Egyptian Mongoose (Herpestes ichneumon) Gut Microbiota: Taxonomical and Functional Differences across Sex and Age Classes



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Egyptian Mongoose Specimens
2.2. DNA Extraction, Quantification, Sequencing and Reads Processing
2.3. Estimation of Alpha-Diversity Indices
2.4. Comparison between Subpopulations Using Beta-Diversity Indices
2.5. Taxonomic and Functional Biomarker Discovery
2.6. Abiotic and Biotic Data Integration
2.7. Data Analysis
3. Results
3.1. Analysis of the Sequencing Data
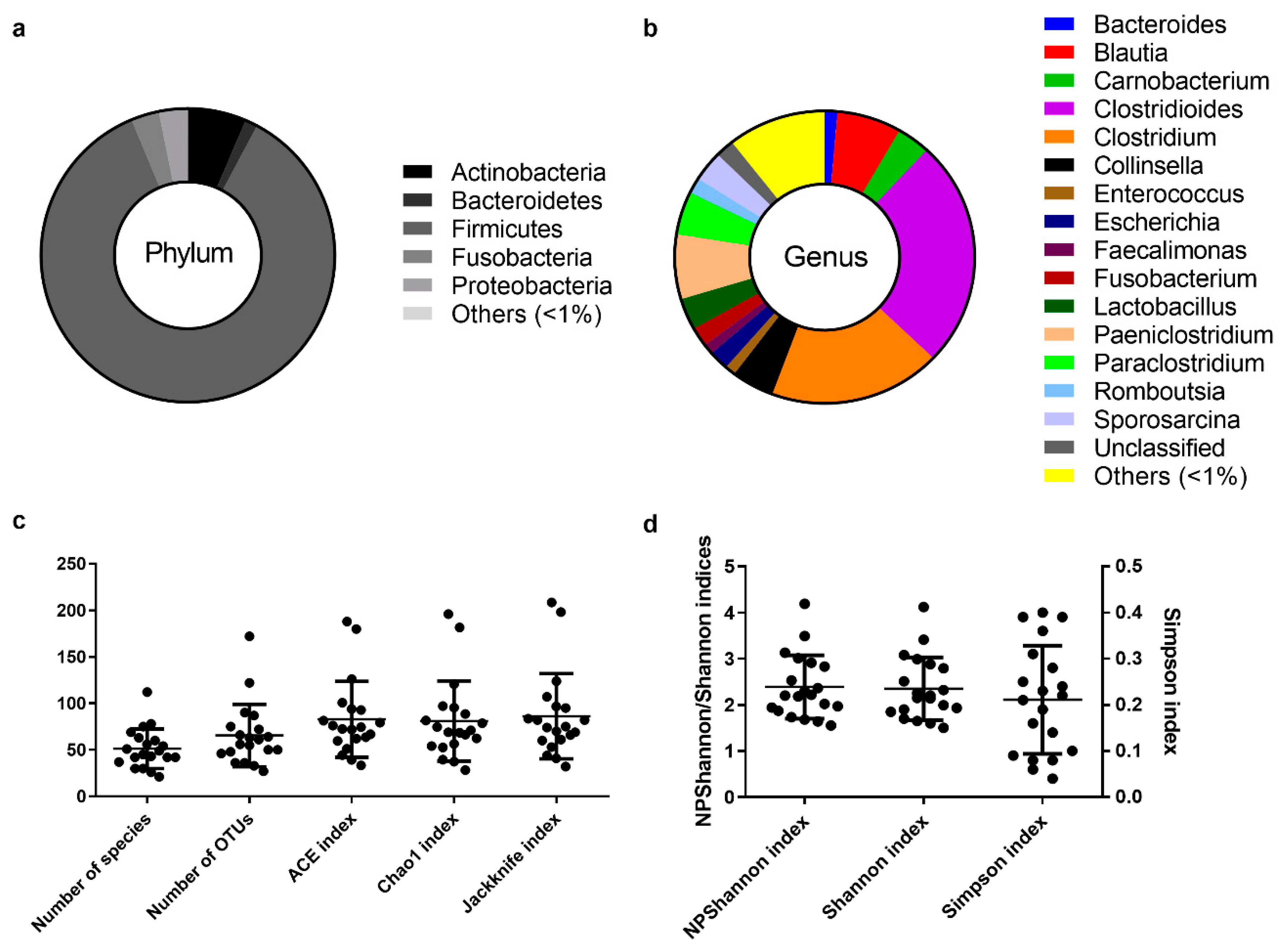
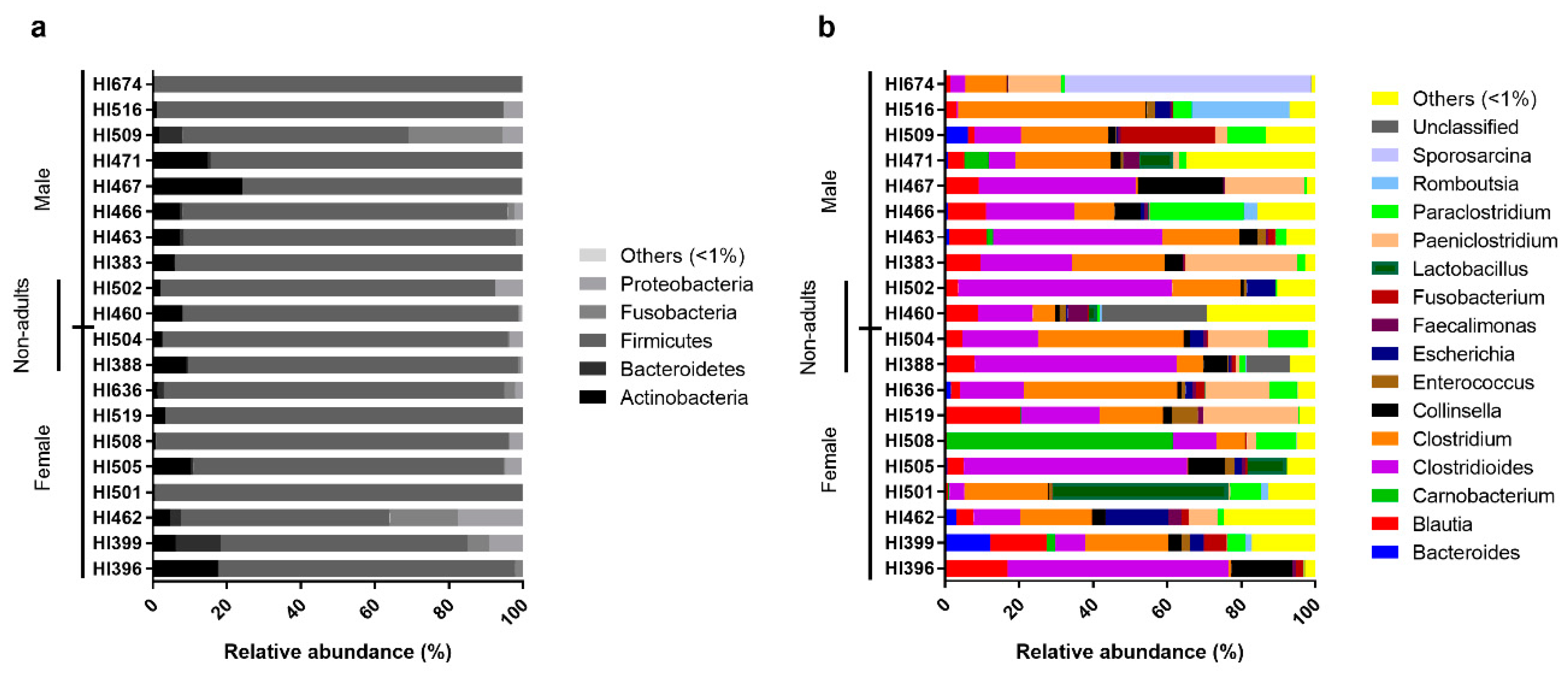
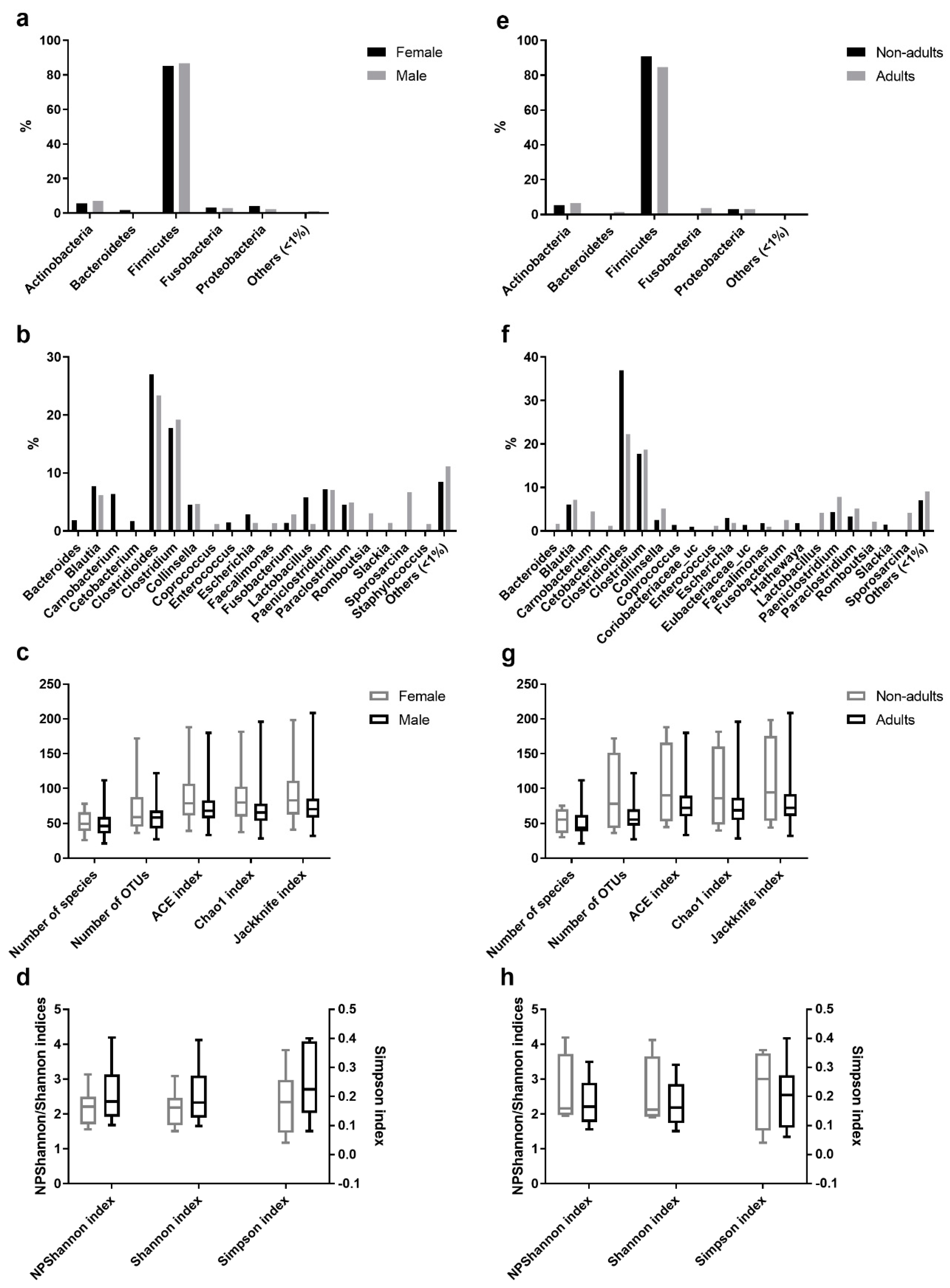
3.2. Bacterial Composition of the Egyptian Mongoose Gut on a Population and Individual Level
3.3. Estimation of Alpha Diversity Indices
3.4. Comparison between Subpopulations Using Beta-Diversity Analysis
3.5. Taxonomic Biomarker Discovery
3.6. Functional Biomarker Discovery
3.7. Abiotic and Biotic Data Integration
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jones, K.E.; Safi, K. Ecology and evolution of mammalian biodiversity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 2451–2461. [Google Scholar] [CrossRef] [PubMed]
