Comparative Genomic and Proteomic Analyses of Three Widespread Phytophthora Species: Phytophthora chlamydospora, Phytophthora gonapodyides and Phytophthora pseudosyringae


Abstract
1. Introduction
2. Materials and Methods
2.1. Isolation and Culturing
2.2. DNA Extraction and Sequencing
2.3. Genome Assembly
2.4. Gene Annotation
2.5. Identification of Cytoplasmic Effectors
2.6. Phylogenomics
2.7. Phylostratigraphy
2.8. Culturing Conditions and Extraction of Phytophthora Extracellular Proteins
2.9. Culturing Conditions and Extraction of Phytophthora Mycelial Proteins
2.10. Protein Digestion and LC-MS/MS Identification of Phytophthora Proteins
2.11. Data Deposition
3. Results and Discussion
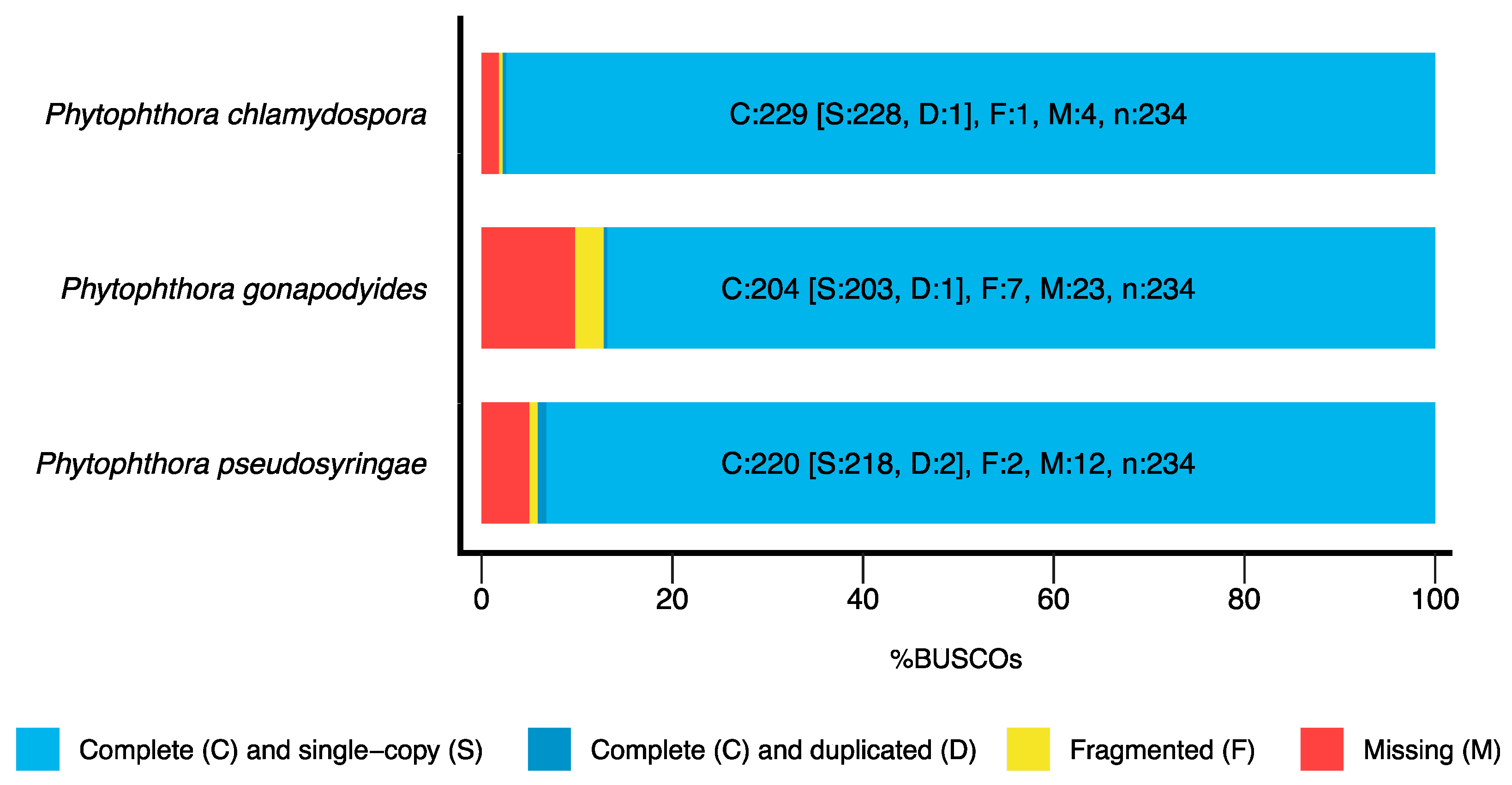
3.1. Genome Sequencing and Assembly
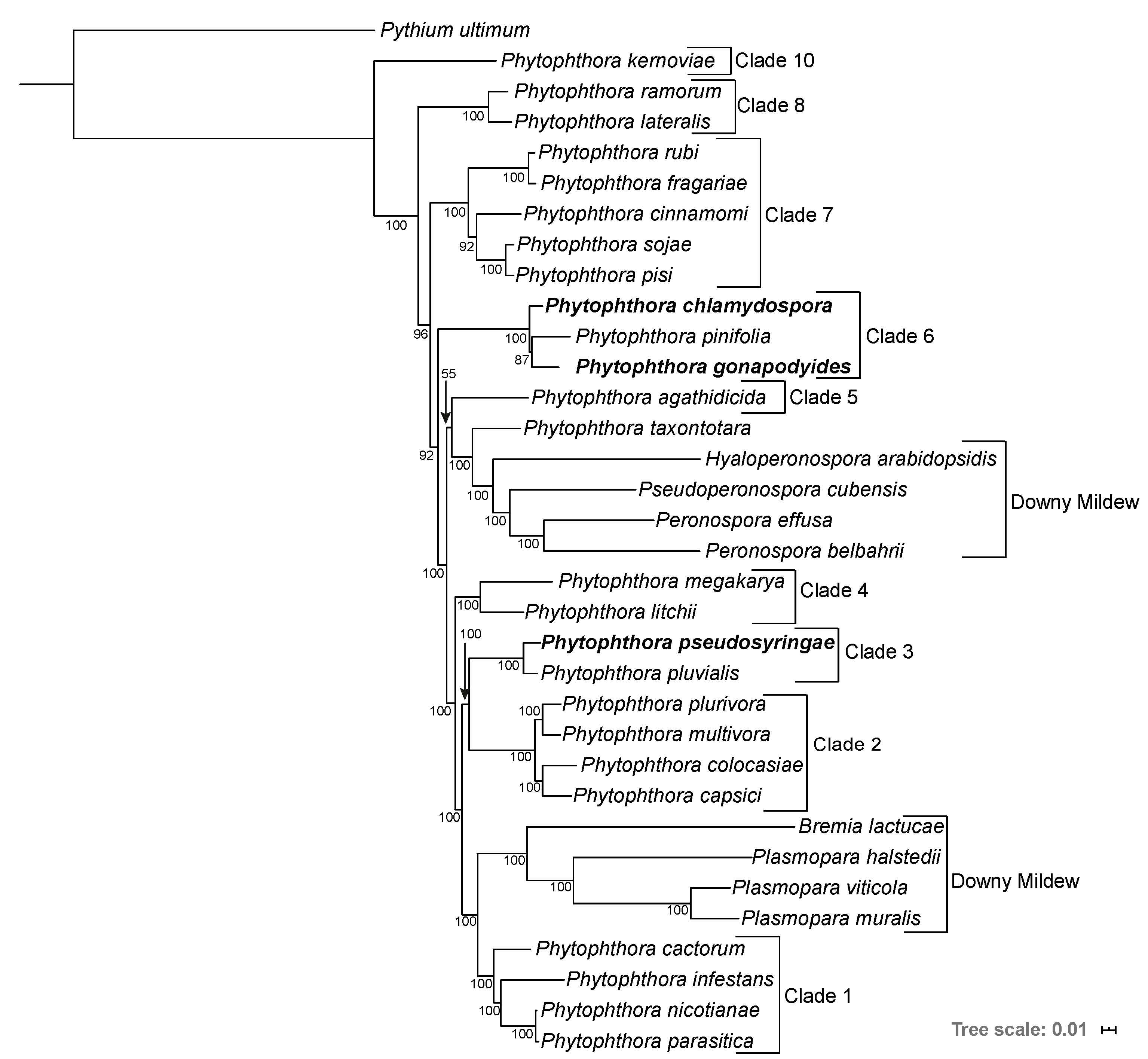
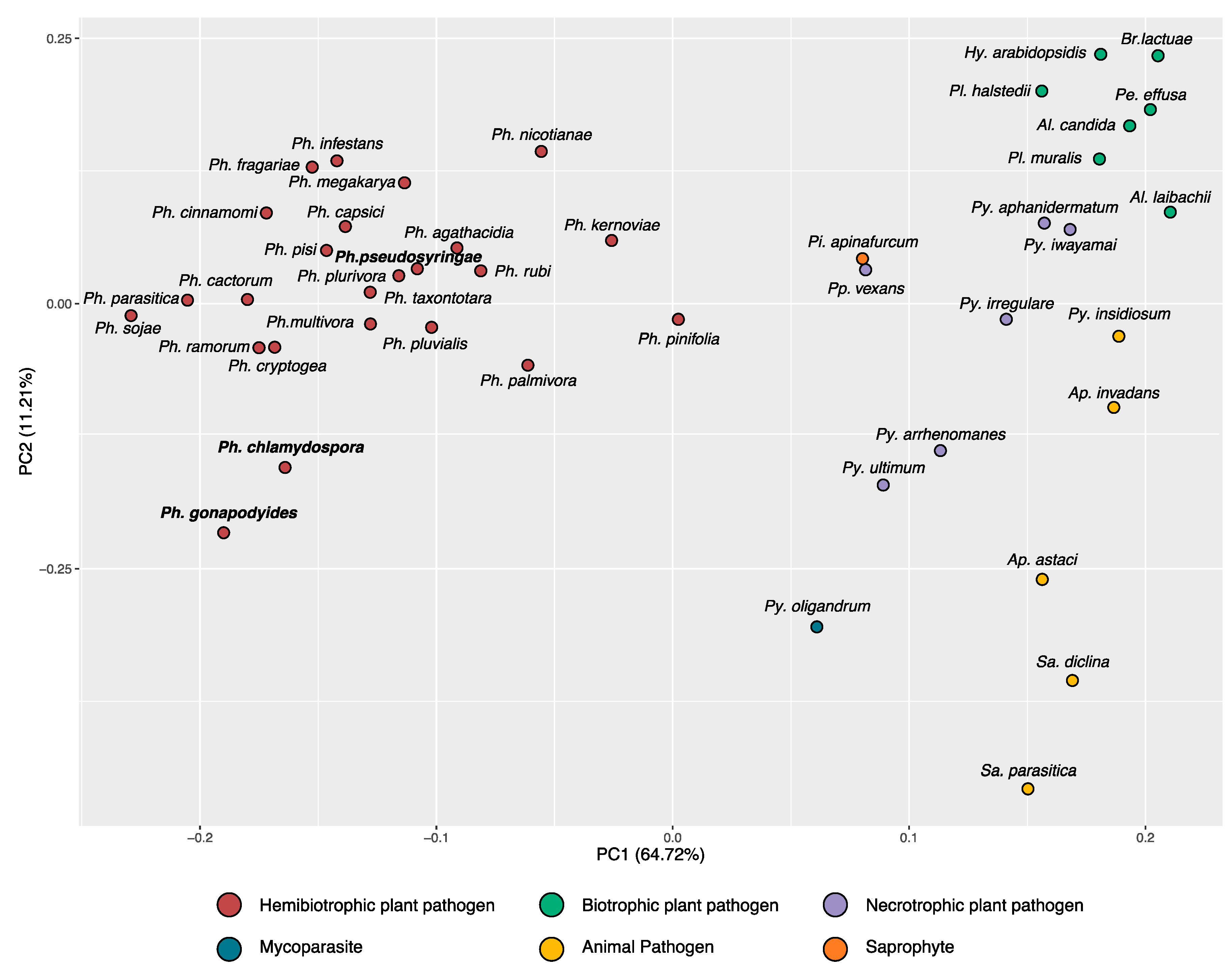
3.2. Phylogenomics Analysis
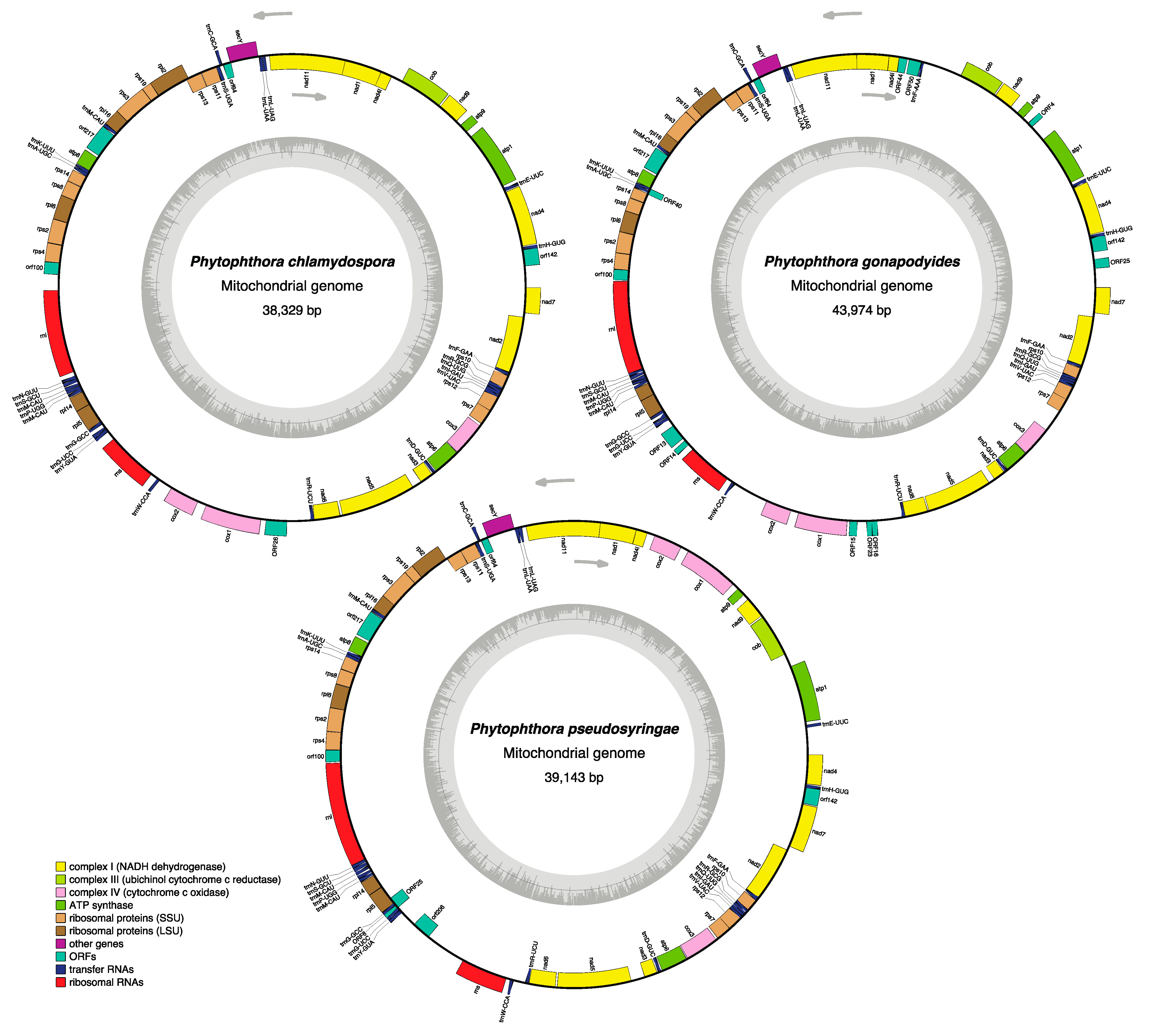
3.3. Phytophthora Mitochondrial Genomes
3.4. Bioinformatic Characterisation of Phytophthora Effector Arsenals
3.5. Carbohydrate Active Enzymes
3.6. Tandemly Duplicated Genes
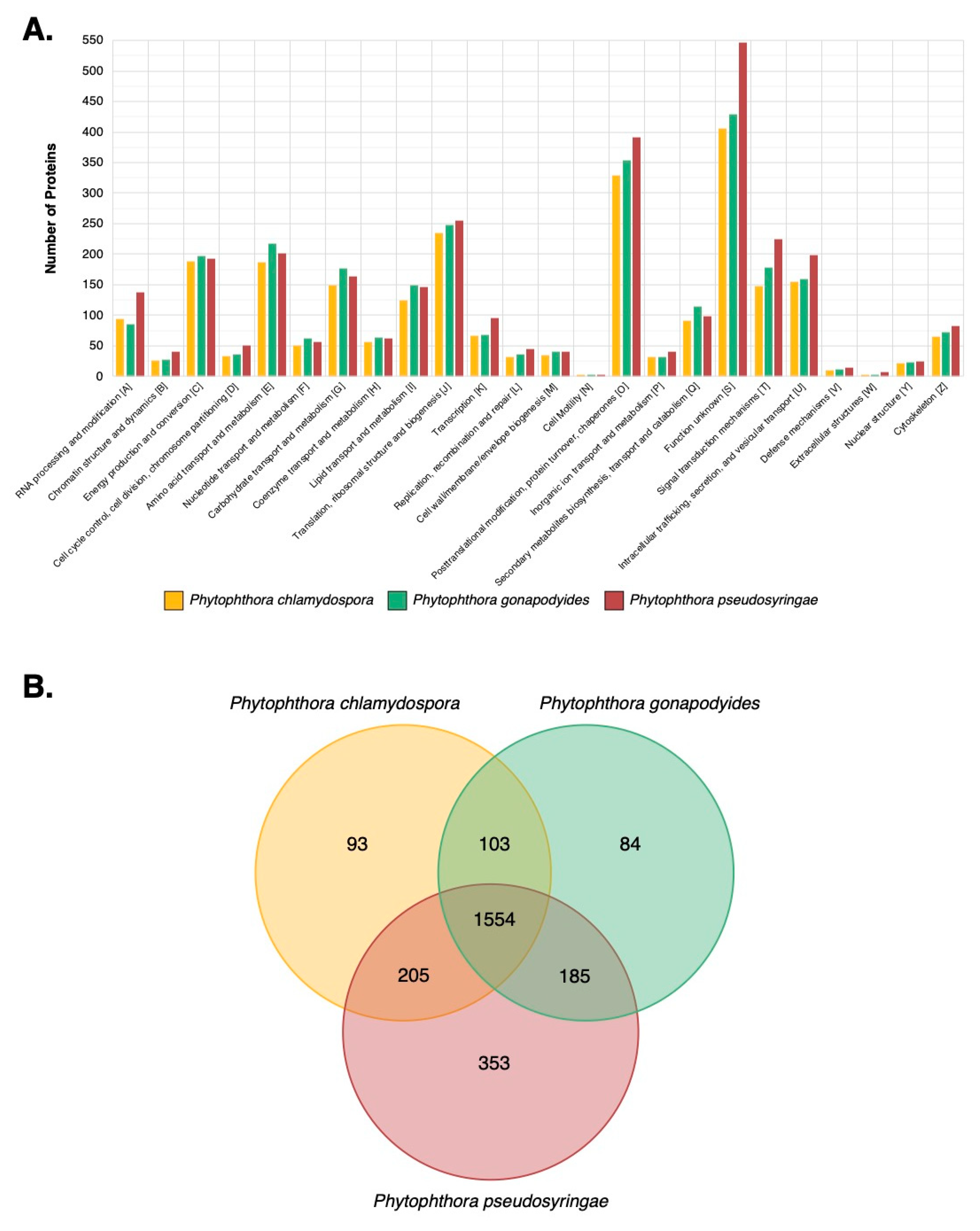
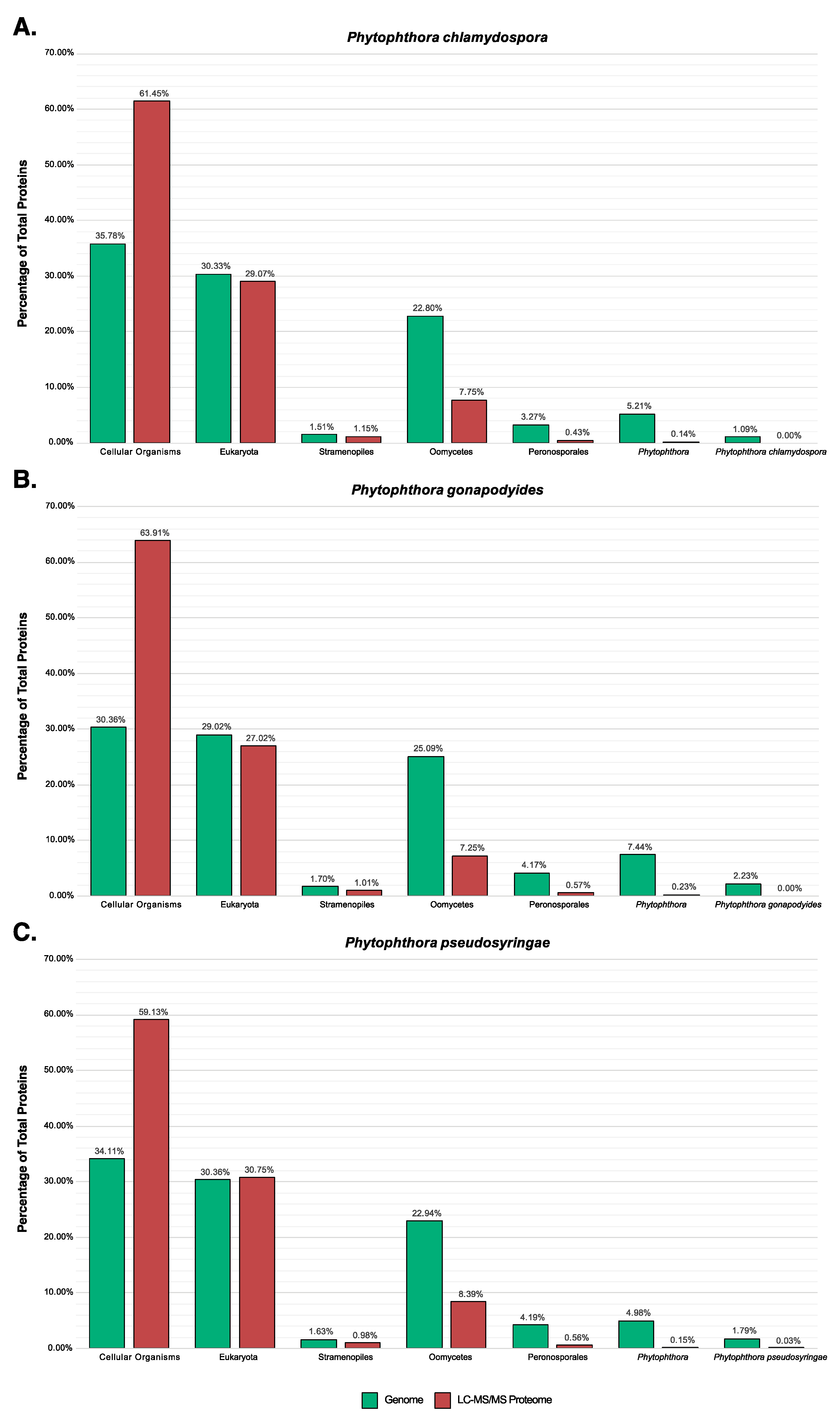
3.7. LC-MS/MS Characterisation of Phytophthora Extracellular Proteomes
3.8. LC-MS/MS Identification of Mycelial Proteins
3.9. Phylostratigraphy Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Burki, F.; Roger, A.J.; Brown, M.W.; Simpson, A.G.B. The New Tree of Eukaryotes. Trends Ecol. Evol. 2020, 35, 43–55. [Google Scholar] [PubMed]
- Yang, X.; Tyler, B.M.; Hong, C. An expanded phylogeny for the genus Phytophthora. IMA Fungus 2017, 8, 355–384. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Kamoun, S.; Zody, M.C.; Jiang, R.H.Y.; Handsaker, R.E.; Cano, L.M.; Grabherr, M.; Kodira, C.D.; Raffaele, S.; Torto-Alalibo, T.; et al. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature 2009, 461, 393–398. [Google Scholar] [PubMed]
- Tyler, B.M. Phytophthora Genome Sequences Uncover Evolutionary Origins and Mechanisms of Pathogenesis. Science 2006, 313, 1261–1266. [Google Scholar]
- Vetukuri, R.R.; Tripathy, S.; Mathu, M.C.; Panda, A.; Kushwaha, S.K.; Chawade, A.; Andreasson, E.; Grenville-Briggs, L.J.; Whisson, S.C. Draft genome sequence for the tree pathogen phytophthora plurivora. Genome Biol. Evol. 2018, 10, 2432–2442. [Google Scholar]
- Armitage, A.D.; Lysøe, E.; Nellist, C.F.; Lewis, L.A.; Cano, L.M.; Harrison, R.J.; Brurberg, M.B. Bioinformatic characterisation of the effector repertoire of the strawberry pathogen Phytophthora cactorum. PLoS ONE 2018, 13, 1–24. [Google Scholar]
- Feau, N.; Taylor, G.; Dale, A.L.; Dhillon, B.; Bilodeau, G.J.; Birol, I.; Jones, S.J.M.; Hamelin, R.C. Genome sequences of six Phytophthora species threatening forest ecosystems. Genomics Data 2016, 10, 85–88. [Google Scholar] [CrossRef]
- Studholme, D.J.; McDougal, R.L.; Sambles, C.; Hansen, E.; Hardy, G.; Grant, M.; Ganley, R.J.; Williams, N.M. Genome sequences of six Phytophthora species associated with forests in New Zealand. Genomics Data 2015, 7, 54–56. [Google Scholar]
- Ali, S.S.; Shao, J.; Lary, D.J.; Kronmiller, B.A.; Shen, D.; Strem, M.D.; Amoako-Attah, I.; Akrofi, A.Y.; Begoude, B.A.D.; ten Hoopen, G.M.; et al. Phytophthora megakarya and Phytophthora palmivora, Closely Related Causal Agents of Cacao Black Pod Rot, Underwent Increases in Genome Sizes and Gene Numbers by Different Mechanisms. Genome Biol. Evol. 2017, 9, 536–557. [Google Scholar]
- Richards, T.A.; Soanes, D.M.; Jones, M.D.M.; Vasieva, O.; Leonard, G.; Paszkiewicz, K.; Foster, P.G.; Hall, N.; Talbot, N.J. Horizontal gene transfer facilitated the evolution of plant parasitic mechanisms in the oomycetes. Proc. Natl. Acad. Sci. USA 2011, 108, 15258–15263. [Google Scholar]
- Kamoun, S. A catalogue of the effector secretome of plant pathogenic oomycetes. Annu. Rev. Phytopathol. 2006, 44, 41–60. [Google Scholar] [CrossRef] [PubMed]
- McGowan, J.; Fitzpatrick, D.A. Genomic, Network, and Phylogenetic Analysis of the Oomycete Effector Arsenal. mSphere 2017, 2, e00408-17. [Google Scholar] [PubMed]
