Metatranscriptomic Analysis of Oil-Exposed Seawater Bacterial Communities Archived by an Environmental Sample Processor (ESP)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Setup
2.2. Sampling
2.2.1. Bacterioplankton Analysis
2.2.2. Chemical Analysis
2.3. Nucleic Acid Extractions and Sequencing
2.3.1. Metatranscriptomics
2.3.2. Amplicon Sequencing
2.4. Data Analysis
2.4.1. Metatranscriptomics
2.4.2. Amplicon Sequencing
3. Results
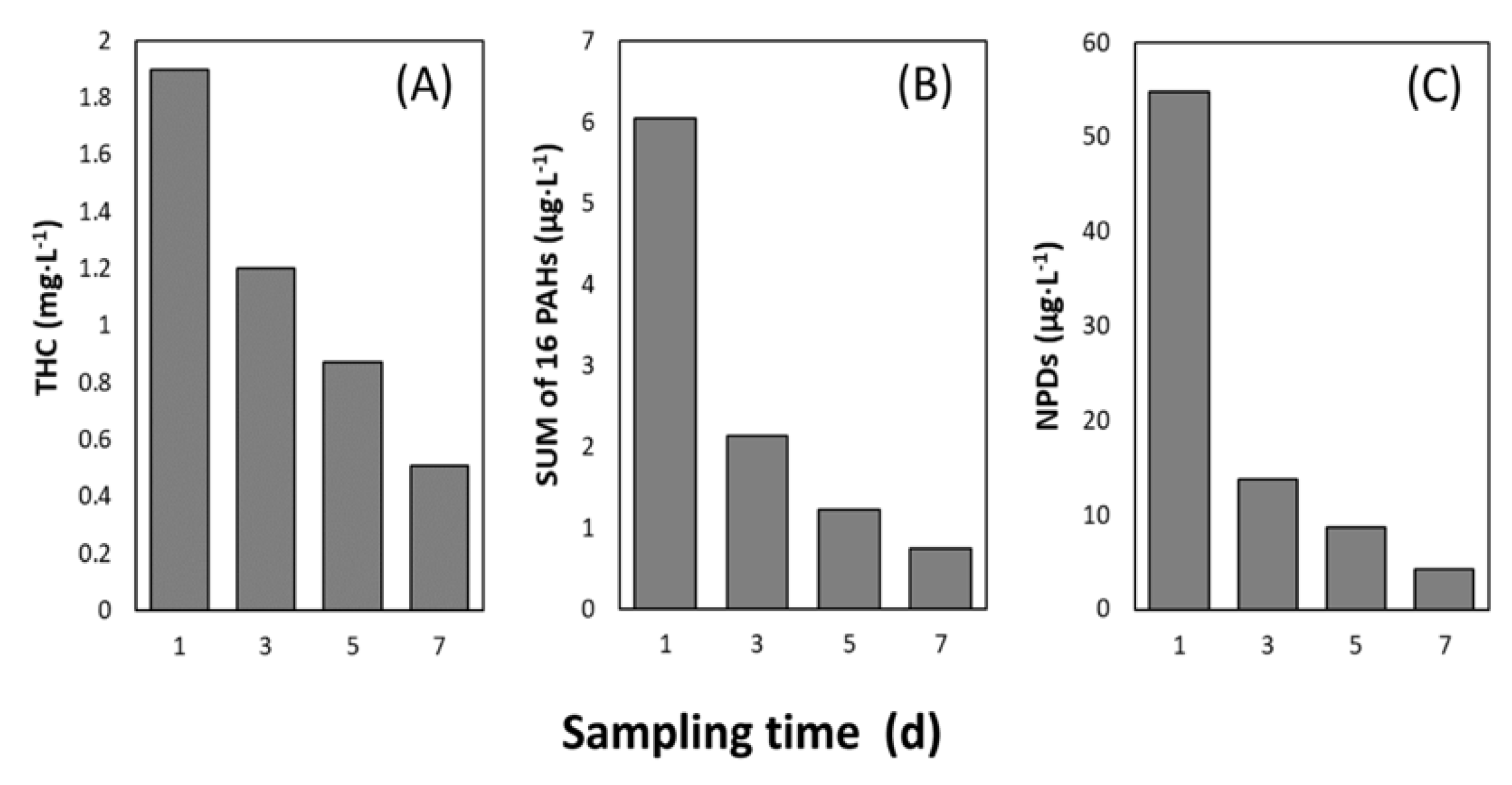
3.1. Total Hydrocarbon Content and PAH Concentration
3.2. Sequencing Statistics
3.2.1. RNA-Sequencing
3.2.2. Amplicon Sequencing
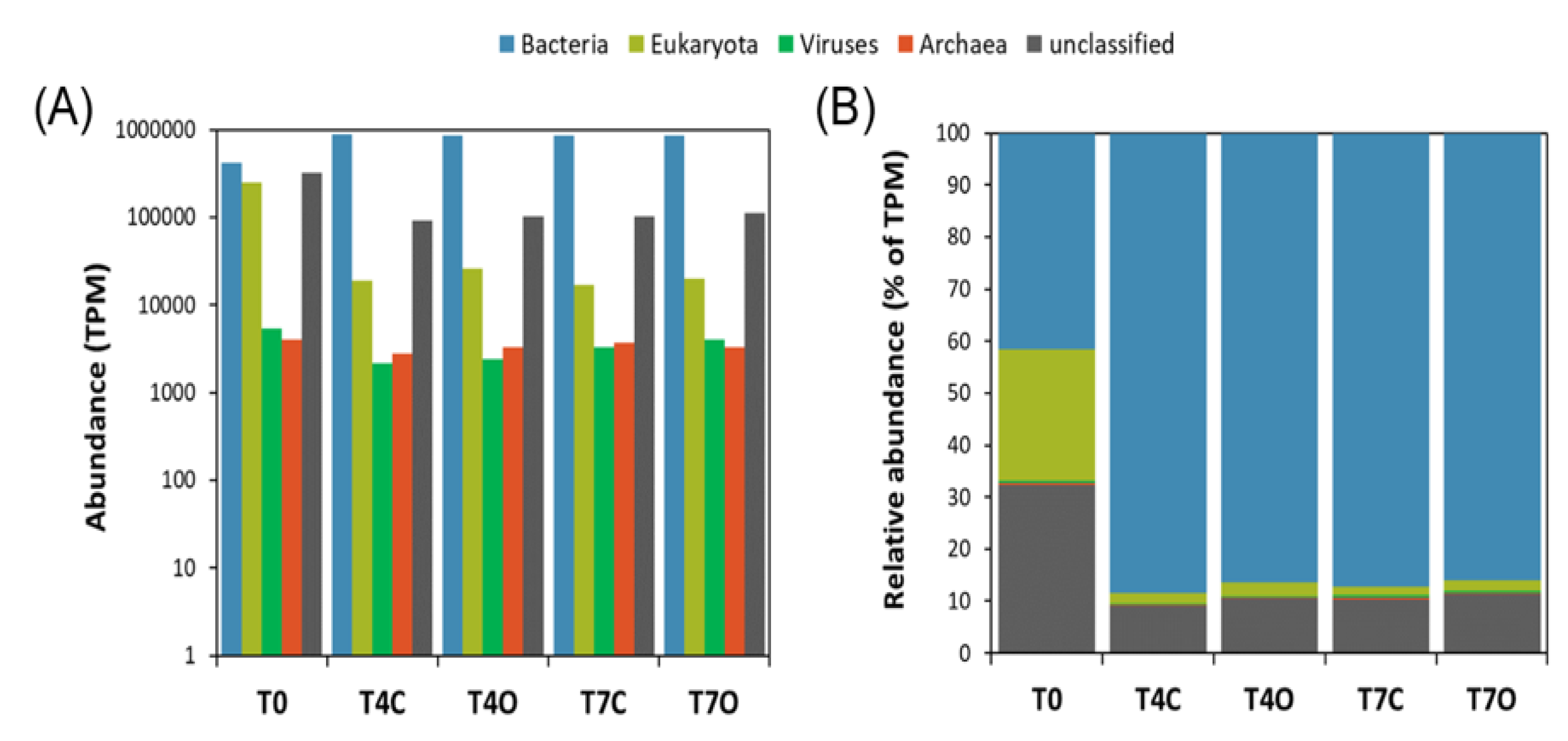
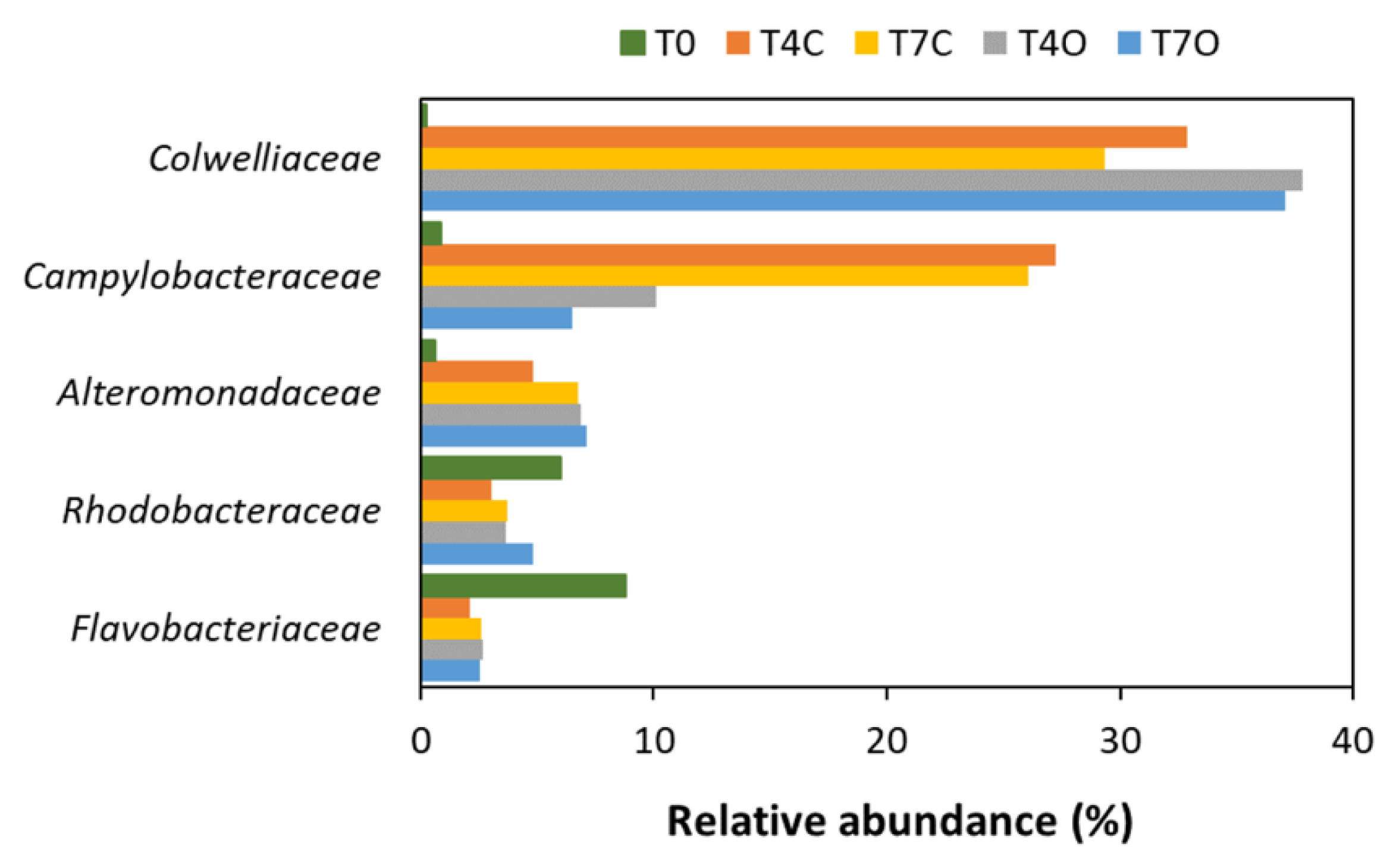
3.3. Taxonomic Analysis of the Microbial Communities
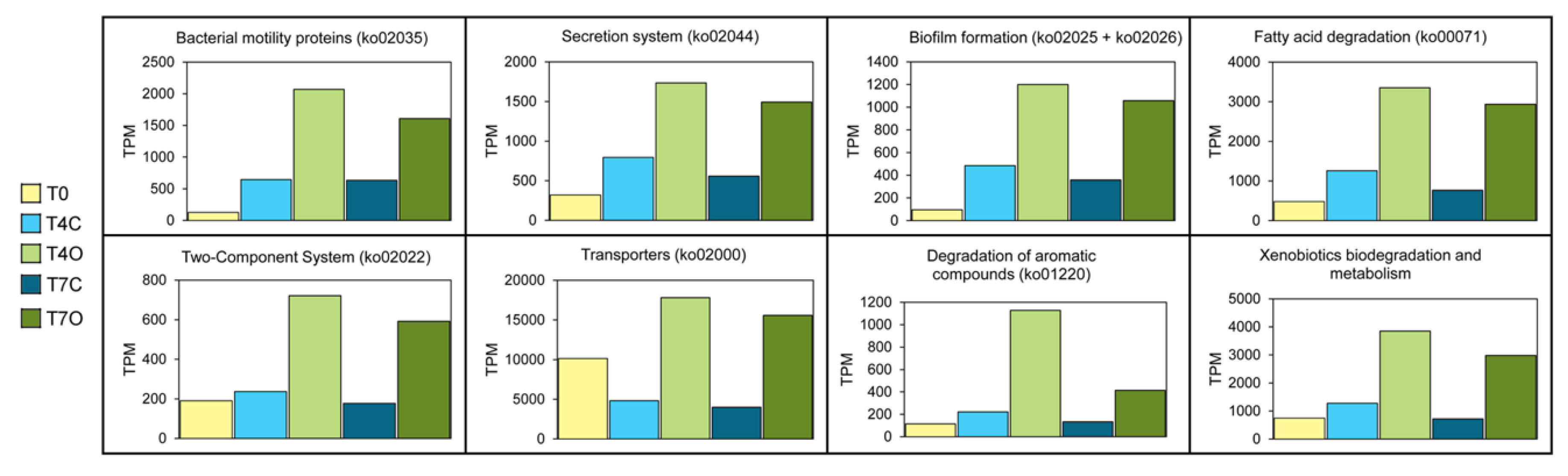
3.4. Functional Analysis of the Microbial Communities
3.5. Upregulated Functions
3.5.1. Upstream Pathways: Motility, Secretion, and Transporters
3.5.2. Alkane Degradation
3.5.3. Aromatic Degradation
4. Discussion
4.1. Rapid Decrease of Petroleum Hydrocarbon Concentrations
4.2. Microbial Community and Activities in the Acclimated Seawater
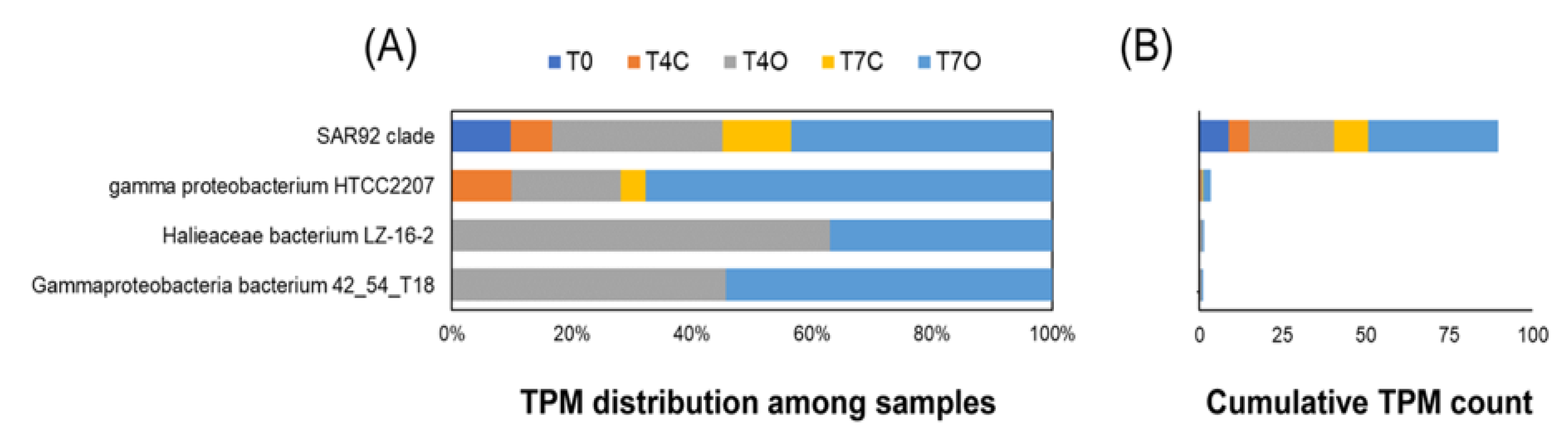
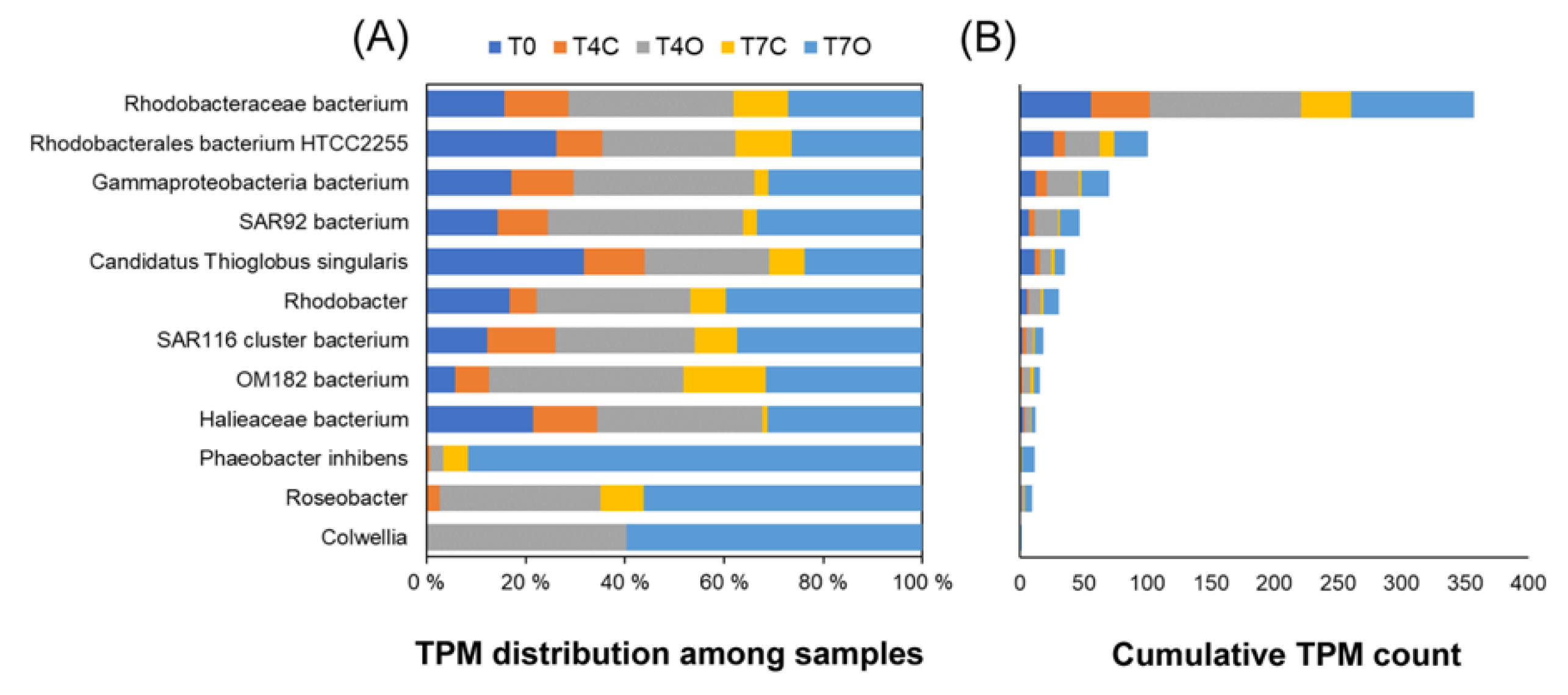
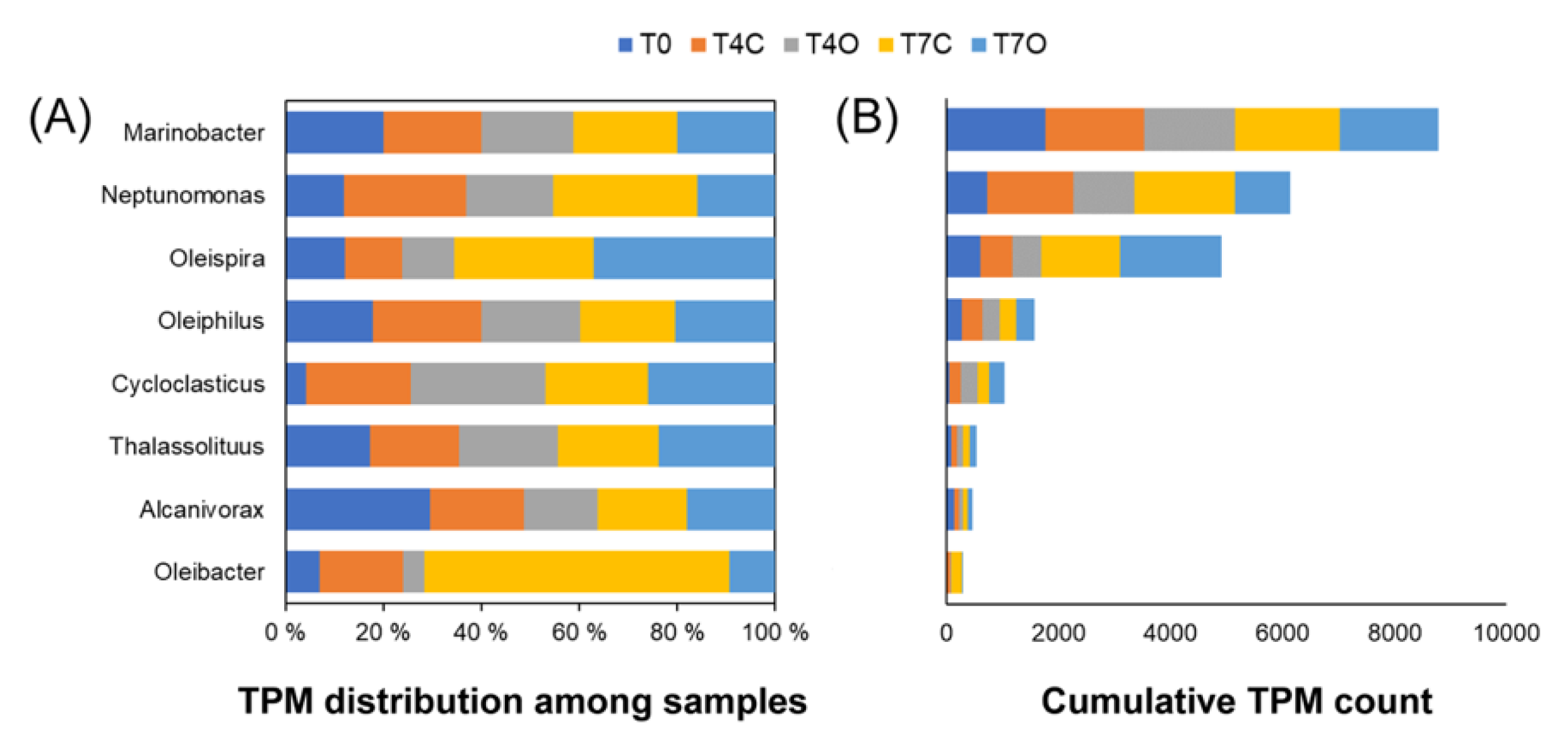
4.3. Dominant Generalists and Phytoplankton-Associated Taxa
4.4. Dominant Functions Reflect the “Tank Effect”
4.5. Upregulated Functions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ussler, W.; Preston, C.; Tavormina, P.; Pargett, D.; Jensen, S.; Roman, B.; Marin, R.; Shah, S.R.; Girguis, P.; Birch, J.M.; et al. Autonomous Application of Quantitative PCR in the Deep Sea: In Situ Surveys of Aerobic Methanotrophs Using the Deep-Sea Environmental Sample Processor. Environ. Sci. Technol. 2013, 47, 9339–9346. [Google Scholar] [CrossRef] [PubMed]
- Danovaro, R.; Carugati, L.; Berzano, M.; Cahill, A.E.; Carvalho, S.; Chenuil, A.; Corinaldesi, C.; Cristina, S.; David, R.; Dell’Anno, A.; et al. Implementing and Innovating Marine Monitoring Approaches for Assessing Marine Environmental Status. Front. Mar. Sci. 2016, 3, 233. [Google Scholar] [CrossRef]
- Ottesen, E.A. Probing the living ocean with ecogenomic sensors. Curr. Opin. Microbiol. 2016, 31, 132–139. [Google Scholar] [CrossRef]
- Jones, D.O.B.; Gates, A.; Huvenne, V.A.; Phillips, A.B.; Bett, B.J. Autonomous marine environmental monitoring: Application in decommissioned oil fields. Sci. Total Environ. 2019, 668, 835–853. [Google Scholar] [CrossRef] [PubMed]
- Palchetti, I.; Bettazzi, F.; Baussant, T. Chapter 12-Electrochemical Hybridization-Based Biosensor in Environmental Monitoring. In Nanotechnology and Biosensors; Nikolelis, D.P., Nikoleli, G.-P., Eds.; Elsevier: Amsterdam, The Netherland, 2018. [Google Scholar] [CrossRef]
- Bagi, A.; Knapik, K.; Nilsen, M.M.; Krolicka, A.; Boccadoro, C.; Baussant, T. Environmental Surveillance of Marine Systems Using Genosensors: Application to Offshore Activities. In Oilfield Microbiology; Skovhus, T.L., Whitby, C., Eds.; CRC Press: Boca Raton, FL, USA, 2019; pp. 235–246. [Google Scholar]
- Nogales, B.; Lanfranconi, M.P.; Piña-Villalonga, J.M.; Bosch, R. Anthropogenic perturbations in marine microbial communities. FEMS Microbiol. Rev. 2011, 35, 275–298. [Google Scholar] [CrossRef]
- Laroche, O.; Wood, S.A.; Tremblay, L.; Ellis, J.; Lear, G.; Pochon, X. A cross-taxa study using environmental DNA/RNA metabarcoding to measure biological impacts of offshore oil and gas drilling and production operations. Mar. Pollut. Bull. 2018, 127, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.; Ng, C.; Nshimyimana, J.P.; Loh, L.L.; Gin, K.Y.-H.; Thompson, J.R. Next-generation sequencing (NGS) for assessment of microbial water quality: Current progress, challenges, and future opportunities. Front. Microbiol. 2015, 6, 2783. [Google Scholar] [CrossRef]