- Bandeira, V.; Virgós, E.; Carvalho, J.; Barros, T.; Cunha, M.; Fonseca, C. Diet footprint of Egyptian mongoose along ecological gradients: Effects of primary productivity and life history traits. Mamm. Biol. 2017, 88, 16–25. [Google Scholar] [CrossRef]
- Kingdon, J. The kingdon field guide to african mammals. J. Mammal 1999, 80, 692–693. [Google Scholar]
- Gaubert, P.; López-Bao, J.V.; Machordom, A.; Morales, A.; Palomares, F.; Tània, B.; Veron, G. Comparative phylogeography of two african carnivorans presumably introduced into europe: Disentangling natural versus human-mediated dispersal across the strait of gibraltar. Blackwell Publ. 2011, 38, 341–358. [Google Scholar] [CrossRef]
- Detry, C.; Bicho, N.; Fernandes, H.; Fernandes, C. The emirate of Córdoba (756–929 ad) and the introduction of the Egyptian mongoose (Herpestes ichneumon) in Iberia: The remains from Muge, Portugal. J. Archaeol. Sci. 2011, 38, 3518–3523. [Google Scholar] [CrossRef]
- Borralho, R.; Rego, F.; Palomares, F.; Hora, A. The distribution of the Egyptian mongoose Herpestes ichneumon (L.) in Portugal. Mammal Rev. 1996, 26, 1–8. [Google Scholar] [CrossRef]
- Balmori, A.; Carbonell, R. Expansion and distribution of the Egyptian mongoose (Herpestes ichneumon) in the Iberian Peninsula. Galemys 2012, 24, 83–85. [Google Scholar] [CrossRef]
