The Human Oral Microbiome in Health and Disease: From Sequences to Ecosystems

Abstract
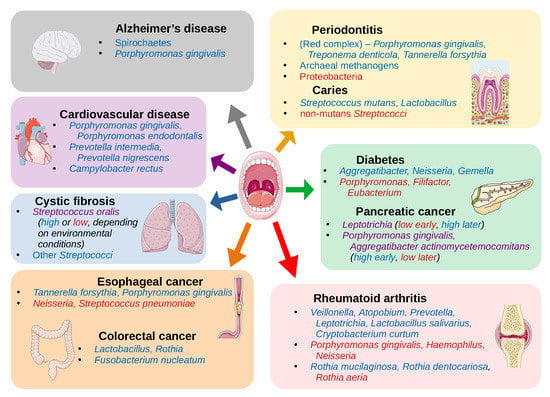
1. Introduction
2. Technical Approaches to Study the Oral Microbiome
3. The Oral Cavity and its Microbial Niches
4. The Healthy Oral Microbiome and Definition of Stomatotypes
5. Non-Bacterial Oral Microbes
6. Oral Microbiome and Oral Diseases
7. Oral Microbiome and Non-Oral Diseases
8. Clinical Potential of the Oral Microbiome/Manipulations and Perturbations of the Oral Microbiome
9. Conclusions and Future Outlook
Funding
Conflicts of Interest
References
- Ursell, L.K.; Metcalf, J.L.; Parfrey, L.W.; Knight, R. Defining the human microbiome. Nutr. Rev. 2012, 70, S38–S44. [Google Scholar] [CrossRef]
- Integrative HMP (iHMP) Research Network Consortium. The Integrative Human Microbiome Project: Dynamic analysis of microbiome-host omics profiles during periods of human health and disease. Cell Host Microbe 2014, 16, 276–289. [Google Scholar] [CrossRef]
- Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Bihan, M.; Yooseph, S.; Methé, B.A. Analyses of the microbial diversity across the human microbiome. PLoS ONE 2012, 7, e32118. [Google Scholar] [CrossRef] [PubMed]
- Marsh, P.D.; Do, T.; Beighton, D.; Devine, D.A. Influence of saliva on the oral microbiota. Periodontology 2000, 70, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Dewhirst, F.E.; Chen, T.; Izard, J.; Paster, B.J.; Tanner, A.C.; Yu, W.H.; Lakshmanan, A.; Wade, W.G. The human oral microbiome. J. Bacteriol. 2010, 192, 5002–5017. [Google Scholar] [CrossRef]
- Paster, B.J.; Olsen, I.; Aas, J.A.; Dewhirst, F.E. The breadth of bacterial diversity in the human periodontal pocket and other oral sites. Periodontology 2000, 42, 80–87. [Google Scholar] [CrossRef]
- Segata, N.; Haake, S.K.; Mannon, P.; Lemon, K.P.; Waldron, L.; Gevers, D.; Huttenhower, C.; Izard, J. Composition of the adult digestive tract bacterial microbiome based on seven mouth surfaces, tonsils, throat and stool samples. Genome Biol. 2012, 13, R42. [Google Scholar] [CrossRef]
- Xu, X.; He, J.; Xue, J.; Wang, Y.; Li, K.; Zhang, K.; Guo, Q.; Liu, X.; Zhou, Y.; Cheng, L.; et al. Oral microbiome differs by age and location. Environ. Microbiol. 2015, 17, 699–710. [Google Scholar] [CrossRef]
- Willis, J.R.; González-Torres, P.; Pittis, A.A.; Bejarano, L.A.; Cozzuto, L.; Andreu-Somavilla, N.; Alloza-Trabado, M.; Valentín, A.; Ksiezopolska, E.; Company, C.; et al. Citizen science charts two major “stomatotypes” in the oral microbiome of adolescents and reveals links with habits and drinking water composition. Microbiome 2018, 6, 218. [Google Scholar] [CrossRef]
- Wang, H.; Altemus, J.; Niazi, F.; Green, H.; Calhoun, B.C.; Sturgis, C.; Grobmyer, S.R.; Eng, C. Breast tissue, oral and urinary microbiomes in breast cancer. Oncotarget 2017, 8, 88122–88138. [Google Scholar] [CrossRef] [PubMed]
- Kato, I.; Vasquez, A.A.; Moyerbrailean, G.; Land, S.; Sun, J.; Lin, H.S.; Ram, J.L. Oral microbiome and history of smoking and colorectal cancer. J. Epidemiol. Res. 2016, 2, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Escapa, I.F.; Chen, T.; Huang, Y.; Gajare, P.; Dewhirst, F.E.; Lemon, K.P. New insights into human nostril microbiome from the expanded Human Oral Microbiome Database (eHOMD): A resource for the microbiome of the human aerodigestive tract. MSystems 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Matarazzo, F.; Ribeiro, A.C.; Feres, M.; Faveri, M.; Mayer, M.P.A. Diversity and quantitative analysis of Archaea in aggressive periodontitis and periodontally healthy subjects. J. Clin. Periodontol. 2011, 38, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Lepp, P.W.; Brinig, M.M.; Ouverney, C.C.; Palm, K.; Armitage, G.C.; Relman, D.A. Methanogenic Archaea and human periodontal disease. Proc. Natl. Acad. Sci. USA 2004, 101, 6176–6181. [Google Scholar] [CrossRef]
- Griffen, A.L.; Beall, C.J.; Campbell, J.H.; Firestone, N.D.; Kumar, P.S.; Yang, Z.K.; Podar, M.; Leys, E.J. Distinct and complex bacterial profiles in human periodontitis and health revealed by 16S pyrosequencing. ISME J. 2012, 6, 1176–1185. [Google Scholar] [CrossRef]