- Hering, D.; Borja, A.; Jones, J.I.; Pont, D.; Boets, P.; Bouchez, A.; Bruce, K.; Drakare, S.; Hänfling, B.; Kahlert, M.; et al. Implementation options for DNA-based identification into ecological status assessment under the European Water Framework Directive. Water Res. 2018, 138, 192–205. [Google Scholar] [CrossRef]
- Cordier, T.; Lanzén, A.; Apothéloz-Perret-Gentil, L.; Stoeck, T.; Pawlowski, J. Embracing Environmental Genomics and Machine Learning for Routine Biomonitoring. Trends Microbiol. 2018, 27, 387–397. [Google Scholar] [CrossRef]
- Lozada, M.; Marcos, M.S.; Commendatore, M.G.; Gil, M.N.; Dionisi, H. The Bacterial Community Structure of Hydrocarbon-Polluted Marine Environments as the Basis for the Definition of an Ecological Index of Hydrocarbon Exposure. Microbes Environ. 2014, 29, 269–276. [Google Scholar] [CrossRef]
- Scholin, C.A.; Birch, J.; Jensen, S.; Iii, R.M.; Massion, E.; Pargett, D.; Preston, C.; Roman, B.; Iii, W.U. The Quest to Develop Ecogenomic Sensors: A 25-Year History of the Environmental Sample Processor (ESP) as a Case Study. Oceanography 2017, 30, 100–113. [Google Scholar] [CrossRef]
- Pargett, D.M.; Birch, J.M.; Preston, C.M.; Ryan, J.P.; Zhang, Y.; Scholin, C.A. Development of a mobile ecogenomic sensor. In Proceedings of the Oceans 2015-MTS/IEEE, Washington, DC, USA, 19–22 October 2015; pp. 1–6. [Google Scholar]
- A Ottesen, E.; Marin, R.; Preston, C.M.; Young, C.R.; Ryan, J.; A Scholin, C.; Delong, E. Metatranscriptomic analysis of autonomously collected and preserved marine bacterioplankton. ISME J. 2011, 5, 1881–1895. [Google Scholar] [CrossRef] [PubMed]
- Krolicka, A.; Boccadoro, C.; Mæland, M.; Preston, C.M.; Birch, J.; Scholin, C.; Baussant, T. Detection of oil leaks by quantifying hydrocarbonoclastic bacteria in cold marine environments using the Environmental Sample Processor. In Proceedings of the 37th AMOP Technical Seminar on Environmental Contamination and Response, Canmore, AB, Canada, 3–5 June 2014; pp. 791–807. [Google Scholar]
- Krolicka, A.; Boccadoro, C.; Nilsen, M.M.; Demir-Hilton, E.; Birch, J.; Preston, C.; Scholin, C.; Baussant, T.; Demi-Hilton, E.; Preston, C. Identification of microbial key-indicators of oil contamination at sea through tracking of oil biotransformation: An Arctic field and laboratory study. Sci. Total Environ. 2019, 696, 133715. [Google Scholar] [CrossRef] [PubMed]
- McGenity, T.J.; Folwell, B.D.; McKew, B.; O Sanni, G. Marine crude-oil biodegradation: A central role for interspecies interactions. Aquat. Biosyst. 2012, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Kostka, J.E.; Teske, A.P.; Joye, S.B.; Head, I.M. The metabolic pathways and environmental controls of hydrocarbon biodegradation in marine ecosystems. Front. Microbiol. 2014, 5, 5. [Google Scholar] [CrossRef]
- Brakstad, O.G.; Throne-Holst, M.; Netzer, R.; Stoeckel, D.; Atlas, R.M. Microbial communities related to biodegradation of dispersed Macondo oil at low seawater temperature with Norwegian coastal seawater. Microb. Biotechnol. 2015, 8, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, R.; Borglin, S.E.; Dubinsky, E.; Andersen, G.; Hazen, T.C. Microbial Response to the MC-252 Oil and Corexit 9500 in the Gulf of Mexico. Front. Microbiol. 2012, 3, 357. [Google Scholar] [CrossRef]
- Harayama, S.; Kasai, Y.; Hara, A. Microbial communities in oil-contaminated seawater. Curr. Opin. Biotechnol. 2004, 15, 205–214. [Google Scholar] [CrossRef]
- Ribicic, D.; Netzer, R.; Hazen, T.C.; Techtmann, S.M.; Drabløs, F.; Brakstad, O.G. Microbial community and metagenome dynamics during biodegradation of dispersed oil reveals potential key-players in cold Norwegian seawater. Mar. Pollut. Bull. 2018, 129, 370–378. [Google Scholar] [CrossRef]