- Barros, T.; Carvalho, J.; Pereira, M.J.R.; Ferreira, J.P.; Fonseca, C. Following the trail: Factors underlying the sudden expansion of the Egyptian mongoose (Herpestes ichneumon) in Portugal. PLoS ONE 2015, 10, e0133768. [Google Scholar] [CrossRef]
- Do Linh San, E.; Maddock, A.H.; Gaubert, P.; Palomares, F. Herpestes ichneumon. The IUCN red list of threatened species 2016. IUCN Red List Threat. Species 2016. [Google Scholar] [CrossRef]
- Bandeira, V.; Virgós, E.; Barros, T.; Cunha, M.V.; Fonseca, C. Geographic variation and sexual dimorphism in body size of the Egyptian mongoose, Herpestes ichneumon in the western limit of its European distribution. Zoologischer Anzeiger J. Comp. Zool. 2016, 264, 1–10. [Google Scholar] [CrossRef]
- Schneider, T.C.; Kappeler, P.M. Social systems and life-history characteristics of mongooses. Biol. Rev. 2014, 89, 173–198. [Google Scholar] [CrossRef]
- Cunha, M.V.; Albuquerque, T.; Themudo, P.; Fonseca, C.; Bandeira, V.; Rosalino, L.M. Implications of Egyptian mongoose microbiome in the management of Mediterranean carnivores. Ecohealth, 3 February 2020; under review. [Google Scholar]
- Pereira, A.C.; Bandeira, V.; Fonseca, C.; Cunha, M.V. Understanding the cross-talk between microbiota, host life-history, and the environment using Egyptian mongoose (Herpestes ichneumon) as a model. (manuscript under preparation for submission).