- Vartoukian, S.R.; Palmer, R.M.; Wade, W.G. Diversity and morphology of members of the phylum “synergistetes” in periodontal health and disease. Appl. Environ. Microbiol. 2009, 75, 3777–3786. [Google Scholar] [CrossRef] [PubMed]
- Costalonga, M.; Herzberg, M.C. The oral microbiome and the immunobiology of periodontal disease and caries. Immunol. Lett. 2014, 162, 22–38. [Google Scholar] [CrossRef]
- Liu, B.; Faller, L.L.; Klitgord, N.; Mazumdar, V.; Ghodsi, M.; Sommer, D.D.; Gibbons, T.R.; Treangen, T.J.; Chang, Y.C.; Li, S.; et al. Deep sequencing of the oral microbiome reveals signatures of periodontal disease. PLoS ONE 2012, 7, e37919. [Google Scholar] [CrossRef]
- Jorth, P.; Turner, K.H.; Gumus, P.; Nizam, N.; Buduneli, N.; Whiteley, M. Metatranscriptomics of the human oral microbiome during health and disease. MBio 2014, 5, e01012–e01014. [Google Scholar] [CrossRef]
- HAUBEK, D. The highly leukotoxic JP2 clone of Aggregatibacter actinomycetemcomitans: Evolutionary aspects, epidemiology and etiological role in aggressive periodontitis. APMIS 2010, 118. [Google Scholar] [CrossRef] [PubMed]
- Gross, E.L.; Leys, E.J.; Gasparovich, S.R.; Firestone, N.D.; Schwartzbaum, J.A.; Janies, D.A.; Asnani, K.; Griffen, A.L. Bacterial 16S sequence analysis of severe caries in young permanent teeth. J. Clin. Microbiol. 2010, 48, 4121–4128. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.; Bowen, W.H. Candida albicans and Streptococcus mutans: A potential synergistic alliance to cause virulent tooth decay in children. Future Microbiol. 2014, 9, 1295–1297. [Google Scholar] [CrossRef] [PubMed]
- Mager, D.L.; Haffajee, A.D.; Devlin, P.M.; Norris, C.M.; Posner, M.R.; Goodson, J.M. The salivary microbiota as a diagnostic indicator of oral cancer: A descriptive, non-randomized study of cancer-free and oral squamous cell carcinoma subjects. J. Transl. Med. 2005, 3, 27. [Google Scholar] [CrossRef]
- Pushalkar, S.; Ji, X.; Li, Y.; Estilo, C.; Yegnanarayana, R.; Singh, B.; Li, X.; Saxena, D. Comparison of oral microbiota in tumor and non-tumor tissues of patients with oral squamous cell carcinoma. BMC Microbiol. 2012, 12, 144. [Google Scholar] [CrossRef]
- Wang, L.; Ganly, I. The oral microbiome and oral cancer. Clin. Lab. Med. 2014, 34, 711–719. [Google Scholar] [CrossRef]
- Peters, B.A.; Wu, J.; Pei, Z.; Yang, L.; Purdue, M.P.; Freedman, N.D.; Jacobs, E.J.; Gapstur, S.M.; Hayes, R.B.; Ahn, J. Oral microbiome composition reflects prospective risk for esophageal cancers. Cancer Res. 2017, 77, 6777–6787. [Google Scholar] [CrossRef]
- Broecker, F.; Russo, G.; Klumpp, J.; Moelling, K. Stable core virome despite variable microbiome after fecal transfer. Gut Microbes 2017, 8, 214–220. [Google Scholar] [CrossRef]
- Oh, J.; Byrd, A.L.; Park, M.; Kong, H.H.; Segre, J.A. Temporal stability of the human skin microbiome. Cell 2016, 165, 854–866. [Google Scholar] [CrossRef]
- Wantland, W.W.; Wantland, E.M.; Remo, J.W.; Winquist, D.L. Studies on Human Mouth Protozoa. J. Dent. Res. 1958, 37, 949–950. [Google Scholar] [CrossRef]
- Fan, X.; Alekseyenko, A.V.; Wu, J.; Peters, B.A.; Jacobs, E.J.; Gapstur, S.M.; Purdue, M.P.; Abnet, C.C.; Stolzenberg-Solomon, R.; Miller, G.; et al. Human oral microbiome and prospective risk for pancreatic cancer: A population-based nested case-control study. Gut 2018, 67, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Torres, P.J.; Fletcher, E.M.; Gibbons, S.M.; Bouvet, M.; Doran, K.S.; Kelley, S.T. Characterization of the salivary microbiome in patients with pancreatic cancer. PeerJ 2015, 3, e1373. [Google Scholar] [CrossRef]
- Whiley, R.A.; Fleming, E.V.; Makhija, R.; Waite, R.D. Environment and colonisation sequence are key parameters driving cooperation and competition between Pseudomonas aeruginosa cystic fibrosis strains and oral commensal streptococci. PLoS ONE 2015, 10, e0115513. [Google Scholar] [CrossRef] [PubMed]
