Proteome Cold-Shock Response in the Extremely Acidophilic Archaeon, Cuniculiplasma divulgatum

 ,
,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Culture Conditions
2.2. Protein Extraction
2.3. Protein Digest
2.4. Analytical Instrumentation and Data Acquisition
2.5. Peptide Identification and Quantification
2.6. Bioinformatics Analysis
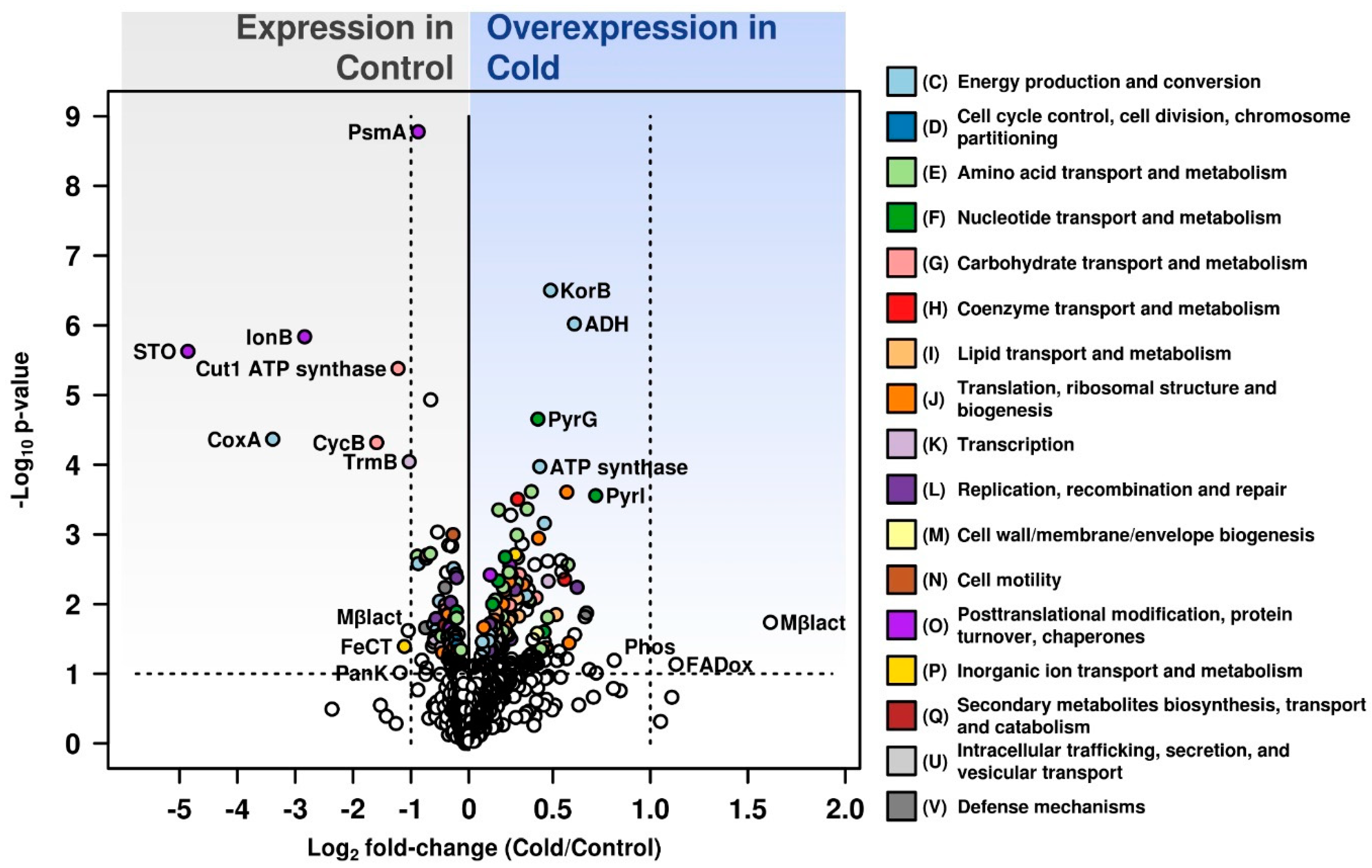
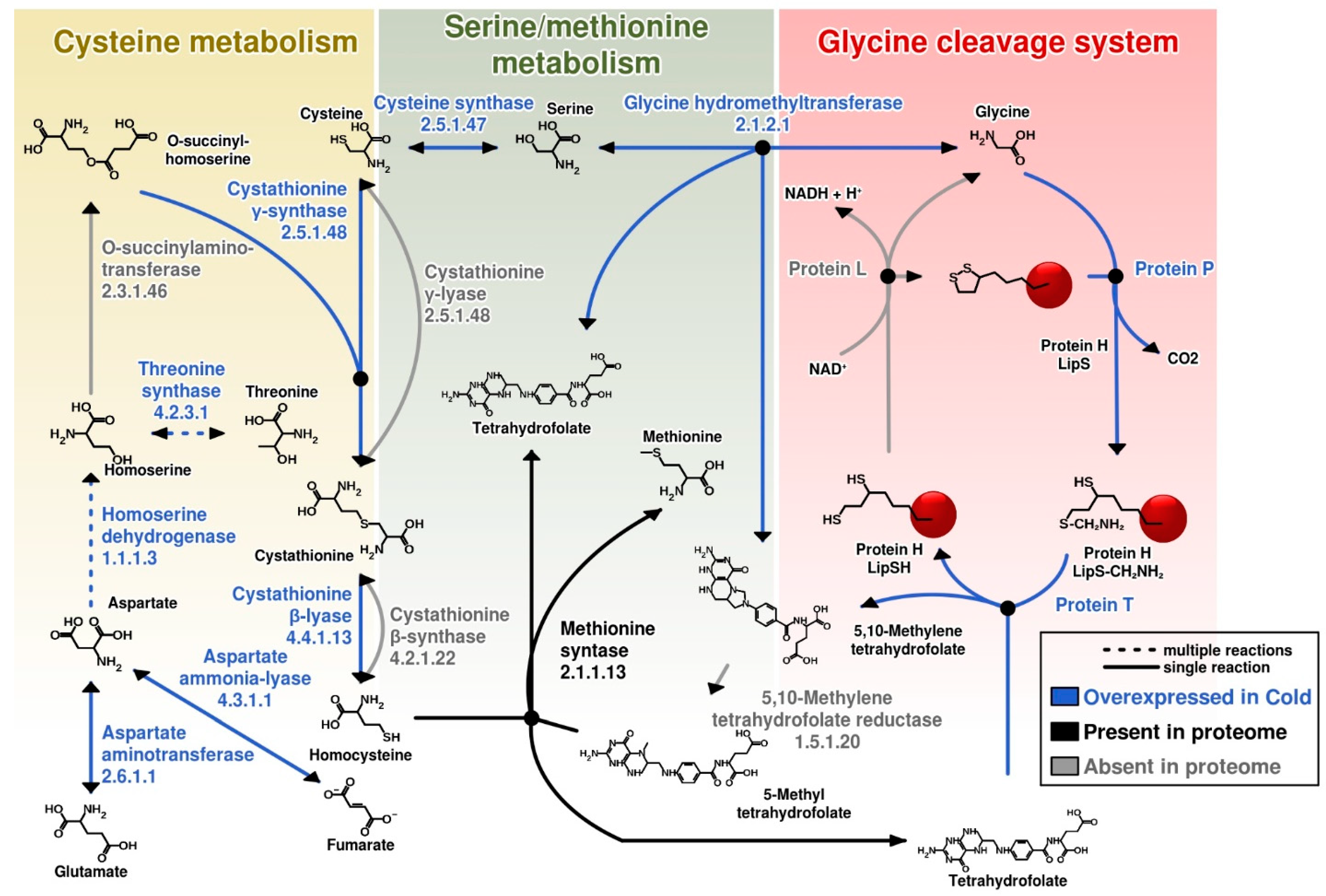
3. Results and Discussion
3.1. Analysis of the Total Intracellular Proteome of C. divulgatum Cells
3.2. Proteins Expressed under Optimal Growth Conditions
3.3. Proteins Overexpressed under Cold Shock Conditions
3.4. Differential Expression under Optimal Growth and Cold Shock Conditions of Functional Categories of Proteins
3.5. Most Abundant Proteins
4. Conclusions
Data
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Golyshina, O.V.; Lünsdorf, H.; Kublanov, I.V.; Goldenstein, N.I.; Hinrichs, K.U.; Golyshin, P.N. The novel extremely acidophilic, cell-wall-deficient archaeon Cuniculiplasma divulgatum gen. nov., sp. nov. represents a new family, Cuniculiplasmataceae fam. nov., of the order Thermoplasmatales. Int. J. Syst. Evol. Microbiol. 2016, 66, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.J.; Banfield, J.F. Microbial communities in acid mine drainage. FEMS Microbiol. Ecol. 2003, 44, 139–152. [Google Scholar] [CrossRef]
- Chen, L.X.; Méndez-García, C.; Dombrowski, N.; Servín-Garcidueñas, L.E.; Eloe-Fadrosh, E.A.; Fang, B.Z.; Luo, Z.H.; Tan, S.; Zhi, X.Y.; Hua, Z.S.; et al. Metabolic versatility of small archaea Micrarchaeota and Parvarchaeota. ISME J. 2018, 12, 756–775. [Google Scholar] [CrossRef] [PubMed]
- Golyshina, O.V.; Bargiela, R.; Golyshin, P.N. Cuniculiplasmataceae, their ecogenomic and metabolic patterns, and interactions with ‘ARMAN’. Extremophiles 2019, 23, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mueller, R.S.; Denef, V.J.; Kalnejais, L.H.; Suttle, K.B.; Thomas, B.C.; Wilmes, P.; Smith, R.L.; Nordstrom, D.K.; McCleskey, R.B.; Shah, M.B.; et al. Ecological distribution and population physiology defined by proteomics in a natural microbial community. Mol. Syst. Biol. 2010, 6, 374. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.S.; Albrecht, H.L.; Dawson, K.S.; Schaperdoth, I.; Freeman, K.H.; Pi, Y.; Pearson, A.; Macalady, J.L. Community genomic analysis of an extremely acidophilic sulfur-oxidizing biofilm. ISME J. 2012, 6, 158–170. [Google Scholar] [CrossRef]