- Van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The third revolution in sequencing technology. Trends Genet. 2018, 34, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16s ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.-H.; Ha, S.-M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing ezbiocloud: A taxonomically united database of 16s rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than blast. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Lee, B.; Moon, T.; Yoon, S.; Weissman, T. Dude-seq: Fast, flexible, and robust denoising for targeted amplicon sequencing. PLoS ONE 2017, 12, e0181463. [Google Scholar] [CrossRef]
- Yarza, P.; Yilmaz, P.; Pruesse, E.; Glockner, F.O.; Ludwig, W.; Schleifer, K.H.; Whitman, W.B.; Euzeby, J.; Amann, R.; Rossello-Mora, R. Uniting the classification of cultured and uncultured bacteria and archaea using 16s rRNA gene sequences. Nat. Rev. Microbiol. 2014, 12, 635–645. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. Uchime improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef]
- Good, I.J. The population frequencies of species and the estimation of population parameters. Biometrika 1953, 40, 237–264. [Google Scholar] [CrossRef]
- Chao, A.; Lee, S.-M. Estimating the number of classes via sample coverage. J. Am. Stat. Assoc. 1992, 87, 210–217. [Google Scholar] [CrossRef]
- Chao, A. Nonparametric estimation of the number of classes in a population. Scand. J. Stat. 1984, 265–270. [Google Scholar]
- Burnham, K.P.; Overton, W.S. Robust estimation of population size when capture probabilities vary among animals. Ecology 1979, 60, 927–936. [Google Scholar] [CrossRef]
- Magurran, A.E. Measuring Biological Diversity; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013. [Google Scholar]
- Chao, A.; Shen, T.-J. Nonparametric prediction in species sampling. J. Agric. Biol. Environ. Stat. 2004, 9, 253–269. [Google Scholar] [CrossRef]
- Heck, K.L.; van Belle, G.; Simberloff, D. Explicit calculation of the rarefaction diversity measurement and the determination of sufficient sample size. Ecology 1975, 56, 1459–1461. [Google Scholar] [CrossRef]
- Whittaker, R.H. Dominance and diversity in land plant communities: Numerical relations of species express the importance of competition in community function and evolution. Sci. N. Y. 1965, 147, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.; Knight, R. Unifrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef]
- Chen, J.; Bittinger, K.; Charlson, E.S.; Hoffmann, C.; Lewis, J.; Wu, G.D.; Collman, R.G.; Bushman, F.D.; Li, H. Associating microbiome composition with environmental covariates using generalized unifrac distances. Bioinformatics 2012, 28, 2106–2113. [Google Scholar] [CrossRef]
- Bray, J.R.; Curtis, J.T. An ordination of the upland forest communities of southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Endres, D.M.; Schindelin, J.E. A new metric for probability distributions. IEEE Trans. Inf. Theory 2003, 49, 1858–1860. [Google Scholar] [CrossRef]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16s rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019, 47, D590–D595. [Google Scholar] [CrossRef] [PubMed]
- Colston, T.J.; Jackson, C.R. Microbiome evolution along divergent branches of the vertebrate tree of life: What is known and unknown. Mol. Ecol. 2016, 25, 3776–3800. [Google Scholar] [CrossRef] [PubMed]
- Schwab, C.; Cristescu, B.; Northrup, J.M.; Stenhouse, G.B.; Gänzle, M. Diet and environment shape fecal bacterial microbiota composition and enteric pathogen load of grizzly bears. PLoS ONE 2011, 6, e27905. [Google Scholar] [CrossRef] [PubMed]