- Teles, R.; Wang, C.Y. Mechanisms involved in the association between periodontal diseases and cardiovascular disease. Oral Dis. 2011, 17, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Chhibber-Goel, J.; Singhal, V.; Bhowmik, D.; Vivek, R.; Parakh, N.; Bhargava, B.; Sharma, A. Linkages between oral commensal bacteria and atherosclerotic plaques in coronary artery disease patients. NPJ Biofilms Microbiomes 2016, 2. [Google Scholar] [CrossRef]
- Roszyk, E.; Puszczewicz, M. Role of human microbiome and selected bacterial infections in the pathogenesis of rheumatoid arthritis. Reumatologia 2017, 55, 242–250. [Google Scholar] [CrossRef]
- Scher, J.U.; Ubeda, C.; Equinda, M.; Khanin, R.; Buischi, Y.; Viale, A.; Lipuma, L.; Attur, M.; Pillinger, M.H.; Weissmann, G.; et al. Periodontal disease and the oral microbiota in new-onset rheumatoid arthritis. Arthritis Rheum. 2012, 64, 3083–3094. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, D.; Jia, H.; Feng, Q.; Wang, D.; Liang, D.; Wu, X.; Li, J.; Tang, L.; Li, Y.; et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat. Med. 2015, 21, 895–905. [Google Scholar] [CrossRef]
- Brusca, S.B.; Abramson, S.B.; Scher, J.U. Microbiome and mucosal inflammation as extra-articular triggers for rheumatoid arthritis and autoimmunity. Curr. Opin. Rheumatol. 2014, 26, 101–107. [Google Scholar] [CrossRef]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5. [Google Scholar] [CrossRef]
- Miklossy, J. Bacterial amyloid and DNA are important constituents of senile plaques: Further evidence of the spirochetal and biofilm nature of senile plaques. J. Alzheimers Dis. 2016, 53, 1459–1473. [Google Scholar] [CrossRef] [PubMed]
- Aguayo, S.; Schuh, C.M.A.P.; Vicente, B.; Aguayo, L.G. Association between alzheimer’s disease and oral and gut microbiota: Are pore forming proteins the missing link? J. Alzheimers Dis. 2018, 65, 29–46. [Google Scholar] [CrossRef] [PubMed]
- Casarin, R.C.V.; Barbagallo, A.; Meulman, T.; Santos, V.R.; Sallum, E.A.; Nociti, F.H.; Duarte, P.M.; Casati, M.Z.; Gonçalves, R.B. Subgingival biodiversity in subjects with uncontrolled type-2 diabetes and chronic periodontitis. J. Periodontal Res. 2013, 48, 30–36. [Google Scholar] [CrossRef]
- Pasolli, E.; Asnicar, F.; Manara, S.; Zolfo, M.; Karcher, N.; Armanini, F.; Beghini, F.; Manghi, P.; Tett, A.; Ghensi, P.; et al. Extensive Unexplored Human Microbiome Diversity Revealed by Over 150,000 Genomes from Metagenomes Spanning Age, Geography, and Lifestyle. Cell 2019, 176, 649–662. [Google Scholar] [CrossRef]
- Clemente, J.C.; Pehrsson, E.C.; Blaser, M.J.; Sandhu, K.; Gao, Z.; Wang, B.; Magris, M.; Hidalgo, G.; Contreras, M.; Noya-Alarcón, Ó.; et al. The microbiome of uncontacted Amerindians. Sci. Adv. 2015, 1, e1500183. [Google Scholar] [CrossRef] [PubMed]
- Henrich, J.; Heine, S.; Norenzayan, A. The weirdest people in the world? Behav. Brain Sci. 2010, 33, 61–83. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef]
- Zaura, E.; Brandt, B.W.; Prodan, A.; Teixeira de Mattos, M.J.; Imangaliyev, S.; Kool, J.; Buijs, M.J.; Jagers, F.L.; Hennequin-Hoenderdos, N.L.; Slot, D.E.; et al. On the ecosystemic network of saliva in healthy young adults. ISME J. 2017, 11, 1218–1231. [Google Scholar] [CrossRef]
- De Filippis, F.; Vannini, L.; La Storia, A.; Laghi, L.; Piombino, P.; Stellato, G.; Serrazanetti, D.I.; Gozzi, G.; Turroni, S.; Ferrocino, I.; et al. The same microbiota and a potentially discriminant metabolome in the saliva of omnivore, ovo-lacto-vegetarian and Vegan individuals. PLoS ONE 2014, 9, e112373. [Google Scholar] [CrossRef]
- Ding, T.; Schloss, P.D. Dynamics and associations of microbial community types across the human body. Nature 2014, 509, 357–360. [Google Scholar] [CrossRef]
- Takeshita, T.; Kageyama, S.; Furuta, M.; Tsuboi, H.; Takeuchi, K.; Shibata, Y.; Shimazaki, Y.; Akifusa, S.; Ninomiya, T.; Kiyohara, Y.; et al. Bacterial diversity in saliva and oral health-related conditions: The Hisayama Study. Sci. Rep. 2016, 6, 22164. [Google Scholar] [CrossRef] [PubMed]
- Malla, M.A.; Dubey, A.; Kumar, A.; Yadav, S.; Hashem, A.; Allah, E.F. Exploring the Human Microbiome: The Potential Future Role of Next-Generation Sequencing in Disease Diagnosis and Treatment. Front. Immunol. 2019, 9, 2868. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S. Shotgun DNA sequencing using cloned DNase I-generated fragments. Nucleic Acids Res. 1981, 9, 3015–3027. [Google Scholar] [CrossRef]