- Krause, S.; Bremges, A.; Münch, P.C.; McHardy, A.C.; Gescher, J. Characterisation of a stable laboratory co-culture of acidophilic nanoorganisms. Sci. Rep. 2017, 7, 3289. [Google Scholar] [CrossRef]
- Golyshina, O.V.; Bargiela, R.; Toshchakov, S.V.; Chernyh, N.A.; Ramayah, S.; Korzhenkov, A.A.; Kublanov, I.V.; Golyshin, P.N. Diversity of “Ca. Micrarchaeota” in Two Distinct Types of Acidic Environments and Their Associations with Thermoplasmatales. Genes 2019, 10, 461. [Google Scholar] [CrossRef]
- Golyshina, O.V.; Toshchakov, S.V.; Makarova, K.S.; Gavrilov, S.N.; Korzhenkov, A.A.; La Cono, V.; Arcadi, E.; Nechitaylo, T.Y.; Ferrer, M.; Kublanov, I.V.; et al. ‘ARMAN’ archaea depend on association with euryarchaeal host in culture and in situ. Nat. Commun. 2017, 8, 60. [Google Scholar] [CrossRef]
- Huber, H.; Stetter, K.O. The Order Thermoplasmatales. In The Prokaryotes: A Handbook on the Biology of Bacteria, 3rd ed.; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.-H., Stackebrandt, E., Eds.; Springer: New York, NY, USA, 2006; Volume 3, pp. 101–112. [Google Scholar]
- Cavicchioli, R. Cold-adapted archaea. Nat. Rev. Microbiol. 2006, 4, 331–343. [Google Scholar] [CrossRef]
- Saunders, N.F.W.; Thomas, T.; Curmi, P.M.G.; Mattick, M.S.; Kuczek, E.; Slade, R.; Davis, J.; Franzmann, P.D.; Boone, D.; Rusterholtz, K.; et al. Mechanisms of thermal adaptation revealed from the genomes of the Antarctic Archaea, Methanogenium frigidum and Methanococcoides burtonii. Genome Res. 2003, 13, 1580–1588. [Google Scholar] [CrossRef] [PubMed]
- Siliakus, M.F.; van der Oost, J.; Kengen, S.W.M. Adaptations of archaeal and bacterial membranes to variations in temperature, pH and pressure. Extremophiles 2017, 21, 651–670. [Google Scholar] [CrossRef] [PubMed]
- De Maayer, P.; Anderson, D.; Cary, C.; Cowan, D.A. Some like it cold: Understanding the survival strategies of psychrophiles. EMBO Rep. 2014, 15, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Tribelli, P.M.; López, N.I. Reporting Key Features in Cold-Adapted Bacteria. Life 2018, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Kube, M.; Chernikova, T.N.; Al-Ramahi, Y.; Beloqui, A.; Lopez-Cortez, N.; Guazzaroni, M.-E.; Heipieper, H.J.; Klages, S.; Kotsyurbenko, O.R.; Langer, I.; et al. Genome sequence and functional genomic analysis of the oil-degrading bacterium Oleispira antarctica. Nat. Commun. 2013, 4, 2156. [Google Scholar] [CrossRef] [PubMed]