- McFarland, L.V.; Mulligan, M.E.; Kwok, R.Y.; Stamm, W.E. Nosocomial acquisition of clostridium difficile infection. N. Engl. J. Med. 1989, 320, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Glad, T.; Bernhardsen, P.; Nielsen, K.M.; Brusetti, L.; Andersen, M.; Aars, J.; Sundset, M.A. Bacterial diversity in faeces from polar bear (Ursus maritimus) in arctic Svalbard. BMC Microbiol. 2010, 10, 10. [Google Scholar] [CrossRef]
- Rosalino, L.M.; Santos, M.J.; Pereira, I.; Santos-Reis, M. Sex-driven differences in Egyptian mongoose’s (Herpestes ichneumon) diet in its Northwestern European range. Eur. J. Wildl. Res. 2009, 55, 293. [Google Scholar] [CrossRef]
- Maurice, C.F.; Cl Knowles, S.; Ladau, J.; Pollard, K.S.; Fenton, A.; Pedersen, A.B.; Turnbaugh, P.J. Marked seasonal variation in the wild mouse gut microbiota. ISME J. 2015, 9, 2423–2434. [Google Scholar] [CrossRef]
- Walter, J. Ecological role of lactobacilli in the gastrointestinal tract: Implications for fundamental and biomedical research. Appl. Environ. Microbiol. 2008, 74, 4985–4996. [Google Scholar] [CrossRef]
- Lahti, L.; Salonen, A.; Kekkonen, R.A.; Salojärvi, J.; Jalanka-Tuovinen, J.; Palva, A.; Orešič, M.; de Vos, W.M. Associations between the human intestinal microbiota, Lactobacillus rhamnosus GG and serum lipids indicated by integrated analysis of high-throughput profiling data. PeerJ 2013, 1, e32. [Google Scholar] [CrossRef] [PubMed]
- Den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.-J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [PubMed]
- Schwab, C.; Ganzle, M. Comparative analysis of fecal microbiota and intestinal microbial metabolic activity in captive polar bears. Can. J. Microbiol. 2011, 57, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Xue, Z.; Zhang, W.; Wang, L.; Hou, R.; Zhang, M.; Fei, L.; Zhang, X.; Huang, H.; Bridgewater, L.C.; Jiang, Y.; et al. The bamboo-eating giant panda harbors a carnivore-like gut microbiota, with excessive seasonal variations. mBio 2015, 6, e00022-15. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Fox, S.; Pemberton, D.; Hogg, C.; Papenfuss, A.T.; Belov, K. The tasmanian devil microbiome—Implications for conservation and management. Microbiome 2015, 3, 76. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Hugon, P.; Lagier, J.-C.; Robert, C.; Lepolard, C.; Papazian, L.; Musso, D.; Vialettes, B.; Raoult, D. Molecular studies neglect apparently gram-negative populations in the human gut microbiota. J. Clin. Microbiol. 2013, 51, 3286–3293. [Google Scholar] [CrossRef]
- Hugon, P.; Lagier, J.C.; Colson, P.; Bittar, F.; Raoult, D. Repertoire of human gut microbes. Microb. Pathog. 2017, 106, 103–112. [Google Scholar] [CrossRef]
- Lagier, J.-C.; Hugon, P.; Khelaifia, S.; Fournier, P.-E.; La Scola, B.; Raoult, D. The rebirth of culture in microbiology through the example of culturomics to study human gut microbiota. Clin. Microbiol. Rev. 2015, 28, 237–264. [Google Scholar] [CrossRef]
- Lagier, J.C.; Armougom, F.; Million, M.; Hugon, P.; Pagnier, I.; Robert, C.; Bittar, F.; Fournous, G.; Gimenez, G.; Maraninchi, M.; et al. Microbial culturomics: Paradigm shift in the human gut microbiome study. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2012, 18, 1185–1193. [Google Scholar] [CrossRef]
- Lagier, J.C.; Million, M.; Hugon, P.; Armougom, F.; Raoult, D. Human gut microbiota: Repertoire and variations. Front. Cell. Infect. Microbiol. 2012, 2, 136. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Hamady, M.; Lozupone, C.; Turnbaugh, P.J.; Ramey, R.R.; Bircher, J.S.; Schlegel, M.L.; Tucker, T.A.; Schrenzel, M.D.; Knight, R.; et al. Evolution of mammals and their gut microbes. Sci. N.Y. 2008, 320, 1647–1651. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Lozupone, C.A.; Hamady, M.; Knight, R.; Gordon, J.I. Worlds within worlds: Evolution of the vertebrate gut microbiota. Nat. Rev. Microbiol. 2008, 6, 776–788. [Google Scholar] [CrossRef] [PubMed]