- Weisburg, W.G.; Barns, S.M.; Pelletier, D.A.; Lane, D.J. 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 1991, 173, 697–703. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Holmes, S.P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017, 11, 2639–2643. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Amir, A.; McDonald, D.; Navas-Molina, J.A.; Kopylova, E.; Morton, J.T.; Zech, X.Z.; Kightley, E.P.; Thompson, L.R.; Hyde, E.R.; Gonzalez, A.; et al. Deblur rapidly resolves single-nucleotide community sequence patterns. MSystems 2017, 2. [Google Scholar] [CrossRef]
- Kembel, S.W.; Wu, M.; Eisen, J.A.; Green, J.L. Incorporating 16S gene copy number information improves estimates of microbial diversity and abundance. PLoS Comput. Biol. 2012, 8, e1002743. [Google Scholar] [CrossRef]
- Louca, S.; Doebeli, M.; Parfrey, L.W. Correcting for 16S rRNA gene copy numbers in microbiome surveys remains an unsolved problem. Microbiome 2018, 6, 41. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.; Quince, C.; Pijl, A.S.; de Hollander, M.; Kowalchuk, G.A. A comparison of rpoB and 16S rRNA as markers in pyrosequencing studies of bacterial diversity. PLoS ONE 2012, 7, e30600. [Google Scholar] [CrossRef] [PubMed]
- Escudié, F.; Auer, L.; Bernard, M.; Mariadassou, M.; Cauquil, L.; Vidal, K.; Maman, S.; Hernandez-Raquet, G.; Combes, S.; Pascal, G. FROGS: Find, rapidly, OTUs with galaxy solution. Bioinformatics 2018, 34, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- Ogier, J.C.; Pagès, S.; Galan, M.; Barret, M.; Gaudriault, S. rpoB, a promising marker for analyzing the diversity of bacterial communities by amplicon sequencing. BMC Microbiol. 2019, 19, 171. [Google Scholar] [CrossRef]
- Martens, M.; Dawyndt, P.; Coopman, R.; Gillis, M.; De Vos, P.; Willems, A. Advantages of multilocus sequence analysis for taxonomic studies: A case study using 10 housekeeping genes in the genus Ensifer (including former Sinorhizobium). Int. J. Syst. Evol. Microbiol. 2008. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, R.; Rani, A.; Metwally, A.; McGee, H.S.; Perkins, D.L. Analysis of the microbiome: Advantages of whole genome shotgun versus 16S amplicon sequencing. Biochem. Biophys. Res. Commun. 2016, 469, 967–977. [Google Scholar] [CrossRef]
- Moran, M.A. Metatranscriptomics: Eavesdropping on Complex Microbial Communities. Microbe 2009, 4, 329–335. [Google Scholar] [CrossRef][Green Version]
- Heyer, R.; Schallert, K.; Zoun, R.; Becher, B.; Saake, G.; Benndorf, D. Challenges and perspectives of metaproteomic data analysis. J. Biotechnol. 2017, 261, 24–36. [Google Scholar] [CrossRef]
- Fiehn, O. Metabolomics—The link between genotypes and phenotypes. Plant Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef]
- Bernini, P.; Bertini, I.; Luchinat, C.; Nepi, S.; Saccenti, E.; Schäfer, H.; Schütz, B.; Spraul, M.; Tenori, L. Individual human phenotypes in metabolic space and time. J. Proteome Res. 2009, 8, 4264–4271. [Google Scholar] [CrossRef]
- Peano, C.; Pietrelli, A.; Consolandi, C.; Rossi, E.; Petiti, L.; Tagliabue, L.; De Bellis, G.; Landini, P. An efficient rRNA removal method for RNA sequencing in GC-rich bacteria. Microb. Inform. Exp. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Aguiar-Pulido, V.; Huang, W.; Suarez-Ulloa, V.; Cickovski, T.; Mathee, K.; Narasimhan, G. Metagenomics, metatranscriptomics, and metabolomics approaches for microbiome analysis. Evol. Bioinform. Online 2016, 12, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Easterly, C.W.; Sajulga, R.; Mehta, S.; Johnson, J.; Kumar, P.; Hubler, S.; Mesuere, B.; Rudney, J.; Griffin, T.J.; Jagtap, P.D. metaQuantome: An integrated, quantitative metaproteomics approach reveals connections between taxonomy and protein function in complex microbiomes. Mol. Cell. Proteom. 2019, 18, S82–S91. [Google Scholar] [CrossRef] [PubMed]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Bolchacova, E.; Voigt, K.; Crous, P.W.; et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [PubMed]