- Lawless, C.; Holman, S.W.; Brownridge, P.; Lanthaler, K.; Harman, V.M.; Watkins, R.; Hammond, D.E.; Miller, R.L.; Sims, P.F.; Grant, C.M.; et al. Direct and Absolute Quantification of over 1800 Yeast Proteins via Selected Reaction Monitoring. Mol. Cell Proteom. 2016, 15, 1309–1322. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Silva, J.C.; Gorenstein, M.V.; Li, G.Z.; Vissers, J.P.; Geromanos, S.J. Absolute quantification of proteins by LCMSE: A virtue of parallel MS acquisition. Mol. Cell Proteom. 2006, 5, 144–156. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Deutsch, E.W.; Csordas, A.; Sun, Z.; Jarnuczak, A.; Perez-Riverol, Y.; Ternent, T.; Campbell, D.S.; Bernal-Llinares, M.; Okuda, S.; Kawano, S.; et al. The ProteomeXchange Consortium in 2017: Supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2017, 54, D1100–D1106. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Xu, Q.W.; Wang, R.; Uszkoreit, J.; Griss, J.; Sanchez, A.; Reisinger, F.; Csordas, A.; Ternent, T.; del Toro, N.; et al. PRIDE Inspector Toolsuite: Moving towards a universal visualization tool for proteomics data standard formats and quality assessment of ProteomeXchange datasets. Mol. Cell Proteom. 2016, 15, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Maupin-Furlow, J.A. Proteolytic systems of archaea: Slicing, dicing, and mincing in the extreme. Emerg. Top. Life Sci. 2018, 2, 561–580. [Google Scholar]
- Pei, J.; Yan, J.; Jiang, Y. Characterization of the ATP-Dependent Lon-Like Protease in Methanobrevibacter smithii. Archaea 2016, 2016, 5759765. [Google Scholar] [CrossRef] [PubMed]
- Besche, H.; Tamura, N.; Tamura, T.; Zwickl, P. Mutational analysis of conserved AAA+ residues in the archaeal Lon protease from Thermoplasma acidophilum. FEBS Lett. 2004, 574, 161–166. [Google Scholar] [CrossRef]
- Cerletti, M.; Martínez, M.J.; Giménez, M.I.; Sastre, D.E.; Paggi, R.A.; De Castro, R.E. The LonB protease controls membrane lipids composition and is essential for viability in the extremophilic haloarchaeon Haloferax volcanii. Environ. Microbiol. 2014, 16, 1779–1792. [Google Scholar] [CrossRef]