- Gerritsen, J.; Fuentes, S.; Grievink, W.; van Niftrik, L.; Tindall, B.J.; Timmerman, H.M.; Rijkers, G.T.; Smidt, H. Characterization of Romboutsia ilealis gen. nov., sp. nov., isolated from the gastro-intestinal tract of a rat, and proposal for the reclassification of five closely related members of the genus Clostridium into the genera Romboutsia gen. nov., Intestinibacter gen. nov., Terrisporobacter gen. nov. and Asaccharospora gen. nov. Int. J. Syst. Evol. Microbiol. 2014, 64, 1600–1616. [Google Scholar]
- Ricaboni, D.; Mailhe, M.; Khelaifia, S.; Raoult, D.; Million, M. Romboutsia timonensis, a new species isolated from human gut. New Microbes New Infect. 2016, 12, 6–7. [Google Scholar] [CrossRef]
- Yang, L.; Liu, S.; Ding, J.; Dai, R.; He, C.; Xu, K.; Honaker, C.F.; Zhang, Y.; Siegel, P.; Meng, H. Gut microbiota co-microevolution with selection for host humoral immunity. Front. Microbiol. 2017, 8, 1243. [Google Scholar] [CrossRef]
- Leisner, J.J.; Laursen, B.G.; Prévost, H.; Drider, D.; Dalgaard, P. Carnobacterium: Positive and negative effects in the environment and in foods. FEMS Microbiol. Rev. 2007, 31, 592–613. [Google Scholar] [CrossRef]
- Sousa, M.; Goncalves, A.; Silva, N.; Serra, R.; Alcaide, E.; Zorrilla, I.; Torres, C.; Canica, M.; Igrejas, G.; Poeta, P. Acquired antibiotic resistance among wild animals: The case of Iberian Lynx (Lynx pardinus). Vet. Q. 2014, 34, 105–112. [Google Scholar] [CrossRef]
- Poeta, P.; Costa, D.; Saenz, Y.; Klibi, N.; Ruiz-Larrea, F.; Rodrigues, J.; Torres, C. Characterization of antibiotic resistance genes and virulence factors in faecal enterococci of wild animals in Portugal. J. Vet. Med. B Infect. Dis. Vet. Public Health 2005, 52, 396–402. [Google Scholar] [CrossRef]
- Lebreton, F.; Willems, R.J.L.; Gilmore, M.S. Enterococcus diversity, origins in nature, and gut colonization. In Enterococci: From Commensals to Leading Causes of Drug Resistant Infection; Gilmore, M.S., Clewell, D.B., Ike, Y., Shankar, N., Eds.; Massachusetts Eye and Ear Infirmary: Boston, MA, USA, 2014. [Google Scholar]
- Seedorf, H.; Griffin, N.W.; Ridaura, V.K.; Reyes, A.; Cheng, J.; Rey, F.E.; Smith, M.I.; Simon, G.M.; Scheffrahn, R.H.; Woebken, D.; et al. Bacteria from diverse habitats colonize and compete in the mouse gut. Cell 2014, 159, 253–266. [Google Scholar] [CrossRef]
- Nakashima, K.; Kimura, S.; Ogawa, Y.; Watanabe, S.; Soma, S.; Kaneko, T.; Yamada, L.; Sawada, H.; Tung, C.-H.; Lu, T.-M.; et al. Chitin-based barrier immunity and its loss predated mucus-colonization by indigenous gut microbiota. Nat. Commun. 2018, 9, 3402. [Google Scholar] [CrossRef]
- Tsuchiya, C.; Sakata, T.; Sugita, H. Novel ecological niche of Cetobacterium somerae, an anaerobic bacterium in the intestinal tracts of freshwater fish. Lett. Appl. Microbiol. 2008, 46, 43–48. [Google Scholar] [CrossRef]
- Finegold, S.M.; Vaisanen, M.L.; Molitoris, D.R.; Tomzynski, T.J.; Song, Y.; Liu, C.; Collins, M.D.; Lawson, P.A. Cetobacterium somerae sp. nov. from human feces and emended description of the genus Cetobacterium. Syst. Appl. Microbiol. 2003, 26, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Hand, D.; Wallis, C.; Colyer, A.; Penn, C.W. Pyrosequencing the canine faecal microbiota: Breadth and depth of biodiversity. PLoS ONE 2013, 8, e53115. [Google Scholar] [CrossRef] [PubMed]
- Keijser, B.J.; Zaura, E.; Huse, S.M.; van der Vossen, J.M.; Schuren, F.H.; Montijn, R.C.; ten Cate, J.M.; Crielaard, W. Pyrosequencing analysis of the oral microflora of healthy adults. J. Dent. Res. 2008, 87, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- Mazmanian, L. Has the microbiota played a critical role in the evolution of the adaptive immune system? Sci. N.Y. 2010, 330, 1768–1773. [Google Scholar]
- Wexler, H.M. Bacteroides: The good, the bad, and the nitty-gritty. Clin. Microbiol. Rev. 2007, 20, 593–621. [Google Scholar] [CrossRef]