- Wylie, K.M.; Weinstock, G.M.; Storch, G.A. Emerging view of the human virome. Transl. Res. 2012, 160, 283–290. [Google Scholar] [CrossRef]
- Thurber, R.; Haynes, M.; Breitbart, M.; Wegley, L.; Rohwer, F. Laboratory procedures to generate viral metagenomes. Nat. Protoc. 2009, 4, 470–483. [Google Scholar] [CrossRef]
- Allen, L.Z.; Ishoey, T.; Novotny, M.A.; McLean, J.S.; Lasken, R.S.; Williamson, S.J. Single virus genomics: A new tool for virus discovery. PLoS ONE 2011, 6, e17722. [Google Scholar] [CrossRef]
- Lim, Y.; Totsika, M.; Morrison, M.; Punyadeera, C. The saliva microbiome profiles are minimally affected by collection method or DNA extraction protocols. Sci. Rep. 2017, 7, 8523. [Google Scholar] [CrossRef]
- Fan, X.; Peters, B.A.; Min, D.; Ahn, J.; Hayes, R.B. Comparison of the oral microbiome in mouthwash and whole saliva samples. PLoS ONE 2018, 13, e0194729. [Google Scholar] [CrossRef]
- Mallick, H.; Ma, S.; Franzosa, E.A.; Vatanen, T.; Morgan, X.C.; Huttenhower, C. Experimental design and quantitative analysis of microbial community multiomics. Genome Biol. 2017, 18, 228. [Google Scholar] [CrossRef]
- Knight, R.; Vrbanac, A.; Taylor, B.C.; Aksenov, A.; Callewaert, C.; Debelius, J.; Gonzalez, A.; Kosciolek, T.; McCall, L.I.; McDonald, D.; et al. Best practices for analysing microbiomes. Nat. Rev. Microbiol. 2018, 16, 410–422. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Susan Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Leo, L.; Shetty, S. Microbiome R Package. Available online: http://microbiome.github.io (accessed on 18 February 2020).
- Jari, O.F.; Guillaume, B.; Michael, F.; Roeland, K.; Pierre, L.; Dan, M.; Peter, R.M.; Minchin, P.R.; O’Hara, R.B.; Gavin, L.S.; et al. Vegan: Community Ecology Package; R Package Version, 2019. Available online: https://CRAN.R-project.org/package=vegan (accessed on 18 February 2020).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019; Available online: https://www.R-project.org/ (accessed on 18 February 2020).
- Douglas, B.; Martin, M.; Ben, B.; Steve, W. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 2015, 67. [Google Scholar] [CrossRef]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Aßhauer, K.P.; Wemheuer, B.; Daniel, R.; Meinicke, P. Tax4Fun: Predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 2015, 31, 2882–2884. [Google Scholar] [CrossRef]
- Zhou, Y.H.; Gallins, P. A Review and Tutorial of Machine Learning Methods for Microbiome Host Trait Prediction. Front. Genet. 2019, 10, 579. [Google Scholar] [CrossRef] [PubMed]
- Pasolli, E.; Truong, D.T.; Malik, F.; Waldron, L.; Segata, N. Machine Learning Meta-analysis of Large Metagenomic Datasets: Tools and Biological Insights. PLoS Comput. Biol. 2016, 12, e1004977. [Google Scholar] [CrossRef] [PubMed]
- Duvallet, C.; Gibbons, S.M.; Gurry, T.; Irizarry, R.A.; Alm, E.J. Meta-analysis of gut microbiome studies identifies disease-specific and shared responses. Nat. Commun. 2017, 8, 1784. [Google Scholar] [CrossRef] [PubMed]
- Flemer, B.; Warren, R.D.; Barrett, M.P.; Cisek, K.; Das, A.; Jeffery, I.B.; Hurley, E.; O’Riordain, M.; Shanahan, F.; O’Toole, P.W. The oral microbiota in colorectal cancer is distinctive and predictive. Gut 2018, 67, 1454–1463. [Google Scholar] [CrossRef] [PubMed]
- Vandeputte, D.; Kathagen, G.; D’hoe, K.; Vieira-Silva, S.; Valles-Colomer, M.; Sabino, J.; Wang, J.; Tito, R.Y.; De Commer, L.; Darzi, Y.; et al. Quantitative microbiome profiling links gut community variation to microbial load. Nature 2017, 551, 507–511. [Google Scholar] [CrossRef]
- Gloor, G.B.; Macklaim, J.M.; Pawlowsky-Glahn, V.; Egozcue, J.J. Microbiome Datasets Are Compositional: And This Is Not Optional. Front. Microbiol. 2017, 8, 2224. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. Waste not, want not: Why rarefying microbiome data is inadmissible. PLoS Comput. Biol. 2014, 10, e1003531. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M. Bacteriology of Humans an Ecological Perspective; Blackwell Publishing Ltd.: Hoboken, NJ, USA, 2008. [Google Scholar]