- Akopian, T.N.; Kisselev, A.F.; Goldberg, A.L. Processive degradation of proteins and other catalytic properties of the proteasome from Thermoplasma acidophilum. J. Biol. Chem. 1997, 272, 1791–1798. [Google Scholar] [CrossRef][Green Version]
- Zhang, Q.; Iwasaki, T.; Wakagi, T.; Oshima, T. 2-oxoacid:ferredoxin oxidoreductase from the thermoacidophilic archaeon, Sulfolobus sp. strain 7. J. Biochem. 1996, 120, 587–599. [Google Scholar] [CrossRef]
- Golyshina, O.V.; Kublanov, I.V.; Tran, H.; Korzhenkov, A.A.; Lünsdorf, H.; Nechitaylo, T.Y.; Gavrilov, S.N.; Toshchakov, S.V.; Golyshin, P.N. Biology of archaea from a novel family Cuniculiplasmataceae (Thermoplasmata) ubiquitous in hyperacidic environments. Sci. Rep. 2016, 6, 39034. [Google Scholar] [CrossRef]
- Golyshina, O.V.; Tran, H.; Reva, O.N.; Lemak, S.; Yakunin, A.F.; Goesmann, A.; Nechitaylo, T.Y.; LaCono, V.; Smedile, F.; Slesarev, A.; et al. Metabolic and evolutionary patterns in the extremely acidophilic archaeon Ferroplasma acidiphilum YT. Sci. Rep. 2017, 7, 3682. [Google Scholar] [CrossRef]
- Grüber, G.; Subramanian Manimekalai, M.S.; Mayer, F.; Müller, V. ATP synthases from archaea: The beauty of a molecular motor. Biochim. Biophys. Acta (BBA) Bioenerg. 2014, 1837, 940–952. [Google Scholar]
- Siddiqui, K.S.; Cavicchioli, R. Cold-Adapted Enzymes. Ann. Rev. Biochem. 2006, 75, 403–433. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, M.V.; Schut, G.J.; Brehm, S.; Datta, S.; Adams, M.W. Cold shock of a hyperthermophilic archaeon: Pyrococcus furiosus exhibits multiple responses to a suboptimal growth temperature with a key role for membrane-bound glycoproteins. J. Bacteriol. 2005, 187, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Nichols, D.S.; Miller, M.R.; Davies, N.W.; Goodchild, A.; Raftery, M.; Cavicchioli, R. Cold adaptation in the Antarctic Archaeon Methanococcoides burtonii involves membrane lipid unsaturation. J. Bacteriol. 2004, 186, 8508–8515. [Google Scholar] [CrossRef] [PubMed]
- Péter, M.; Glatz, A.; Gudmann, P.; Gombos, I.; Török, Z.; Horváth, I.; Vígh, L.; Balogh, G. Metabolic crosstalk between membrane and storage lipids facilitates heat stress management in Schizosaccharomyces pombe. PLoS ONE 2017, 12, e0173739. [Google Scholar] [CrossRef]