- Alipour, M.J.; Jalanka, J.; Pessa-Morikawa, T.; Kokkonen, T.; Satokari, R.; Hynönen, U.; Iivanainen, A.; Niku, M. The composition of the perinatal intestinal microbiota in cattle. Sci. Rep. 2018, 8, 10437. [Google Scholar] [CrossRef]
- Navarro, M.; Dutra, F.; Uzal, F.A. Bacillary Hemoglobinuria. In Clostridial Diseases of Animals, 1st ed.; Uzal, F.A., Songer, W.G., Prescott, J.F., Eds.; Willey & Blackwell: Ames, IA, USA; Hoboken, NJ, USA, 2016; pp. 265–274. [Google Scholar]
- Ndongo, S.; Cassir, N.; Delerce, J.; Raoult, D.; La Scola, B. ‘Clostridium mediterraneense,’ a new bacterial species isolated from the human gut. New Microbes New Infect. 2017, 20, 25–26. [Google Scholar] [CrossRef]
- Bandeira, V.; Virgós, E.; Azevedo, A.; Carvalho, J.; Cunha, M.V.; Fonseca, C. Sex and season explain spleen weight variation in the Egyptian mongoose. Curr. Zool. 2018, 65, 11–20. [Google Scholar] [CrossRef]
- Aivelo, T.; Laakkonen, J.; Jernvall, J. Population- and individual-level dynamics of the intestinal microbiota of a small primate. Appl. Environ. Microbiol. 2016, 82, 3537–3545. [Google Scholar] [CrossRef] [PubMed]
- Degnan, P.H.; Pusey, A.E.; Lonsdorf, E.V.; Goodall, J.; Wroblewski, E.E.; Wilson, M.L.; Rudicell, R.S.; Hahn, B.H.; Ochman, H. Factors associated with the diversification of the gut microbial communities within chimpanzees from Gombe national park. Proc. Natl. Acad. Sci. USA 2012, 109, 13034–13039. [Google Scholar] [CrossRef] [PubMed]
- Amato, K.R.; Leigh, S.R.; Kent, A.; Mackie, R.I.; Yeoman, C.J.; Stumpf, R.M.; Wilson, B.A.; Nelson, K.E.; White, B.A.; Garber, P.A. The role of gut microbes in satisfying the nutritional demands of adult and juvenile wild, black howler monkeys (Alouatta pigra). Am. J. Phys. Anthropol. 2014, 155, 652–664. [Google Scholar] [CrossRef] [PubMed]
- Finlayson-Trick, E.C.L.; Getz, L.J.; Slaine, P.D.; Thornbury, M.; Lamoureux, E.; Cook, J.; Langille, M.G.I.; Murray, L.E.; McCormick, C.; Rohde, J.R.; et al. Taxonomic differences of gut microbiomes drive cellulolytic enzymatic potential within hind-gut fermenting mammals. PLoS ONE 2017, 12, e0189404. [Google Scholar] [CrossRef]
- Clavel, T.; Lepage, P.; Charrier, C. The Family Coriobacteriaceae; Springer Nature: Basel, Switzerland, 2014; Volume 49, pp. 201–238. [Google Scholar]
- Compo, N.R.; Gomez, D.E.; Tapscott, B.; Weese, J.S.; Turner, P.V. Fecal bacterial microbiota of Canadian commercial mink (Neovison vison): Yearly, life stage, and seasonal comparisons. PLoS ONE 2018, 13, e0207111. [Google Scholar] [CrossRef]
- Tidjani Alou, M.; Million, M.; Traore, S.I.; Mouelhi, D.; Khelaifia, S.; Bachar, D.; Caputo, A.; Delerce, J.; Brah, S.; Alhousseini, D.; et al. Gut bacteria missing in severe acute malnutrition, can we identify potential probiotics by culturomics? Front. Microbiol. 2017, 8, 899. [Google Scholar] [CrossRef]
- Goodrich, J.K.; Waters, J.L.; Poole, A.C.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.T.; et al. Human genetics shape the gut microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef]
- Kaakoush, N.O. Insights into the role of Erysipelotrichaceae in the human host. Front. Cell. Infect. Microbiol. 2015, 5, 84. [Google Scholar] [CrossRef]
- Clarke, S.F.; Murphy, E.F.; O’Sullivan, O.; Ross, R.P.; O’Toole, P.W.; Shanahan, F.; Cotter, P.D. Targeting the microbiota to address diet-induced obesity: A time dependent challenge. PLoS ONE 2013, 8, e65790. [Google Scholar] [CrossRef]
- Hu, X.; Liu, G.; Li, Y.; Wei, Y.; Lin, S.; Liu, S.; Zheng, Y.; Hu, D. High-throughput analysis reveals seasonal variation of the gut microbiota composition within forest musk deer (Moschus berezovskii). Front. Microbiol. 2018, 9, 1674. [Google Scholar] [CrossRef]