- Dubinsky, E.; Conrad, M.; Chakraborty, R.; Bill, M.; Borglin, S.E.; Hollibaugh, J.T.; Mason, O.U.; Piceno, Y.; Reid, F.C.; Stringfellow, W.; et al. Succession of Hydrocarbon-Degrading Bacteria in the Aftermath of the Deepwater Horizon Oil Spill in the Gulf of Mexico. Environ. Sci. Technol. 2013, 47, 10860–10867. [Google Scholar] [CrossRef]
- Hazen, T.C.; Dubinsky, E.; DeSantis, T.Z.; Andersen, G.; Piceno, Y.; Singh, N.; Jansson, J.K.; Probst, A.J.; Borglin, S.E.; Fortney, J.L.; et al. Deep-Sea Oil Plume Enriches Indigenous Oil-Degrading Bacteria. Science 2010, 330, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Gontikaki, E.; Potts, L.; A Anderson, J.; Witte, U. Hydrocarbon-degrading bacteria in deep-water subarctic sediments (Faroe-Shetland Channel). J. Appl. Microbiol. 2018, 125, 1040–1053. [Google Scholar] [CrossRef] [PubMed]
- Yakimov, M.; Timmis, K.N.; Golyshin, P.N. Obligate oil-degrading marine bacteria. Curr. Opin. Biotechnol. 2007, 18, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Hu, Z.; Wang, H. A Retrospective Review of Microbiological Methods Applied in Studies Following the Deepwater Horizon Oil Spill. Front. Microbiol. 2018, 9, 520. [Google Scholar] [CrossRef]
- Scoma, A.; Hernandez-Sanabria, E.; Lacoere, T.; Junca, H.; Boon, N.; Pieper, D.H.; Vilchez-Vargas, R. Primers: Bacterial Genes Encoding Enzymes for Aerobic Hydrocarbon Degradation. In Immunity in Insects; Springer Science and Business Media LLC: Berlin, Germany, 2015; pp. 23–37. [Google Scholar]
- Cébron, A.; Norini, M.-P.; Beguiristain, T.; Leyval, C. Real-Time PCR quantification of PAH-ring hydroxylating dioxygenase (PAH-RHDα) genes from Gram positive and Gram negative bacteria in soil and sediment samples. J. Microbiol. Methods 2008, 73, 148–159. [Google Scholar] [CrossRef]
- Shahsavari, E.; Aburto-Medina, A.; Taha, M.; Ball, A.S. A quantitative PCR approach for quantification of functional genes involved in the degradation of polycyclic aromatic hydrocarbons in contaminated soils. MethodsX 2016, 3, 205–211. [Google Scholar] [CrossRef]
- Lu, Z.; Deng, Y.; Van Nostrand, J.D.; He, Z.; Voordeckers, J.; Zhou, A.; Lee, Y.-J.; Mason, O.U.; Dubinsky, E.; Chavarria, K.L.; et al. Microbial gene functions enriched in the Deepwater Horizon deep-sea oil plume. ISME J. 2011, 6, 451–460. [Google Scholar] [CrossRef]
- Vilchez-Vargas, R.; Geffers, R.; Suarez-Diez, M.; Conte, I.; Waliczek, A.; Kaser, V.S.; Kralova, M.; Junca, H.; Pieper, D.H. Analysis of the microbial gene landscape and transcriptome for aromatic pollutants and alkane degradation using a novel internally calibrated microarray system. Environ. Microbiol. 2012, 15, 1016–1039. [Google Scholar] [CrossRef]
- Bargiela, R.; Yakimov, M.; Golyshin, P.N.; Ferrer, M. Distribution of Hydrocarbon Degradation Pathways in the Sea. In Consequences of Microbial Interactions with Hydrocarbons, Oils, and Lipids: Production of Fuels and Chemicals; Springer Science and Business Media LLC: Berlin, Germany, 2017; pp. 629–651. [Google Scholar]
- Yergeau, E.; Maynard, C.; Sanschagrin, S.; Champagne, J.; Juck, D.; Lee, K.; Greer, C.W. Microbial Community Composition, Functions, and Activities in the Gulf of Mexico 1 Year after the Deepwater Horizon Accident. Appl. Environ. Microbiol. 2015, 81, 5855–5866. [Google Scholar] [CrossRef]
- Tremblay, J.; Fortin, N.; Elias, M.; Wasserscheid, J.; King, T.L.; Lee, K.; Greer, C.W. Metagenomic and metatranscriptomic responses of natural oil degrading bacteria in the presence of dispersants. Environ. Microbiol. 2019, 21, 2307–2319. [Google Scholar] [CrossRef]
- Tremblay, J.; Yergeau, E.; Fortin, N.; Cobanli, S.; Elias, M.; King, T.L.; Lee, K.; Greer, C.W. Chemical dispersants enhance the activity of oil- and gas condensate-degrading marine bacteria. ISME J. 2017, 11, 2793–2808. [Google Scholar] [CrossRef] [PubMed]