- Barrero-Sicilia, C.; Silvestre, S.; Haslam, R.P.; Michaelson, L.V. Lipid remodelling: Unravelling the response to cold stress in Arabidopsis and its extremophile relative Eutrema salsugineum. Plant Sci. 2017, 263, 194–200. [Google Scholar] [CrossRef]
- Cavicchioli, R.; Thomas, T.; Curmi, P.M.G. Cold stress response in Archaea. Extremophiles 2000, 4, 321–331. [Google Scholar] [CrossRef]
- Motoyama, K.; Unno, H.; Hattori, A.; Takaoka, T.; Ishikita, H.; Kawaide, H.; Yoshimura, T.; Hemmi, H. A Single Amino Acid Mutation Converts (R)-5-Diphosphomevalonate Decarboxylase into a Kinase. J. Biol. Chem. 2017, 292, 2457–2469. [Google Scholar] [CrossRef]
- Pamnani, V.; Tamura, T.; Lupas, A.; Peters, J.; Cejka, Z.; Ashraf, W.; Baumeister, W. Cloning, sequencing and expression of VAT, a CDC48/p97 ATPase homologue from the archaeon Thermoplasma acidophilum. FEBS Lett. 1997, 404, 263–268. [Google Scholar] [CrossRef]
- Koonin, E.V.; Aravind, L.; Galperin, M.Y. A comparative-genomic view of the microbial stress response. In Bacterial Stress Responses; Storz, G., Hengge-Aronis, R., Eds.; ASM Press: Washington, DC, USA, 2000; pp. 417–444. [Google Scholar]
- Remelli, W.; Cereda, A.; Papenbrock, J.; Forlani, F.; Pagani, S. The rhodanese RhdA helps Azotobacter vinelandii in maintaining cellular redoxbalance. Biol. Chem. 2010, 391, 777–784. [Google Scholar] [CrossRef]
- Aussignargues, C.; Giuliani, M.C.; Infossi, P.; Lojou, E.; Guiral, M.; Giudici-Orticoni, M.T.; Ilbert, M. Rhodanese functions as sulfur supplier for key enzymes in sulfur energy metabolism. J. Biol. Chem. 2012, 287, 19936–19948. [Google Scholar] [CrossRef]
- Cipollone, R.; Frangipani, E.; Tiburzi, F.; Imperi, F.; Ascenzi, P.; Visca, P. Involvement of Pseudomonas aeruginosa rhodanese in protection from cyanide toxicity. Appl. Environ. Microbiol. 2007, 73, 390–398. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chaston, J.J.; Smits, C.; Aragão, D.; Wong, A.S.; Ahsan, B.; Sandin, S.; Molugu, S.K.; Molugu, S.K.; Bernal, R.A.; Stock, D.; et al. Structural and Functional Insights into the Evolution and Stress Adaptation of Type II Chaperonins. Structure 2016, 24, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Plaitakis, A.; Kalef-Ezra, E.; Kotzamani, D.; Zaganas, I.; Spanaki, C. The Glutamate Dehydrogenase Pathway and Its Roles in Cell and Tissue Biology in Health and Disease. Biology 2017, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Cabello, P.; Roldán, M.D.; Moreno-Vivián, C. Nitrate reduction and the nitrogen cycle in archaea. Microbiology 2004, 150, 3527–3546. [Google Scholar] [CrossRef]