- Zhao, G.; Zhou, L.; Dong, Y.; Cheng, Y.; Song, Y. The gut microbiome of hooded cranes (Grus monacha) wintering at Shengjin lake, China. Microbiol. Open 2017, 6, e00447. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.-Y.; Zolnik, C.P.; Wang, Z.; Qiu, Y.; Usyk, M.; Wang, T.; Kizer, J.R.; Landay, A.L.; Kurland, I.J.; Anastos, K.; et al. Gut microbiota and plasma metabolites associated with diabetes in women with, or at high risk for, HIV infection. EBioMedicine 2018, 37, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Neis, E.P.; Dejong, C.H.; Rensen, S.S. The role of microbial amino acid metabolism in host metabolism. Nutrients 2015, 7, 2930–2946. [Google Scholar] [CrossRef] [PubMed]
- Agus, A.; Planchais, J.; Sokol, H. Gut microbiota regulation of tryptophan metabolism in health and disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef]
- Hobbs, M.E.; Williams, H.J.; Hillerich, B.; Almo, S.C.; Raushel, F.M. l-galactose metabolism in Bacteroides vulgatus from the human gut microbiota. Biochemistry 2014, 53, 4661–4670. [Google Scholar] [CrossRef]
- Brown, K.L.; Hancock, R.E. Cationic host defense (antimicrobial) peptides. Curr. Opin. Immunol. 2006, 18, 24–30. [Google Scholar] [CrossRef]
- Trevelline, B.K.; Fontaine, S.S.; Hartup, B.K.; Kohl, K.D. Conservation biology needs a microbial renaissance: A call for the consideration of host-associated microbiota in wildlife management practices. Proc. R. Soc. B Biol. Sci. 2019, 286, 20182448. [Google Scholar] [CrossRef]
- Amato, K.R.; Yeoman, C.J.; Kent, A.; Righini, N.; Carbonero, F.; Estrada, A.; Rex Gaskins, H.; Stumpf, R.M.; Yildirim, S.; Torralba, M.; et al. Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. ISME J. 2013, 7, 1344. [Google Scholar] [CrossRef]
- Barelli, C.; Albanese, D.; Donati, C.; Pindo, M.; Dallago, C.; Rovero, F.; Cavalieri, D.; Tuohy, K.M.; Hauffe, H.C.; de Filippo, C. Habitat fragmentation is associated to gut microbiota diversity of an endangered primate: Implications for conservation. Sci. Rep. 2015, 5, 14862. [Google Scholar] [CrossRef]
- Menke, S.; Meier, M.; Mfune, J.K.E.; Melzheimer, J.; Wachter, B.; Sommer, S. Effects of host traits and land-use changes on the gut microbiota of the Namibian black-backed jackal (Canis mesomelas). FEMS Microbiol. Ecol. 2017, 93. [Google Scholar] [CrossRef]
- Kikuchi, Y.; Tada, A.; Musolin, D.L.; Hari, N.; Hosokawa, T.; Fujisaki, K.; Fukatsu, T. Collapse of insect gut symbiosis under simulated climate change. mBio 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Bestion, E.; Jacob, S.; Zinger, L.; Di Gesu, L.; Richard, M.; White, J.; Cote, J. Climate warming reduces gut microbiota diversity in a vertebrate ectotherm. Nat. Ecol. Amp. Evol. 2017, 1, 0161. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, S.S.; Novarro, A.J.; Kohl, K.D. Environmental temperature alters the digestive performance and gut microbiota of a terrestrial amphibian. J. Exp. Biol. 2018, 221. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pereira, A.C.; Bandeira, V.; Fonseca, C.; Cunha, M.V. Egyptian Mongoose (Herpestes ichneumon) Gut Microbiota: Taxonomical and Functional Differences across Sex and Age Classes. Microorganisms 2020, 8, 392. https://doi.org/10.3390/microorganisms8030392
Pereira AC, Bandeira V, Fonseca C, Cunha MV. Egyptian Mongoose (Herpestes ichneumon) Gut Microbiota: Taxonomical and Functional Differences across Sex and Age Classes. Microorganisms. 2020; 8(3):392. https://doi.org/10.3390/microorganisms8030392
Chicago/Turabian StylePereira, André C., Victor Bandeira, Carlos Fonseca, and Mónica V. Cunha. 2020. "Egyptian Mongoose (Herpestes ichneumon) Gut Microbiota: Taxonomical and Functional Differences across Sex and Age Classes" Microorganisms 8, no. 3: 392. https://doi.org/10.3390/microorganisms8030392
APA StylePereira, A. C., Bandeira, V., Fonseca, C., & Cunha, M. V. (2020). Egyptian Mongoose (Herpestes ichneumon) Gut Microbiota: Taxonomical and Functional Differences across Sex and Age Classes. Microorganisms, 8(3), 392. https://doi.org/10.3390/microorganisms8030392