- Wawra, S.; Belmonte, R.; Löbach, L.; Saraiva, M.; Willems, A.; van West, P. Secretion, delivery and function of oomycete effector proteins. Curr. Opin. Microbiol. 2012, 15, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Stassen, J.H.M.; Van den Ackerveken, G. How do oomycete effectors interfere with plant life? Curr. Opin. Plant Biol. 2011, 14, 407–414. [Google Scholar] [CrossRef]
- Meijer, H.J.G.; Mancuso, F.M.; Espadas, G.; Seidl, M.F.; Chiva, C.; Govers, F.; Sabido, E. Profiling the Secretome and Extracellular Proteome of the Potato Late Blight Pathogen Phytophthora infestans. Mol. Cell. Proteomics 2014, 13, 2101–2113. [Google Scholar] [CrossRef]
- Severino, V.; Farina, A.; Fleischmann, F.; Dalio, R.J.D.; Di Maro, A.; Scognamiglio, M.; Fiorentino, A.; Parente, A.; Osswald, W.; Chambery, A. Molecular profiling of the Phytophthora plurivora secretome: A step towards understanding the cross-talk between plant pathogenic oomycetes and their hosts. PLoS ONE 2014, 9, e112317. [Google Scholar] [CrossRef]
- Hosseini, S.; Resjö, S.; Liu, Y.; Durling, M.; Heyman, F.; Levander, F.; Liu, Y.; Elfstrand, M.; Funck Jensen, D.; Andreasson, E.; et al. Comparative proteomic analysis of hyphae and germinating cysts of Phytophthora pisi and Phytophthora sojae. J. Proteomics 2015, 117, 24–40. [Google Scholar]
- Resjö, S.; Brus, M.; Ali, A.; Meijer, H.J.G.; Sandin, M.; Govers, F.; Levander, F.; Grenville-Briggs, L.; Andreasson, E. Proteomic Analysis of Phytophthora infestans Reveals the Importance of Cell Wall Proteins in Pathogenicity. Mol. Cell. Proteomics 2017, 16, 1958–1971. [Google Scholar] [CrossRef]
- Savidor, A.; Donahoo, R.S.; Hurtado-Gonzales, O.; Land, M.L.; Shah, M.B.; Lamour, K.H.; McDonald, W.H. Cross-species Global Proteomics Reveals Conserved and Unique Processes in Phytophthora sojae and Phytophthora ramorum. Mol. Cell. Proteomics 2008, 7, 1501–1516. [Google Scholar]
- Jung, T.; Nechwatal, J.; Cooke, D.E.L.; Hartmann, G.; Blaschke, M.; Oßwald, W.F.; Duncan, J.M.; Delatour, C. Phytophthora pseudosyringae sp. nov., a new species causing root and collar rot of deciduous tree species in Europe. Mycol. Res. 2003, 107, 772–789. [Google Scholar] [CrossRef]
- Hansen, E.M.; Reeser, P.W.; Sutton, W. Ecology and pathology of Phytophthora ITS clade 3 species in forests in western Oregon, USA. Mycologia 2017, 109, 100–114. [Google Scholar] [CrossRef] [PubMed]
- O’Hanlon, R.; Choiseul, J.; Corrigan, M.; Catarame, T.; Destefanis, M. Diversity and detections of Phytophthora species from trade and non-trade environments in Ireland. EPPO Bull. 2016, 46, 594–602. [Google Scholar]
- O’Hanlon, R.; McCracken, A.; Cooke, L. Diversity and ecology of Phytophthora species on the island of Ireland. Biol. Environ. Proc. R. Irish Acad. 2016, 116B, 27. [Google Scholar]
- Fajardo, S.N.; Valenzuela, S.; Dos Santos, A.F.; González, M.P.; Sanfuentes, E.A. Phytophthora pseudosyringae associated with the mortality of Nothofagus obliqua in a pure stand in central-southern Chile. For. Pathol. 2017, 47, e12361. [Google Scholar]
- Varela, C.P.; Vázquez, J.P.M.; Casal, O.A.; Martínez, C.R. First Report of Phytophthora pseudosyringae on Chestnut Nursery Stock in Spain. Plant Dis. 2007, 91, 1517. [Google Scholar] [CrossRef]
- Redondo, M.Á.; Boberg, J.; Stenlid, J.; Oliva, J. First Report of Phytophthora pseudosyringae Causing Basal Cankers on Horse Chestnut in Sweden. Plant Dis. 2016, 100, 1024. [Google Scholar] [CrossRef]
- Scanu, B.; Webber, J.F. Dieback and mortality of Nothofagus in Britain: Ecology, pathogenicity and sporulation potential of the causal agent Phytophthora pseudosyringae. Plant Pathol. 2016, 65, 26–36. [Google Scholar]
- Riddell, C.E.; Frederickson-Matika, D.; Armstrong, A.C.; Elliot, M.; Forster, J.; Hedley, P.E.; Morris, J.; Thorpe, P.; EL Cooke, D.; Pritchard, L.; et al. Metabarcoding reveals a high diversity of woody host-associated Phytophthora spp. in soils at public gardens and amenity woodlands in Britain. PeerJ 2019, 7, e6931. [Google Scholar] [CrossRef]
- Brasier, C.M.; Cooke, D.E.L.; Duncan, J.M.; Hansen, E.M. Multiple new phenotypic taxa from trees and riparian ecosystems in Phytophthora gonapodyides-P. megasperma ITS Clade 6, which tend to be high-temperature tolerant and either inbreeding or sterile. Mycol. Res. 2003, 107, 277–290. [Google Scholar] [CrossRef]