- Laubichler, M.D.; Renn, J. Extended evolution: A conceptual framework for integrating regulatory networks and niche construction. J. Exp. Zool. B Mol. Dev. Evol. 2015, 324, 565–577. [Google Scholar] [CrossRef] [PubMed]
- van der Meulen, T.A.; Harmsen, H.J.M.; Bootsma, H.; Liefers, S.C.; Vich, V.A.; Zhernakova, A.; Fu, J.; Wijmenga, C.; Spijkervet, F.K.L.; Kroese, F.G.M.; et al. Dysbiosis of the buccal mucosa microbiome in primary Sjögren’s syndrome patients. Rheumatology 2018, 57, 2225–2234. [Google Scholar] [CrossRef]
- Ganesan, S.M.; Joshi, V.; Fellows, M.; Dabdoub, S.M.; Nagaraja, H.N.; O’Donnell, B.; Deshpande, N.R.; Kumar, P.S. A tale of two risks: Smoking, diabetes and the subgingival microbiome. ISME J. 2017, 11, 2075–2089. [Google Scholar] [CrossRef]
- Abusleme, L.; Dupuy, A.K.; Dutzan, N.; Silva, N.; Burleson, J.A.; Strausbaugh, L.D.; Gamonal, J.; Diaz, P.I. The subgingival microbiome in health and periodontitis and its relationship with community biomass and inflammation. ISME J. 2013, 7, 1016–1025. [Google Scholar] [CrossRef]
- Moutsopoulos, N.M.; Konkel, J.E. Tissue-Specific Immunity at the Oral Mucosal Barrier. Trends Immunol. 2018, 39, 276–287. [Google Scholar] [CrossRef]
- Mark, W.J.L.; Rossetti, B.J.; Rieken, C.W.; Dewhirst, F.E.; Borisy, G.G. Biogeography of a human oral microbiome at the micron scale. Proc. Natl. Acad. Sci. USA 2016, 113, E791–E800. [Google Scholar] [CrossRef]
- Wei, Y.; Shi, M.; Zhen, M.; Wang, C.; Hu, W.; Nie, Y.; Wu, X. Comparison of subgingival and buccal mucosa microbiome in chronic and aggressive periodontitis: A pilot study. Front. Cell. Infect. Microbiol. 2019, 9, 53. [Google Scholar] [CrossRef]
- Asakawa, M.; Takeshita, T.; Furuta, M.; Kageyama, S.; Takeuchi, K.; Hata, J.; Ninomiya, T.; Yamashita, Y. Tongue Microbiota and Oral Health Status in Community-Dwelling Elderly Adults. mSphere 2018, 3. [Google Scholar] [CrossRef]
- Lu, H.; Ren, Z.; Li, A.; Li, J.; Xu, S.; Zhang, H.; Jiang, J.; Yang, J.; Luo, Q.; Zhou, K.; et al. Tongue coating microbiome data distinguish patients with pancreatic head cancer from healthy controls. J. Oral Microbiol. 2019, 11, 1563409. [Google Scholar] [CrossRef] [PubMed]
- Fukui, Y.; Aoki, K.; Ishii, Y.; Tateda, K. The palatine tonsil bacteriome, but not the mycobiome, is altered in HIV infection. BMC Microbiol. 2018, 18, 127. [Google Scholar] [CrossRef] [PubMed]
- Koren, O.; Knights, D.; Gonzalez, A.; Waldron, L.; Segata, N.; Knight, R.; Huttenhower, C.; Ley, R.E. A guide to enterotypes across the human body: Meta-analysis of microbial community structures in human microbiome datasets. PLoS Comput. Biol. 2013, 9, e1002863. [Google Scholar] [CrossRef] [PubMed]
- Debelius, J.; Song, S.J.; Vazquez-Baeza, Y.; Xu, Z.Z.; Gonzalez, A.; Knight, R. Tiny microbes, enormous impacts: What matters in gut microbiome studies? Genome Biol. 2016, 17, 217. [Google Scholar] [CrossRef]
- Costello, E.K.; Lauber, C.L.; Hamady, M.; Fierer, N.; Gordon, J.I.; Knight, R. Bacterial community variation in human body habitats across space and time. Science 2009, 326, 1694–1697. [Google Scholar] [CrossRef]
- Vázquez-Baeza, Y.; Gonzalez, A.; Smarr, L.; McDonald, D.; Morton, J.T.; Navas-Molina, J.A.; Knight, R. Bringing the dynamic microbiome to life with animations. Cell Host Microbe 2017, 21, 7–10. [Google Scholar] [CrossRef]
- Eren, A.M.; Borisy, G.G.; Huse, S.M.; Mark Welch, J.L. Oligotyping analysis of the human oral microbiome. Proc. Natl. Acad. Sci. USA 2014, 111, E2875–E2884. [Google Scholar] [CrossRef]
- Lim, Y.; Fukuma, N.; Totsika, M.; Kenny, L.; Morrison, M.; Punyadeera, C. The performance of an oral microbiome biomarker panel in predicting oral cavity and oropharyngeal cancers. Front. Cell. Infect. Microbiol. 2018, 8, 267. [Google Scholar] [CrossRef]
- Woo, J.S.; Lu, D.Y. Procurement, transportation, and storage of saliva, buccal swab, and oral wash specimens. In Methods in Molecular Biology; Yong, W., Ed.; Humana Press: New York, NY, USA, 2019; Volume 1897. [Google Scholar]
- XIT Genomic DNA from Buccal Cells for Extraction of Genomic DNA from Buccal/Cheek Cells. G-Biosciences. Available online: https://www.gbiosciences.com/image/pdfs/protocol/786-341_protocol.pdf (accessed on 18 February 2020).