- Mason, O.U.; Hazen, T.C.; Borglin, S.; Chain, P.; Dubinsky, E.; Fortney, J.L.; Han, J.; Holman, H.-Y.N.; Hultman, J.; Lamendella, R.; et al. Metagenome, metatranscriptome and single-cell sequencing reveal microbial response to Deepwater Horizon oil spill. ISME J. 2012, 6, 1715–1727. [Google Scholar] [CrossRef] [PubMed]
- Rivers, A.; Sharma, S.; Tringe, S.G.; Martin, J.; Joye, S.; Moran, M.A. Transcriptional response of bathypelagic marine bacterioplankton to the Deepwater Horizon oil spill. ISME J. 2013, 7, 2315–2329. [Google Scholar] [CrossRef] [PubMed]
- Vikram, A.; Lipus, D.; Bibby, K. Metatranscriptome analysis of active microbial communities in produced water samples from the Marcellus Shale. Microb. Ecol. 2016, 72, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Arnberg, M.; Keitel-Gröner, F.; Westerlund, S.; Ramanand, S.; Bechmann, R.K.; Baussant, T. Exposure to chemically-dispersed oil is more harmful to early developmental stages of the Northern shrimp Pandalus borealis than mechanically-dispersed oil. Mar. Pollut. Bull. 2019, 145, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Preston, C.M.; Harris, A.; Ryan, J.; Roman, B.; Marin, R.; Jensen, S.; Everlove, C.; Birch, J.; Dzenitis, J.M.; Pargett, D.; et al. Underwater Application of Quantitative PCR on an Ocean Mooring. PLoS ONE 2011, 6, e22522. [Google Scholar] [CrossRef]
- Keith, L.H. The Source of U.S. EPA’s Sixteen PAH Priority Pollutants. Polycycl. Aromat. Compd. 2014, 35, 147–160. [Google Scholar] [CrossRef]
- Mizrahi-Man, O.; Davenport, E.; Gilad, Y. Taxonomic Classification of Bacterial 16S rRNA Genes Using Short Sequencing Reads: Evaluation of Effective Study Designs. PLoS ONE 2013, 8, e53608. [Google Scholar] [CrossRef]
- Sinclair, L.; Osman, O.A.; Bertilsson, S.; Eiler, A. Microbial Community Composition and Diversity via 16S rRNA Gene Amplicons: Evaluating the Illumina Platform. PLoS ONE 2015, 10, e0116955. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R.A. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Kopylova, E.; Noé, L.; Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef] [PubMed]
- Burge, S.W.; Daub, J.; Eberhardt, R.; Tate, J.; Barquist, L.; Nawrocki, E.P.; Eddy, S.R.; Gardner, P.P.; Bateman, A. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res. 2012, 41, D226–D232. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Peng, Y.; Leung, H.C.M.; Yiu, S.-M.; Lv, M.-J.; Zhu, X.-G.; Chin, F.Y. IDBA-tran: A more robust de novo de Bruijn graph assembler for transcriptomes with uneven expression levels. Bioinformatics 2013, 29, i326–i334. [Google Scholar] [CrossRef]
- Toseland, A.; Moxon, S.; Mock, T.; Moulton, V. Metatranscriptomes from diverse microbial communities: Assessment of data reduction techniques for rigorous annotation. BMC Genom. 2014, 15, 901. [Google Scholar] [CrossRef]
- Zhu, W.; Lomsadze, A.; Borodovsky, M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 2010, 38, e132. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012, 131, 281–285. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2014, 12, 59–60. [Google Scholar] [CrossRef]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Boil. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics 2011, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- I Love, M.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Boil. 2014, 15, 31. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Cerro-Gálvez, E.; Casal, P.; Lundin, D.; Piña, B.; Pinhassi, J.; Dachs, J.; Vila-Costa, M. Microbial responses to anthropogenic dissolved organic carbon in the Arctic and Antarctic coastal seawaters. Environ. Microbiol. 2019, 21, 1466–1481. [Google Scholar] [CrossRef]