- Hansen, E.M.; Reeser, P.W.; Sutton, W. Phytophthora Beyond Agriculture. Annu. Rev. Phytopathol. 2012, 50, 359–378. [Google Scholar]
- Hansen, E.M.; Reeser, P.; Sutton, W.; Brasier, C.M. Redesignation of Phytophthora taxon Pgchlamydo as Phytophthora chlamydospora sp. nov. North Am. Fungi 2015, 10, 1–14. [Google Scholar]
- Kurbetli, İ.; Aydoğdu, M.; Sülü, G. Phytophthora chlamydospora and P. megasperma associated with root and crown rot of sour cherry in Turkey. J. Plant Dis. Prot. 2017, 124, 403–406. [Google Scholar] [CrossRef]
- Ginetti, B.; Carmignani, S.; Ragazzi, A.; Moricca, S. Phytophthora Taxon Pgchlamydo is a Cause of Shoot Blight and Root and Collar Rot of Viburnum tinus in Italy. Plant Dis. 2014, 98, 1432. [Google Scholar] [CrossRef] [PubMed]
- Blomquist, C.L.; Yakabe, L.E.; Soriano, M.C.; Negrete, M.A. First Report of Leaf Spot Caused by Phytophthora taxon Pgchlamydo on Evergreen Nursery Stock in California. Plant Dis. 2012, 96, 1691. [Google Scholar]
- Derviş, S.; Türkölmez, Ş.; Çiftçi, O.; Ulubaş Serçe, Ç. First Report of Phytophthora chlamydospora Causing Root Rot on Walnut (Juglans regia) Trees in Turkey. Plant Dis. 2016, 100, 2336. [Google Scholar] [CrossRef]
- Türkölmez, Ş.; Derviş, S.; Çiftçi, O.; Ulubaş Serçe, Ç. First Report of Phytophthora chlamydospora Causing Root and Crown Rot on Almond (Prunus dulcis) Trees in Turkey. Plant Dis. 2016, 100, 1796. [Google Scholar] [CrossRef]
- Latorre, B.A.; Rioja, M.E.; Wilcox, W.F. Phytophthora Species Associated with Crown and Root Rot of Apple in Chile. Plant Dis. 2001, 85, 603–606. [Google Scholar]
- Belisario, A.; Luongo, L.; Vitale, S.; Galli, M.; Haegi, A. Phytophthora gonapodyides Causes Decline and Death of English (Persian) Walnut (Juglans regia) in Italy. Plant Dis. 2016, 100, 2537. [Google Scholar] [CrossRef]
- Reeser, P.W.; Sutton, W.; Hansen, E.M. Phytophthora Species Causing Tanoak Stem Cankers in Southwestern Oregon. Plant Dis. 2008, 92, 1252. [Google Scholar]
- Corcobado, T.; Cubera, E.; Pérez-Sierra, A.; Jung, T.; Solla, A. First report of Phytophthora gonapodyides involved in the decline of Quercus ilex in xeric conditions in Spain. New Dis. Reports 2010, 22, 33. [Google Scholar]
- O’Hanlon, R.; Choiseul, J.; Grogan, H.; Brennan, J.M. In-vitro characterisation of the four lineages of Phytophthora ramorum. Eur. J. Plant Pathol. 2016, 2. [Google Scholar] [CrossRef]
- O’Hanlon, R.; Choiseul, J.; Brennan, J.M.; Grogan, H. Assessment of the eradication measures applied to Phytophthora ramorum in Irish Larix kaempferi forests. For. Pathol. 2018, 48, e12389. [Google Scholar]
- Jeffers, S.N. Comparison of Two Media Selective for Phytophthora and Pythium Species. Plant Dis. 1986, 70, 1038. [Google Scholar] [CrossRef]
- Werres, S.; Marwitz, R.; Man In’t veld, W.A.; De Cock, A.W.A.M.; Bonants, P.J.M.; De Weerdt, M.; Themann, K.; Ilieva, E.; Baayen, R.P. Phytophthora ramorum sp. nov., a new pathogen on Rhododendron and Viburnum. Mycol. Res. 2001, 105, 1155–1165. [Google Scholar] [CrossRef]
- Gallegly, M.E.; ChuanXue, H. Phytophthora: Identifying Species by Morphology and DNA Fingerprints.; American Phytopathological Society (APS Press): St. Paul, MN, USA, 2008; ISBN 9780890543641. [Google Scholar]
- Erwin, D.C.; Ribeiro, O.K. Phytophthora Diseases Worldwide; American Phytopathological Society (APS Press): St. Paul, MN, USA, 1996; ISBN 0890542120. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef]
- Vurture, G.W.; Sedlazeck, F.J.; Nattestad, M.; Underwood, C.J.; Fang, H.; Gurtowski, J.; Schatz, M.C. GenomeScope: Fast reference-free genome profiling from short reads. Bioinformatics 2017, 33, 2202–2204. [Google Scholar]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.a.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar]
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2011, 27, 578–579. [Google Scholar] [CrossRef]
- Boetzer, M.; Pirovano, W. Toward almost closed genomes with GapFiller. Genome Biol. 2012, 13, R56. [Google Scholar] [CrossRef]