- Pramanik, R.; Thompson, H.; Kistler, J.O.; Wade, W.G.; Galloway, J.; Peakman, T.; Proctor, G.B. Effects of the UK Biobank collection protocol on potential biomarkers in saliva. Int. J. Epidemiol. 2012, 41, 1786–1797. [Google Scholar] [CrossRef]
- Lassalle, F.; Spagnoletti, M.; Fumagalli, M.; Shaw, L.; Dyble, M.; Walker, C.; Thomas, M.G.; Bamberg Migliano, A.; Balloux, F. Oral microbiomes from hunter-gatherers and traditional farmers reveal shifts in commensal balance and pathogen load linked to diet. Mol. Ecol. 2018, 27, 182–195. [Google Scholar] [CrossRef]
- Jeffery, I.; Claesson, M.; O’Toole, P.; Shanahan, F. Categorization of the gut microbiota: Enterotypes or gradients? Nat. Rev. Microbiol. 2012, 10, 591–592. [Google Scholar] [CrossRef] [PubMed]
- Knights, D.; Ward, T.L.; McKinlay, C.E.; Miller, H.; Gonzalez, A.; McDonald, D.; Knight, R. Rethinking “enterotypes”. Cell Host Microbe 2014, 16, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Costea, P.I.; Hildebrand, F.; Arumugam, M.; Miller, H.; Gonzalez, A.; McDonald, D.; Knight, R. Enterotypes in the landscape of gut microbial community composition. Nat. Microbiol. 2018, 3, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.L.; Bor, B.; Agnello, M.; Shi, W.; He, X. Ecology of the oral microbiome: Beyond bacteria. Trends Microbiol. 2017, 25, 362–374. [Google Scholar] [CrossRef]
- Peters, B.A.; Wu, J.; Hayes, R.B.; Ahn, J. The oral fungal mycobiome: Characteristics and relation to periodontitis in a pilot study. BMC Microbiol. 2017, 17, 157. [Google Scholar] [CrossRef]
- Bandara, H.M.H.N.; Panduwawala, C.P.; Samaranayake, L.P. Biodiversity of the human oral mycobiome in health and disease. Oral Dis. 2019, 25, 363–371. [Google Scholar] [CrossRef]
- Dupuy, A.K.; David, M.S.; Li, L.; Heider, T.N.; Peterson, J.D.; Montano, E.A.; Dongari-Bagtzoglou, A.; Diaz, P.I.; Strausbaugh, L.D. Redefining the human oral mycobiome with improved practices in amplicon-based taxonomy: Discovery of Malassezia as a prominent commensal. PLoS ONE 2014, 9, e90899. [Google Scholar] [CrossRef]
- Saunders, C.W.; Scheynius, A.; Heitman, J. Malassezia fungi are specialized to live on skin and associated with dandruff, eczema, and other skin diseases. PLoS Pathog. 2012, 8, e1002701. [Google Scholar] [CrossRef]
- Wu, G.; Zhao, H.; Li, C.; Rajapakse, M.P.; Wong, W.C.; Xu, J.; Saunders, C.W.; Reeder, N.L.; Reilman, R.A.; Scheynius, A.; et al. Genus-wide comparative genomics of malassezia delineates its phylogeny, physiology, and niche adaptation on human skin. PLoS Genet. 2015, 11, e1005614. [Google Scholar] [CrossRef]
- Hibbett, D.; Taylor, J. Fungal systematics: Is a new age of enlightenment at hand? Nat. Rev. Microbiol. 2013, 11, 129–133. [Google Scholar] [CrossRef]
- Donovan, P.D.; Gonzalez, G.; Higgins, D.G.; Butler, G.; Ito, K. Identification of fungi in shotgun metagenomics datasets. PLoS ONE 2018, 13, e0192898. [Google Scholar] [CrossRef] [PubMed]
- de la Cruz Peña, M.J.; Martinez-Hernandez, F.; Garcia-Heredia, I.; Lluesma Gomez, M.; Fornas, Ò.; Martinez-Garcia, M. Deciphering the human virome with single-virus genomics and metagenomics. Viruses 2018, 10, 113. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Brocal, V.; Moya, A. The analysis of the oral DNA virome reveals which viruses are widespread and rare among healthy young adults in Valencia (Spain). PLoS ONE 2018, 13, e0191867. [Google Scholar] [CrossRef] [PubMed]
- Willner, D.; Furlan, M.; Haynes, M.; Schmieder, R.; Angly, F.E.; Silva, J.; Tammadoni, S.; Nosrat, B.; Conrad, D.; Rohwer, F. Metagenomic analysis of respiratory tract DNA viral communities in cystic fibrosis and non-cystic fibrosis individuals. PLoS ONE 2009, 4, e7370. [Google Scholar] [CrossRef] [PubMed]
- Feki, A.; Molet, B.; Haag, R.; Kremer, M. [Protozoa of the human oral cavity (epidemiological correlations and pathogenic possibilities]. J. Biol. Buccale 1981, 9, 155–161. [Google Scholar] [PubMed]
- Chomicz, L.; Piekarczyk, J.; Starościak, B.; Fiedor, P.; Piekarczyk, B.; Szubińska, D.; Zawadzki, P.J.; Walski, M. Comparative studies on the occurrence of protozoans, bacteria and fungi in the oral cavity of patients with systemic disorders. Acta Parasitol. 2002, 47, 147–153. [Google Scholar]