- Radwan, S.S.; Khanafer, M.; Al-Awadhi, H. Ability of the So-Called Obligate Hydrocarbonoclastic Bacteria to Utilize Nonhydrocarbon Substrates Thus Enhancing Their Activities Despite their Misleading Name. BMC Microbiol. 2019, 19, 41. [Google Scholar] [CrossRef]
- Konat, G.K.a.J. The role of phytoplankton in the transport and distribution of polynuclear aromatic hydrocarbons [PAHs] in the southern Baltic environment. Oceanologia 1997, 39, 267–277. [Google Scholar]
- Gonzalez-Gaya, B.; Martínez-Varela, A.; Vila-Costa, M.; Casal, P.; Cerro-Gálvez, E.; Berrojalbiz, N.; Lundin, D.; Vidal, M.; Mompeán, C.; Bode, A.; et al. Biodegradation as an important sink of aromatic hydrocarbons in the oceans. Nat. Geosci. 2019, 12, 119–125. [Google Scholar] [CrossRef]
- Rinta-Kanto, J.; Sun, S.; Sharma, S.; Kiene, R.P.; Moran, M.A. Bacterial community transcription patterns during a marine phytoplankton bloom. Environ. Microbiol. 2011, 14, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Nowinski, B.; Smith, C.B.; Thomas, C.M.; Esson, K.; Marin, R.; Preston, C.M.; Birch, J.M.; Scholin, C.A.; Huntemann, M.; Clum, A.; et al. Microbial metagenomes and metatranscriptomes during a coastal phytoplankton bloom. Sci. Data 2019, 6, 129. [Google Scholar] [CrossRef] [PubMed]
- Klindworth, A.; Mann, A.J.; Huang, S.; Wichels, A.; Quast, C.; Waldmann, J.; Teeling, H.; Glöckner, F.O. Diversity and activity of marine bacterioplankton during a diatom bloom in the North Sea assessed by total RNA and pyrotag sequencing. Mar. Genom. 2014, 18, 185–192. [Google Scholar] [CrossRef]
- Wemheuer, B.; Wemheuer, F.; Hollensteiner, J.; Meyer, F.-D.; Voget, S.; Daniel, R. The green impact: Bacterioplankton response toward a phytoplankton spring bloom in the southern North Sea assessed by comparative metagenomic and metatranscriptomic approaches. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef]
- O Delmont, T.; Hammar, K.M.; Ducklow, H.W.; Yager, P.L.; Post, A.F. Phaeocystis antarctica blooms strongly influence bacterial community structures in the Amundsen Sea polynya. Front. Microbiol. 2014, 5, 646. [Google Scholar] [CrossRef]
- Brakstad, O.G.; Lofthus, S.; Ribicic, D.; Netzer, R. Biodegradation of Petroleum Oil in Cold Marine Environments. In Psychrophiles: From Biodiversity to Biotechnology; Springer Science and Business Media LLC: Berlin, Germany, 2017; pp. 613–644. [Google Scholar]
- Suk, W.A.; Mishamandani, S. Changing exposures in a changing world: Models for reducing the burden of disease. Rev. Environ. Health 2016, 31, 93–96. [Google Scholar] [CrossRef]
- Thompson, H.; Bowler, B.; Angelova, A.; Jones, M.; Gutierrez, T. Enhanced crude oil biodegradative potential of natural phytoplankton-associated hydrocarbonoclastic bacteria. Environ. Microbiol. 2017, 19, 2843–2861. [Google Scholar] [CrossRef]
- Binark, N.; Güven, K.C.; Gezgin, T.; Unlu, S. Oil Pollution of Marine Algae. Bull. Environ. Contam. Toxicol. 2000, 64, 866–872. [Google Scholar] [CrossRef]
- Kowalewska, G. Phytoplankton—The main factorresponsible for transport of polynuclear aromatic hydrocarbons from water sediments in the Southern Baltic ecosys-tem. ICES J. Mar. Sci. 1999, 56, 219–222. [Google Scholar] [CrossRef]
- Dos Santos, A.L.; Gourvil, P.; Rodríguez, F.; Garrido, J.L.; Vaulot, D. Photosynthetic pigments of oceanic Chlorophyta belonging to prasinophytes clade VII. J. Phycol. 2016, 52, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Dupré, C.; Yoneda, K.; Watanabe, M.M.; Legrand, J.; Grizeau, D. Characteristics of extracellular hydrocarbon-rich microalga Botryococcus braunii for biofuels production: Recent advances and opportunities. Process Biochem. 2016, 51, 1866–1875. [Google Scholar] [CrossRef]
- Methé, B.A.; Nelson, K.E.; Deming, J.W.; Momen, B.; Melamud, E.; Zhang, X.; Moult, J.; Madupu, R.; Nelson, K.E.; Dodson, R.J.; et al. The psychrophilic lifestyle as revealed by the genome sequence of Colwellia psychrerythraea 34H through genomic and proteomic analyses. Proc. Natl. Acad. Sci. USA 2005, 102, 10913–10918. [Google Scholar] [CrossRef] [PubMed]
