of functions among clusters, and clusters among locations). Therefore, even if the level of clustering differs between these two methods (expected as the algorithms differ), the conclusions from these two methods do not.



 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