- Mikheenko, A.; Prjibelski, A.; Saveliev, V.; Antipov, D.; Gurevich, A. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 2018, 34, i142–i150. [Google Scholar] [CrossRef]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [PubMed]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2: Automated genomic discovery of transposable element families. bioRxiv 2019. [Google Scholar] [CrossRef] [PubMed]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2016, 45, gkw955. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [PubMed]
- Hoff, K.J.; Lange, S.; Lomsadze, A.; Borodovsky, M.; Stanke, M. BRAKER1: Unsupervised RNA-Seq-Based Genome Annotation with GeneMark-ET and AUGUSTUS: Table 1. Bioinformatics 2016, 32, 767–769. [Google Scholar] [CrossRef] [PubMed]
- Bruna, T.; Lomsadze, A.; Borodovsky, M. GeneMark-EP and -EP+: Automatic eukaryotic gene prediction supported by spliced aligned proteins. bioRxiv 2020, 2019.12.31.891218. [Google Scholar]
- Ter-Hovhannisyan, V.; Lomsadze, A.; Chernoff, Y.O.; Borodovsky, M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008, 18, 1979–1990. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar]
- Iwata, H.; Gotoh, O. Benchmarking spliced alignment programs including Spaln2, an extended version of Spaln that incorporates additional species-specific features. Nucleic Acids Res. 2012, 40, e161. [Google Scholar] [CrossRef]
- Hoff, K.J.; Stanke, M. Predicting Genes in Single Genomes with AUGUSTUS. Curr. Protoc. Bioinforma. 2018, 65, e57. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; von Mering, C.; Bork, P. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef] [PubMed]
- Bendtsen, J.D.; Nielsen, H.; Von Heijne, G.; Brunak, S. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 2004, 340, 783–795. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef]
- Sperschneider, J.; Williams, A.H.; Hane, J.K.; Singh, K.B.; Taylor, J.M. Evaluation of Secretion Prediction Highlights Differing Approaches Needed for Oomycete and Fungal Effectors. Front. Plant Sci. 2015, 6, 1–14. [Google Scholar]
- Raffaele, S.; Win, J.; Cano, L.M.; Kamoun, S. Analyses of genome architecture and gene expression reveal novel candidate virulence factors in the secretome of Phytophthora infestans. BMC Genomics 2010, 11, 637. [Google Scholar] [CrossRef]
- Sperschneider, J.; Dodds, P.N.; Singh, K.B.; Taylor, J.M. ApoplastP: Prediction of effectors and plant proteins in the apoplast using machine learning. New Phytol. 2018, 217, 1764–1778. [Google Scholar] [CrossRef]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef]
- Urban, M.; Cuzick, A.; Rutherford, K.; Irvine, A.; Pedro, H.; Pant, R.; Sadanadan, V.; Khamari, L.; Billal, S.; Mohanty, S.; et al. PHI-base: A new interface and further additions for the multi-species pathogen–host interactions database. Nucleic Acids Res. 2017, 45, D604–D610. [Google Scholar] [CrossRef]
- Enright, A.J.; Van Dongen, S.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef]
- Klopfenstein, D.V.; Zhang, L.; Pedersen, B.S.; Ramírez, F.; Vesztrocy, A.W.; Naldi, A.; Mungall, C.J.; Yunes, J.M.; Botvinnik, O.; Weigel, M.; et al. GOATOOLS: A Python library for Gene Ontology analyses. Sci. Rep. 2018, 8, 1–17. [Google Scholar]
- Win, J.; Morgan, W.; Bos, J.; Krasileva, K.V.; Cano, L.M.; Chaparro-garcia, A.; Ammar, R.; Staskawicz, B.J.; Kamoun, S. Adaptive Evolution Has Targeted the C-Terminal Domain of the RXLR Effectors of Plant Pathogenic Oomycetes © American Society of Plant Biologists Adaptive Evolution Has Targeted the C-Terminal Domain of the RXLR Effectors of Plant Pathogenic Oomycetes. Plant Cell 2007, 19, 2349–2369. [Google Scholar] [CrossRef]
- Eddy, S. Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef]
- Whisson, S.C.; Boevink, P.C.; Moleleki, L.; Avrova, A.O.; Morales, J.G.; Gilroy, E.M.; Armstrong, M.R.; Grouffaud, S.; van West, P.; Chapman, S. A translocation signal for delivery of oomycete effector proteins into host plant cells. Nature 2007, 450, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Boutemy, L.S.; King, S.R.F.F.; Win, J.; Hughes, R.K.; Clarke, T.A.; Blumenschein, T.M.A.A.; Kamoun, S.; Banfield, M.J. Structures of Phytophthora RXLR effector proteins: A conserved but adaptable fold underpins functional diversity. J. Biol. Chem. 2011, 286, 35834–35842. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar]