- Bælum, J.; Borglin, S.; Chakraborty, R.; Fortney, J.L.; Lamendella, R.; Mason, O.U.; Auer, M.; Zemla, M.; Bill, M.; Conrad, M.; et al. Deep-sea bacteria enriched by oil and dispersant from the Deepwater Horizon spill. Environ. Microbiol. 2012, 14, 2405–2416. [Google Scholar] [CrossRef] [PubMed]
- Redmond, M.C.; Valentine, D.L. Natural gas and temperature structured a microbial community response to the Deepwater Horizon oil spill. Proc. Natl. Acad. Sci. USA 2011, 109, 20292–20297. [Google Scholar] [CrossRef] [PubMed]
- Sieradzki, E.T.; Morando, M.; Fuhrman, J.A. Metagenomics and stable isotope probing offer insights into metabolism of polycyclic aromatic hydrocarbons degraders in chronically polluted seawater. bioRxiv 2019. [Google Scholar] [CrossRef]
- Krolicka, A.; Boccadoro, C.; Nilsen, M.M.; Baussant, T. Capturing Early Changes in the Marine Bacterial Community as a Result of Crude Oil Pollution in a Mesocosm Experiment. Microbes Environ. 2017, 32, 358–366. [Google Scholar] [CrossRef]
- Campeão, M.E.; Swings, J.; Silva, B.S.; Otsuki, K.; Thompson, F.L.; Thompson, C.C. “CandidatusColwellia aromaticivorans” sp. nov., “CandidatusHalocyntiibacter alkanivorans” sp. nov., and “CandidatusUlvibacter alkanivorans” sp. nov. Genome Sequences. Microbiol. Resour. Announc. 2019, 8, e00086–e00119. [Google Scholar] [CrossRef]
- Campeão, M.E.; Reis, L.; Leomil, L.; De Oliveira, L.; Otsuki, K.; Gardinali, P.; Pelz, O.; Valle, R.; Thompson, F.L.; Thompson, C.C. The Deep-Sea Microbial Community from the Amazonian Basin Associated with Oil Degradation. Front. Microbiol. 2017, 8, 1019. [Google Scholar] [CrossRef]
- Otth, L.; Solís, G.; Wilson, M.; Fernandez, H. Susceptibility of Arcobacter butzleri to heavy metals. Braz. J. Microbiol. 2005, 36, 286–288. [Google Scholar] [CrossRef][Green Version]
- Allers, E.; Gómez-Consarnau, L.; Pinhassi, J.; Gasol, J.M.; Šimek, K.; Pernthaler, J. Response of Alteromonadaceae and Rhodobacteriaceae to glucose and phosphorus manipulation in marine mesocosms. Environ. Microbiol. 2007, 9, 2417–2429. [Google Scholar] [CrossRef] [PubMed]
- Chronopoulou, M.; Sanni, G.O.; Silas-Olu, D.I.; Van Der Meer, J.R.; Timmis, K.N.; Brussaard, C.P.D.; McGenity, T.J. Generalist hydrocarbon-degrading bacterial communities in the oil-polluted water column of the North Sea. Microb. Biotechnol. 2014, 8, 434–447. [Google Scholar] [CrossRef]
- Von Scheibner, M.; Sommer, U.; Jürgens, K. Tight Coupling of Glaciecola spp. and Diatoms during Cold-Water Phytoplankton Spring Blooms. Front. Microbiol. 2017, 8, 2441. [Google Scholar] [CrossRef] [PubMed]
- Villarreal-Chiu, J.F.; Quinn, J.P.; McGrath, J.W. The Genes and Enzymes of Phosphonate Metabolism by Bacteria, and Their Distribution in the Marine Environment. Front. Microbiol. 2012, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Shivaramu, M.S.; Randive, A.K.; Kumari, R.; Gauns, M.; Ponnapakkam, L.A. Spatiotemporal variation of alkaline phosphatase activity in coastal waters off Trivandrum. Oceanologia 2019, 61, 170–177. [Google Scholar] [CrossRef]
- Baltar, F.; Lundin, D.; Palovaara, J.; Lekunberri, I.; Reinthaler, T.; Herndl, G.J.; Pinhassi, J. Prokaryotic Responses to Ammonium and Organic Carbon Reveal Alternative CO2 Fixation Pathways and Importance of Alkaline Phosphatase in the Mesopelagic North Atlantic. Front. Microbiol. 2016, 7, 788. [Google Scholar] [CrossRef]
- Mezzina, M.P.; Pettinari, M.J. Phasins, Multifaceted Polyhydroxyalkanoate Granule-Associated Proteins. Appl. Environ. Microbiol. 2016, 82, 5060–5067. [Google Scholar] [CrossRef]