- Kobayashi, T.; Higuchi, S.; Kimura, K.; Kudo, T.; Horikoshi, K. Properties of Glutamate Dehydrogenase and Its Involvement in Alanine Production in a Hyperthermophilic Archaeon, Thermococcus profundus. J. Biochem. 1995, 118, 587–592. [Google Scholar] [CrossRef]
- Porcelli, M.; Ilisso, C.P.; De Leo, E.; Cacciapuoti, G. Biochemical characterization of a thermostable adenosylmethionine synthetase from the archaeon Pyrococcus furiosus with high catalytic power. Appl. Biochem. Biotechnol. 2015, 175, 2916–2933. [Google Scholar] [CrossRef]
- Maruyama, T.; Suzuki, R.; Furutani, M. Archaeal peptidyl prolyl cis-trans isomerases (PPIases) update. Front. Biosci. 2004, 9, 1680–1720. [Google Scholar] [CrossRef]
- Belnap, C.P.; Pan, C.; Denef, V.J.; Samatova, N.F.; Hettich, R.L.; Banfield, J.F. Quantitative proteomic analyses of the response of acidophilic microbial communities to different pH conditions. ISME J. 2011, 5, 1152–1161. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bargiela, R.; Lanthaler, K.; Potter, C.M.; Ferrer, M.; Yakunin, A.F.; Paizs, B.; Golyshin, P.N.; Golyshina, O.V. Proteome Cold-Shock Response in the Extremely Acidophilic Archaeon, Cuniculiplasma divulgatum. Microorganisms 2020, 8, 759. https://doi.org/10.3390/microorganisms8050759
Bargiela R, Lanthaler K, Potter CM, Ferrer M, Yakunin AF, Paizs B, Golyshin PN, Golyshina OV. Proteome Cold-Shock Response in the Extremely Acidophilic Archaeon, Cuniculiplasma divulgatum. Microorganisms. 2020; 8(5):759. https://doi.org/10.3390/microorganisms8050759
Chicago/Turabian StyleBargiela, Rafael, Karin Lanthaler, Colin M. Potter, Manuel Ferrer, Alexander F. Yakunin, Bela Paizs, Peter N. Golyshin, and Olga V. Golyshina. 2020. "Proteome Cold-Shock Response in the Extremely Acidophilic Archaeon, Cuniculiplasma divulgatum" Microorganisms 8, no. 5: 759. https://doi.org/10.3390/microorganisms8050759
APA StyleBargiela, R., Lanthaler, K., Potter, C. M., Ferrer, M., Yakunin, A. F., Paizs, B., Golyshin, P. N., & Golyshina, O. V. (2020). Proteome Cold-Shock Response in the Extremely Acidophilic Archaeon, Cuniculiplasma divulgatum. Microorganisms, 8(5), 759. https://doi.org/10.3390/microorganisms8050759