- Lartillot, N.; Rodrigue, N.; Stubbs, D.; Richer, J. PhyloBayes MPI: Phylogenetic Reconstruction with Infinite Mixtures of Profiles in a Parallel Environment. Syst. Biol. 2013, 62, 611–615. [Google Scholar]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed]
- Drost, H.-G.; Gabel, A.; Grosse, I.; Quint, M. Evidence for Active Maintenance of Phylotranscriptomic Hourglass Patterns in Animal and Plant Embryogenesis. Mol. Biol. Evol. 2015, 32, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Quint, M.; Drost, H.-G.; Gabel, A.; Ullrich, K.K.; Bönn, M.; Grosse, I. A transcriptomic hourglass in plant embryogenesis. Nature 2012, 490, 98–101. [Google Scholar] [CrossRef]
- Morrin, S.T.; Owens, R.A.; Le Berre, M.; Gerlach, J.Q.; Joshi, L.; Bode, L.; Irwin, J.A.; Hickey, R.M. Interrogation of Milk-Driven Changes to the Proteome of Intestinal Epithelial Cells by Integrated Proteomics and Glycomics. J. Agric. Food Chem. 2019, 67, 1902–1917. [Google Scholar] [CrossRef]
- Owens, R.A.; O’Keeffe, G.; Smith, E.B.; Dolan, S.K.; Hammel, S.; Sheridan, K.J.; Fitzpatrick, D.A.; Keane, T.M.; Jones, G.W.; Doyle, S. Interplay between Gliotoxin Resistance, Secretion, and the Methyl/Methionine Cycle in Aspergillus fumigatus. Eukaryot. Cell 2015, 14, 941–957. [Google Scholar]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Delgado, J.; Núñez, F.; Asensio, M.A.; Owens, R.A. Quantitative proteomic profiling of ochratoxin A repression in Penicillium nordicum by protective cultures. Int. J. Food Microbiol. 2019, 305, 108243. [Google Scholar]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar]
- Fletcher, K.; Gil, J.; Bertier, L.D.; Kenefick, A.; Wood, K.J.; Zhang, L.; Reyes-Chin-Wo, S.; Cavanaugh, K.; Tsuchida, C.; Wong, J.; et al. Genomic signatures of heterokaryosis in the oomycete pathogen Bremia lactucae. Nat. Commun. 2019, 10, 1–13. [Google Scholar]
- Fang, Y. “Francis”; Coelho, M.A.; Shu, H.; Schotanus, K.; Thimmappa, B.C.; Yadav, V.; Chen, H.; Malc, E.P.; Wang, J.; Mieczkowski, P.A.; et al. Long transposon-rich centromeres in an oomycete reveal divergence of centromere features in Stramenopila-Alveolata-Rhizaria lineages. PLoS Genet. 2020, 16, e1008646. [Google Scholar]
- Malar C, M.; Yuzon, J.D.; Panda, A.; Kasuga, T.; Tripathy, S. Updated Assembly of Phytophthora ramorum pr102 Isolate Incorporating Long Reads from PacBio Sequencing. Mol. Plant-Microbe Interact. 2019, 32, 1472–1474. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, C.G.P.; Fitzpatrick, D.A. Phylogenomic Reconstruction of the Oomycete Phylogeny Derived from 37 Genomes. mSphere 2017, 2, e00095-17. [Google Scholar] [CrossRef] [PubMed]
- Blair, J.E.; Coffey, M.D.; Park, S.Y.; Geiser, D.M.; Kang, S. A multi-locus phylogeny for Phytophthora utilizing markers derived from complete genome sequences. Fungal Genet. Biol. 2008, 45, 266–277. [Google Scholar] [CrossRef]
- Martin, F.N.; Blair, J.E.; Coffey, M.D. A combined mitochondrial and nuclear multilocus phylogeny of the genus Phytophthora. Fungal Genet. Biol. 2014, 66, 19–32. [Google Scholar] [CrossRef]
- Yang, X.; Hong, C. Differential Usefulness of Nine Commonly Used Genetic Markers for Identifying Phytophthora Species. Front. Microbiol. 2018, 9, 2334. [Google Scholar]
- Fitzpatrick, D.A.; Logue, M.E.; Stajich, J.E.; Butler, G. A fungal phylogeny based on 42 complete genomes derived from supertree and combined gene analysis. BMC Evol. Biol. 2006, 6, 99. [Google Scholar]
- Fletcher, K.; Klosterman, S.J.; Derevnina, L.; Martin, F.; Bertier, L.D.; Koike, S.; Reyes-Chin-Wo, S.; Mou, B.; Michelmore, R. Comparative genomics of downy mildews reveals potential adaptations to biotrophy. BMC Genom. 2018, 19, 8–10. [Google Scholar]
- Karlovsky, P.; Fartmann, B. Genetic code and phylogenetic origin of oomycetous mitochondria. J. Mol. Evol. 1992, 34, 254–258. [Google Scholar]
- Martin, F.N.; Bensasson, D.; Tyler, B.M.; Boore, J.L. Mitochondrial genome sequences and comparative genomics of Phytophthora ramorum and P. sojae. Curr. Genet. 2007, 51, 285–296. [Google Scholar] [CrossRef][Green Version]
- Derevnina, L.; Dagdas, Y.F.; De la Concepcion, J.C.; Bialas, A.; Kellner, R.; Petre, B.; Domazakis, E.; Du, J.; Wu, C.; Lin, X.; et al. Nine things to know about elicitins. New Phytol. 2016, 212, 888–895. [Google Scholar] [PubMed]