- Dunn, M.F.; Ramirez-Trujillo, J.A.; Hernández-Lucas, I. Major roles of isocitrate lyase and malate synthase in bacterial and fungal pathogenesis. Microbiology 2009, 155, 3166–3175. [Google Scholar] [CrossRef]
- Tribelli, P.M.; Venero, E.C.S.; Ricardi, M.; Gómez-Lozano, M.; Iustman, L.J.R.; Molin, S.; López, N.I. Novel Essential Role of Ethanol Oxidation Genes at Low Temperature Revealed by Transcriptome Analysis in the Antarctic Bacterium Pseudomonas extremaustralis. PLoS ONE 2015, 10, e0145353. [Google Scholar] [CrossRef]
- Wang, W.; Shao, Z. The long-chain alkane metabolism network of Alcanivorax dieselolei. Nat. Commun. 2014, 5, 5755. [Google Scholar] [CrossRef] [PubMed]
- Blanvillain, S.; Meyer, D.; Boulanger, A.; Lautier, M.; Guynet, C.; Denancé, N.; Vasse, J.; Lauber, E.; Arlat, M. Plant Carbohydrate Scavenging through TonB-Dependent Receptors: A Feature Shared by Phytopathogenic and Aquatic Bacteria. PLoS ONE 2007, 2, e224. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.N.; Lozada, M.; Anselmino, L.E.; Musumeci, M.A.; Henrissat, B.; Jansson, J.K.; Mac Cormack, W.P.; Carroll, J.; Sjöling, S.; Lundgren, L.; et al. Metagenomics unveils the attributes of the alginolytic guilds of sediments from four distant cold coastal environments. Environ. Microbiol. 2016, 18, 4471–4484. [Google Scholar] [CrossRef] [PubMed]
- Cochran, P.; Kellogg, C.; Paul, J.H. Prophage induction of indigenous marine lysogenic bacteria by environmental pollutants. Mar. Ecol. Prog. Ser. 1998, 164, 125–133. [Google Scholar] [CrossRef]
- Jiang, S.C.; Paul, J. Occurrence of lysogenic bacteria in marine microbial communities as determined by prophage induction. Mar. Ecol. Prog. Ser. 1996, 142, 27–38. [Google Scholar] [CrossRef]
- Thingstad, T.F. Elements of a theory for the mechanisms controlling abundance, diversity, and biogeochemical role of lytic bacterial viruses in aquatic systems. Limnol. Oceanogr. 2000, 45, 1320–1328. [Google Scholar] [CrossRef]
- Sauret, C.; Böttjer, D.; Talarmin, A.; Guigue, C.; Conan, P.; Pujo-Pay, M.; Ghiglione, J.-F. Top-Down Control of Diesel-Degrading Prokaryotic Communities. Microb. Ecol. 2015, 70, 445–458. [Google Scholar] [CrossRef]
- Fuhrman, J. Bacterioplankton Roles in Cycling of Organic Matter: The Microbial Food Web. Prim. Product. Biogeochem. Cycles Sea 1992, 43, 361–383. [Google Scholar] [CrossRef]
- Rosenberg, E.; Bittan-Banin, G.; Sharon, G.; Shon, A.; Hershko, G.; Levy, I.; Ron, E.Z. The phage-driven microbial loop in petroleum bioremediation. Microb. Biotechnol. 2010, 3, 467–472. [Google Scholar] [CrossRef]
- Herrick, J.B.; Stuart-Keil, K.G.; Ghiorse, W.C.; Madsen, E.L. Natural horizontal transfer of a naphthalene dioxygenase gene between bacteria native to a coal tar-contaminated field site. Appl. Environ. Microbiol. 1997, 63, 2330–2337. [Google Scholar] [CrossRef]
- Costeira, R.; Doherty, R.; Allen, C.; Larkin, M.J.; Kulakov, L.A. Analysis of viral and bacterial communities in groundwater associated with contaminated land. Sci. Total Environ. 2019, 656, 1413–1426. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, C.M.; Rodriguez-Valera, F.; Kimes, N.E.; Ghai, R. Expanding the Marine Virosphere Using Metagenomics. PLoS Genet. 2013, 9, e1003987. [Google Scholar] [CrossRef] [PubMed]
- Garin-Fernandez, A.; Pereira-Flores, E.; Glöckner, F.O.; Wichels, A. The North Sea goes viral: Occurrence and distribution of North Sea bacteriophages. Mar. Genom. 2018, 41, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Van Hamme, J.D.; Singh, A.; Ward, O.P. Recent Advances in Petroleum Microbiology. Microbiol. Mol. Boil. Rev. 2003, 67, 503–549. [Google Scholar] [CrossRef] [PubMed]
- Joye, S.; Kleindienst, S.; Gilbert, J.; Handley, K.M.; Weisenhorn, P.; Overholt, W.; Kostka, J. Responses of Microbial Communities to Hydrocarbon Exposures. Oceanography 2016, 29, 136–149. [Google Scholar] [CrossRef]