- Jiang, R.H.Y.; Tyler, B.M.; Whisson, S.C.; Hardham, A.R.; Govers, F. Ancient Origin of Elicitin Gene Clusters in Phytophthora Genomes. Mol. Biol. Evol. 2006, 23, 338–351. [Google Scholar] [CrossRef]
- Seidl, M.F.; Van den Ackerveken, G. Activity and Phylogenetics of the Broadly Occurring Family of Microbial Nep1-Like Proteins. Annu. Rev. Phytopathol. 2019, 57, 367–386. [Google Scholar] [PubMed]
- Orsomando, G.; Lorenzi, M.; Raffaelli, N.; Dalla Rizza, M.; Mezzetti, B.; Ruggieri, S. Phytotoxic Protein PcF, Purification, Characterization, and cDNA Sequencing of a Novel Hydroxyproline-containing Factor Secreted by the Strawberry Pathogen Phytophthora cactorum. J. Biol. Chem. 2001, 276, 21578–21584. [Google Scholar] [CrossRef] [PubMed]
- Raaymakers, T.M.; Van den Ackerveken, G. Extracellular Recognition of Oomycetes during Biotrophic Infection of Plants. Front. Plant Sci. 2016, 7, 1–12. [Google Scholar] [CrossRef]
- Brunner, F.; Rosahl, S.; Lee, J.; Rudd, J.J.; Geiler, C.; Kauppinen, S.; Rasmussen, G.; Scheel, D.; Nürnberger, T. Pep-13, a plant defense-inducing pathogen-associated pattern from Phytophthora transglutaminases. EMBO J. 2002, 21, 6681–6688. [Google Scholar] [CrossRef]
- Gaulin, E.; Jauneau, A.; Villalba, F.; Rickauer, M.; Esquerré-Tugayé, M.-T.; Bottin, A. The CBEL glycoprotein of Phytophthora parasitica var-nicotianae is involved in cell wall deposition and adhesion to cellulosic substrates. J. Cell Sci. 2002, 115, 4565–4575. [Google Scholar]
- Schneiter, R.; Di Pietro, A. The CAP protein superfamily: Function in sterol export and fungal virulence. Biomol. Concepts 2013, 4, 519–525. [Google Scholar] [CrossRef]
- Fries, M.; Ihrig, J.; Brocklehurst, K.; Shevchik, V.E.; Pickersgill, R.W. Molecular basis of the activity of the phytopathogen pectin methylesterase. EMBO J. 2007, 26, 3879–3887. [Google Scholar] [CrossRef]
- Vercauteren, I.; de Almeida Engler, J.; De Groodt, R.; Gheysen, G. An Arabidopsis thaliana Pectin Acetylesterase Gene Is Upregulated in Nematode Feeding Sites Induced by Root-knot and Cyst Nematodes. Mol. Plant-Microbe Interact. 2002, 15, 404–407. [Google Scholar] [CrossRef]
- Win, J.; Krasileva, K.V.; Kamoun, S.; Shirasu, K.; Staskawicz, B.J.; Banfield, M.J. Sequence Divergent RXLR Effectors Share a Structural Fold Conserved across Plant Pathogenic Oomycete Species. PLoS Pathog. 2012, 8, e1002400. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Gu, B.; Huang, G.; Tian, Y.; Quan, J.; Lindqvist-Kreuze, H.; Shan, W. Conserved RXLR Effector Genes of Phytophthora infestans Expressed at the Early Stage of Potato Infection Are Suppressive to Host Defense. Front. Plant Sci. 2017, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; McLellan, H.; Bukharova, T.; He, Q.; Murphy, F.; Shi, J.; Sun, S.; van Weymers, P.; Ren, Y.; Thilliez, G.; et al. Phytophthora infestans RXLR effectors act in concert at diverse subcellular locations to enhance host colonization. J. Exp. Bot. 2019, 70, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Schornack, S.; van Damme, M.; Bozkurt, T.O.; Cano, L.M.; Smoker, M.; Thines, M.; Gaulin, E.; Kamoun, S.; Huitema, E. Ancient class of translocated oomycete effectors targets the host nucleus. Proc. Natl. Acad. Sci. USA 2010, 107, 17421–17426. [Google Scholar] [CrossRef]
- Stam, R.; Jupe, J.; Howden, A.J.M.; Morris, J.A.; Boevink, P.C.; Hedley, P.E.; Huitema, E. Identification and Characterisation CRN Effectors in Phytophthora capsici Shows Modularity and Functional Diversity. PLoS ONE 2013, 8, 1–13. [Google Scholar] [CrossRef]
- Zerillo, M.M.; Adhikari, B.N.; Hamilton, J.P.; Buell, C.R.; Lévesque, C.A.; Tisserat, N. Carbohydrate-Active Enzymes in Pythium and Their Role in Plant Cell Wall and Storage Polysaccharide Degradation. PLoS ONE 2013, 8, e72572. [Google Scholar]
- Sharma, R.; Xia, X.; Cano, L.M.; Evangelisti, E.; Kemen, E.; Judelson, H.; Oome, S.; Sambles, C.; van den Hoogen, D.J.; Kitner, M.; et al. Genome analyses of the sunflower pathogen Plasmopara halstedii provide insights into effector evolution in downy mildews and Phytophthora. BMC Genom. 2015, 16, 741. [Google Scholar] [CrossRef]
- Benhamou, N.; le Floch, G.; Vallance, J.; Gerbore, J.; Grizard, D.; Rey, P. Pythium oligandrum: An example of opportunistic success. Microbiol. (United Kingdom) 2012, 158, 2679–2694. [Google Scholar] [CrossRef]
- Rodenburg, S.Y.A.; de Ridder, D.; Govers, F.; Seidl, M.F. Oomycete metabolism is highly dynamic and reflects lifestyle adaptations. bioRxiv 2020, 2020.02.12.941195. [Google Scholar]
- Thines, M.; Sharma, R.; Rodenburg, S.Y.A.; Gogleva, A.; Judelson, H.S.; Xia, X.; van den Hoogen, J.; Kitner, M.; Klein, J.; Neilen, M.; et al. The genome of Peronospora belbahrii reveals high heterozygosity, a low number of canonical effectors and CT-rich promoters. bioRxiv 2019, 721027. [Google Scholar]
- McGowan, J.; Byrne, K.P.; Fitzpatrick, D.A. Comparative Analysis of Oomycete Genome Evolution Using the Oomycete Gene Order Browser (OGOB). Genome Biol. Evol. 2019, 11, 189–206. [Google Scholar] [CrossRef] [PubMed]
- Bendtsen, J.D.; Jensen, L.J.; Blom, N.; von Heijne, G.; Brunak, S. Feature-based prediction of non-classical and leaderless protein secretion. Protein Eng. Des. Sel. 2004, 17, 349–356. [Google Scholar] [CrossRef]
- Pennington, H.G.; Jones, R.; Kwon, S.; Bonciani, G.; Thieron, H.; Chandler, T.; Luong, P.; Morgan, S.N.; Przydacz, M.; Bozkurt, T.; et al. The fungal ribonuclease-like effector protein CSEP0064/BEC1054 represses plant immunity and interferes with degradation of host ribosomal RNA. PLoS Pathog. 2019, 15, e1007620. [Google Scholar]
- Wang, S.; Boevink, P.C.; Welsh, L.; Zhang, R.; Whisson, S.C.; Birch, P.R.J. Delivery of cytoplasmic and apoplastic effectors from Phytophthora infestans haustoria by distinct secretion pathways. New Phytol. 2017, 216, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Welsh, L.; Thorpe, P.; Whisson, S.C.; Boevink, P.C.; Birch, P.R.J. The Phytophthora infestans Haustorium Is a Site for Secretion of Diverse Classes of Infection-Associated Proteins. MBio 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Munro, S.; Pelham, H.R.B. A C-terminal signal prevents secretion of luminal ER proteins. Cell 1987, 48, 899–907. [Google Scholar] [CrossRef]
- Papp, S.; Opas, M. Sub-Cellular Distribution of Calreticulin. In Calreticulin: Second Edition; Eggleton, P., Michalak, M., Eds.; Springer US: Boston, MA, USA, 2003; pp. 38–48. ISBN 978-1-4419-9258-1. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Isolate Number | Source | Date Collected | Location (Latitude, Longitude) | BioProject Accession |
|---|---|---|---|---|---|
| Phytophthora chlamydospora | P17-99 | Rhododendron ponticum leaf baiting of a stream | 08/08/2017 | Tollymore forest, Co. Down, Northern Ireland, UK (54°13’16.9”N, 5°55’49.3”W) | PRJNA599565 |
| Phytophthora gonapodyides | P17-128 | Rhododendron ponticum leaf baiting of a stream | 28/08/2017 | Rostrevor forest, Co. Louth, Ireland (54°07’43.4”N, 6°09’24.2”W) | PRJNA599567 |
| Phytophthora pseudosyringae | PR13-731 | Bleeding bark canker of Fagus sylvatica | 31/07/2015 | Mullaghreelan forest, Co Kildare, Ireland (52°56’00.4”N, 6°52’41.0”W) | PRJNA599564 |
| Genome Assembly | Phytophthora chlamydospora | Phytophthora gonapodyides | Phytophthora pseudosyringae |
|---|---|---|---|
| Estimated Genome Size (bp) | 51,100,498 | 65,211,327 | 51,026,880 |
| Assembly Size (bp) | 45,264,984 | 61,088,431 | 47,882,184 |
| Number of Scaffolds | 4077 | 16,449 | 3627 |
| N50 (bp) | 26,559 | 5455 | 26,492 |
| L50 | 466 | 2927 | 526 |
| GC Content | 55.7% | 55.7% | 54.8% |
| Sequencing Coverage | 98x | 76x | 102x |
| Repeat Masked | 9.0% | 16.1% | 13.3% |
| Estimated Heterozygosity | 0.68% | 1.88% | 0.15% |
| BUSCO Completeness | 97.8% | 87.2% | 94.1% |
| Gene Prediction | |||
| Gene Models | 17,872 | 23,348 | 17,439 |
| CDS density | 56.3% | 43.3% | 49.2% |
| BUSCO Completeness | 97.5% | 88.1% | 94.4% |
| Proteins with Pfam domains | 10,759 (60.2%) | 12,181 (52.2%) | 10,130 (58.1%) |
| Phytophthora chlamydospora | Phytophthora gonapodyides | Phytophthora pseudosyringae | |
|---|---|---|---|
| Secreted Proteins | 1140 (6.38%) | 1291 (5.53%) | 1131 (6.49%) |
| ApoplastP Hits | 554 (48.5%) | 630 (48.8%) | 533 (47.1%) |
| PHI-Base homologs | 249 (21.8%) | 234 (18.1%) | 243 (21.5%) |
| Apoplastic effectors | |||
| Berberine-like proteins | 1 (1) | 2 (1) | 3 (1) |
| Cysteine-rich secretory proteins (CAP) | 34 (22) | 34 (25) | 31 (22) |