- Parales, R.E.; Ditty, J.L. Chemotaxis to Hydrocarbons. In Cellular Ecophysiology of Microbe: Hydrocarbon and Lipid Interactions; Springer Science and Business Media LLC: Berlin, Germany, 2018; pp. 221–239. [Google Scholar]
- Shi, Y.; McCarren, J.; Delong, E. Transcriptional responses of surface water marine microbial assemblages to deep-sea water amendment. Environ. Microbiol. 2011, 14, 191–206. [Google Scholar] [CrossRef]
- Suleiman, M.; Zecher, K.; Yücel, O.; Jagmann, N.; Philipp, B. Interkingdom Cross-Feeding of Ammonium from Marine Methylamine-Degrading Bacteria to the Diatom Phaeodactylum tricornutum. Appl. Environ. Microbiol. 2016, 82, 7113–7122. [Google Scholar] [CrossRef]
- Landa, M.; Burns, A.S.; Durham, B.; Esson, K.; Nowinski, B.; Sharma, S.; Vorobev, A.; Nielsen, T.; Kiene, R.P.; Moran, M.A. Sulfur metabolites that facilitate oceanic phytoplankton-bacteria carbon flux. ISME J. 2019, 13, 2536–2550. [Google Scholar] [CrossRef]
- Clifford, E.L.; Varela, M.M.; De Corte, D.; Bode, A.; Ortiz, V.; Herndl, G.J.; Sintes, E. Taurine Is a Major Carbon and Energy Source for Marine Prokaryotes in the North Atlantic Ocean off the Iberian Peninsula. Microb. Ecol. 2019, 78, 299–312. [Google Scholar] [CrossRef]
- Wasmund, K.; Burns, K.A.; Bourne, D.G. Novel Alkane Hydroxylase Gene (alkB) Diversity in Sediments Associated with Hydrocarbon Seeps in the Timor Sea, Australia. Appl. Environ. Microbiol. 2009, 75, 7391–7398. [Google Scholar] [CrossRef]
- Wang, W.; Wang, L.; Shao, Z. Diversity and Abundance of Oil-Degrading Bacteria and Alkane Hydroxylase (alkB) Genes in the Subtropical Seawater of Xiamen Island. Microb. Ecol. 2010, 60, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xu, W.; Liu, Y.; Cai, M.; Luo, Z.; Li, M. Metagenomics Reveals Microbial Diversity and Metabolic Potentials of Seawater and Surface Sediment from a Hadal Biosphere at the Yap Trench. Front. Microbiol. 2018, 9, 2402. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, H.; Bomberg, M.; Nyyssönen, M.; Reunamo, A.; Jørgensen, K.S.; Vikman, M. Oil degradation potential of microbial communities in water and sediment of Baltic Sea coastal area. PLoS ONE 2019, 14, e0218834. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhong, R.; Shan, D.; Shao, Z. Indigenous oil-degrading bacteria in crude oil-contaminated seawater of the Yellow sea, China. Appl. Microbiol. Biotechnol. 2014, 98, 7253–7269. [Google Scholar] [CrossRef]
- Paissé, S.; Duran, R.; Coulon, F.; Goñi-Urriza, M. Are alkane hydroxylase genes (alkB) relevant to assess petroleum bioremediation processes in chronically polluted coastal sediments? Appl. Microbiol. Biotechnol. 2011, 92, 835–844. [Google Scholar] [CrossRef]











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knapik, K.; Bagi, A.; Krolicka, A.; Baussant, T. Metatranscriptomic Analysis of Oil-Exposed Seawater Bacterial Communities Archived by an Environmental Sample Processor (ESP). Microorganisms 2020, 8, 744. https://doi.org/10.3390/microorganisms8050744
Knapik K, Bagi A, Krolicka A, Baussant T. Metatranscriptomic Analysis of Oil-Exposed Seawater Bacterial Communities Archived by an Environmental Sample Processor (ESP). Microorganisms. 2020; 8(5):744. https://doi.org/10.3390/microorganisms8050744
Chicago/Turabian StyleKnapik, Kamila, Andrea Bagi, Adriana Krolicka, and Thierry Baussant. 2020. "Metatranscriptomic Analysis of Oil-Exposed Seawater Bacterial Communities Archived by an Environmental Sample Processor (ESP)" Microorganisms 8, no. 5: 744. https://doi.org/10.3390/microorganisms8050744
APA StyleKnapik, K., Bagi, A., Krolicka, A., & Baussant, T. (2020). Metatranscriptomic Analysis of Oil-Exposed Seawater Bacterial Communities Archived by an Environmental Sample Processor (ESP). Microorganisms, 8(5), 744. https://doi.org/10.3390/microorganisms8050744