| Elicitins | 57 (45) | 59 (47) | 45 (34) |
| Necrosis-inducing proteins | 25 (19) | 33 (22) | 22 (19) |
| PAN/Apple domain | 25 (21) | 32 (20) | 20 (15) |
| PcF phytotoxins | 1 (0) | 1 (1) | 1 (1) |
| Transglutaminase elicitors | 14 (11) | 16 (11) | 17 (11) |
| Proteases and protease inhibitors | |||
| Aspartyl proteases | 59 (4) | 53 (3) | 26 (2) |
| Papain family cysteine proteases | 19 (8) | 22 (6) | 20 (8) |
| Serine proteases | 13 (6) | 12 (4) | 18 (9) |
| Kazal-type protease inhibitors | 13 (12) | 15 (11) | 16 (10) |
| Cathepsin propeptide inhibitors | 4 (2) | 4 (1) | 4 (3) |
| Cytoplasmic effectors | |||
| Crinklers | 77 (28) | 80 (18) | 90 (37) |
| RxLRs | 132 (132) | 132 (132) | 186 (186) |
| Polysaccharide modifying enzymes | |||
| Cellulases | 30 (7) | 35 (4) | 22 (5) |
| Lytic polysaccharide mono-oxygenases | 6 (5) | 4 (2) | 5 (5) |
| Cutinases | 3 (2) | 4 (2) | 5 (5) |
| Chitinases | 1 (1) | 2 (0) | 2 (1) |
| Fungal cellulose binding domains | 8 (6) | 10 (7) | 8 (8) |
| Pectate lyases | 34 (23) | 25 (14) | 31 (23) |
| Pectin acetylesterases | 6 (4) | 6 (3) | 6 (5) |
| Pectinesterases | 7 (6) | 7 (5) | 15 (9) |
| Phytophthora chlamydospora | Phytophthora gonapodyides | Phytophthora pseudosyringae | |
|---|---|---|---|
| Glycoside Hydrolases | 234 (144) | 243 (123) | 210 (121) |
| Glycosyl Transferases | 125 (11) | 118 (10) | 120 (12) |
| Polysaccharide Lyases | 38 (27) | 33 (22) | 33 (27) |
| Carbohydrate Esterases | 46 (16) | 46 (12) | 53 (22) |
| Auxiliary Activities | 33 (14) | 41 (11) | 29 (11) |
| Carbohydrate-Binding Modules | 10 (2) | 12 (4) | 10 (1) |
| Total CAZymes | 483 (213) | 487 (179) | 453 (194) |
| Phytophthora chlamydospora | Phytophthora gonapodyides | Phytophthora pseudosyringae | |
|---|---|---|---|
| Tandem Clusters | 979 | 833 | 874 |
| Genes in Tandem Clusters | 2513 (14.1%) | 1863 (8.0%) | 2225 (12.8%) |
| Average Number of Genes Per Tandem Cluster | 2.57 | 2.24 | 2.55 |
| Secreted Proteins in Tandem Clusters | 354 (31.1%) | 265 (20.5%) | 328 (29.0%) |
| Phytophthora chlamydospora | Phytophthora gonapodyides | Phytophthora pseudosyringae | |
|---|---|---|---|
| Total protein groups identified | 321 | 246 | 313 |
| Total proteins identified | 351 | 283 | 331 |
| PAN/Apple domain | 10 | 8 | 5 |
| Transglutaminase elicitor | 6 | 6 | 8 |
| Elicitin | 5 | 8 | 4 |
| Cysteine-rich secretory protein family (CAP) | 4 | 4 | 4 |
| Necrosis inducing protein | 2 | 4 | 2 |
| PcF phytotoxin | 0 | 0 | 1 |
| Ribonuclease | 1 | 1 | 3 |
| Berberine-like protein | 1 | 1 | 0 |
| Glycoside hydrolases | 60 | 68 | 61 |
| Polysaccharide lyases | 7 | 9 | 5 |
| Carbohydrate esterases | 4 | 4 | 2 |
| Auxiliary activities | 8 | 5 | 4 |
| Carbohydrate-binding modules | 3 | 4 | 3 |
| PHI-Base homologs | 140 | 96 | 109 |
| Apoplastic proteins | 99 | 90 | 92 |
| Single transmembrane proteins | 33 | 25 | 29 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McGowan, J.; O’Hanlon, R.; Owens, R.A.; Fitzpatrick, D.A. Comparative Genomic and Proteomic Analyses of Three Widespread Phytophthora Species: Phytophthora chlamydospora, Phytophthora gonapodyides and Phytophthora pseudosyringae. Microorganisms 2020, 8, 653. https://doi.org/10.3390/microorganisms8050653
McGowan J, O’Hanlon R, Owens RA, Fitzpatrick DA. Comparative Genomic and Proteomic Analyses of Three Widespread Phytophthora Species: Phytophthora chlamydospora, Phytophthora gonapodyides and Phytophthora pseudosyringae. Microorganisms. 2020; 8(5):653. https://doi.org/10.3390/microorganisms8050653
Chicago/Turabian StyleMcGowan, Jamie, Richard O’Hanlon, Rebecca A. Owens, and David A. Fitzpatrick. 2020. "Comparative Genomic and Proteomic Analyses of Three Widespread Phytophthora Species: Phytophthora chlamydospora, Phytophthora gonapodyides and Phytophthora pseudosyringae" Microorganisms 8, no. 5: 653. https://doi.org/10.3390/microorganisms8050653
APA StyleMcGowan, J., O’Hanlon, R., Owens, R. A., & Fitzpatrick, D. A. (2020). Comparative Genomic and Proteomic Analyses of Three Widespread Phytophthora Species: Phytophthora chlamydospora, Phytophthora gonapodyides and Phytophthora pseudosyringae. Microorganisms, 8(5), 653. https://doi.org/10.3390/microorganisms8050653