- Cielecka, D.; Chomicz, L.; Piekarczyk, J.; Walski, M.; Zawadzki, P.J.; Bednarczyk, A.; Szubińska, D. Oral cavity condition and the occurrence of parasitic protozoans in patients with genetic diseases. Acta Parasitol. 2000, 45, 107–112. [Google Scholar]
- Wade, W.G. The oral microbiome in health and disease. Pharmacol. Res. 2013, 69, 137–143. [Google Scholar] [CrossRef]
- Horz, H.P. Archaeal lineages within the human microbiome: Absent, rare or elusive? Life 2015, 5, 1333–1345. [Google Scholar] [CrossRef]
- Whittaker, R.H. Communities and Ecosystems; MacMillan Publishing Company, Inc.: New York, NY, USA, 1975. [Google Scholar]
- Socransky, S.; Haffajee, A.; Cugini, M.; Smith, C.; Kent, R.L., Jr. Microbial complexes in subgingival plaque. J. Clin. Periodontol. 1998, 25, 134–144. [Google Scholar] [CrossRef]
- Kurkivuori, J.; Salaspuro, V.; Kaihovaara, P.; Kari, K.; Rautemaa, R.; Grönroos, L.; Meurman, J.H.; Salaspuro, M. Acetaldehyde production from ethanol by oral streptococci. Oral Oncol. 2007, 43, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Meurman, J.H. Oral microbiota and cancer. J. Oral Microbiol. 2010, 2. [Google Scholar] [CrossRef] [PubMed]
- Lax, A. Bacterial toxins and cancer—A case to answer? Nat. Rev. Microbiol. 2005, 3, 343–349. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.N.; Araújo-Pérez, F.; Azcárate-Peril, A.; Yeh, J.J.; Sandler, R.S.; Keku, T.O. Fusobacterium is associated with colorectal adenomas. PLoS ONE 2013, 8, e53653. [Google Scholar] [CrossRef]
- Castellarin, M.; Warren, R.L.; Freeman, J.D.; Dreolini, L.; Krzywinski, M.; Strauss, J.; Barnes, R.; Watson, P.; Allen-Vercoe, E.; Moore, R.A.; et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012, 22, 299–306. [Google Scholar] [CrossRef]
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M.; Ojesina, A.I.; Jung, J.; Bass, A.J.; Tabernero, J.; et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012, 22, 292–298. [Google Scholar] [CrossRef]
- Fitzpatrick, S.G.; Katz, J. The association between periodontal disease and cancer: A review of the literature. J. Dent. 2010, 38, 83–95. [Google Scholar] [CrossRef]
- Michaud, D.S.; Fu, Z.; Shi, J.; Chung, M. Periodontal disease, tooth loss, and cancer risk. Epidemiol. Rev. 2017, 39, 49–58. [Google Scholar] [CrossRef]
- Abnet, C.C.; Qiao, Y.L.; Dawsey, S.M.; Dong, Z.W.; Taylor, P.R.; Mark, S.D. Tooth loss is associated with increased risk of total death and death from upper gastrointestinal cancer, heart disease, and stroke in a Chinese population-based cohort. Int. J. Epidemiol. 2005, 34, 467–474. [Google Scholar] [CrossRef]
- Mirvish, S.S. Role of N-nitroso compounds (NOC) and N-nitrosation in etiology of gastric, esophageal, nasopharyngeal and bladder cancer and contribution to cancer of known exposures to NOC. Cancer Lett. 1995, 93, 17–48. [Google Scholar] [CrossRef]
- Chalabi, M.; Moghim, S.; Mogharehabed, A.; Najafi, F.; Rezaie, F. EBV and CMV in chronic periodontitis: A prevalence study. Arch. Virol. 2008, 153, 1917. [Google Scholar] [CrossRef] [PubMed]
- Slots, J.; Sugar, C.; Kamma, J.J. Cytomegalovirus periodontal presence is associated with subgingival Dialister pneumosintes and alveolar bone loss. Oral Microbiol. Immunol. 2002, 17, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Tateno, T.; Ueno, S.; Hiwatashi, K.; Matsumoto, M.; Okumura, H.; Setoyama, T.; Uchikado, Y.; Sakoda, M.; Kubo, F.; Ishigami, S.; et al. Expression of receptor for advanced glycation end products (RAGE) is related to prognosis in patients with esophageal squamous cell carcinoma. Ann. Surg. Oncol. 2009, 16, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.W.; Shi, W.; Huang, G.T.; Kinder Haake, S.; Park, N.H.; Kuramitsu, H.; Genco, R.J. Interactions between periodontal bacteria and human oral epithelial cells: Fusobacterium nucleatum adheres to and invades epithelial cells. Infect. Immun. 2000, 68, 3140–3146. [Google Scholar] [CrossRef] [PubMed]
- Bizzarro, S.; Loos, B.G.; Laine, M.L.; Crielaard, W.; Zaura, E. Subgingival microbiome in smokers and non-smokers in periodontitis: An exploratory study using traditional targeted techniques and a next-generation sequencing. J. Clin. Periodontol. 2013, 40, 483–492. [Google Scholar] [CrossRef]
- Moon, J.H.; Lee, J.H.; Lee, J.Y. Subgingival microbiome in smokers and non-smokers in Korean chronic periodontitis patients. Mol. Oral Microbiol. 2015, 30, 227–241. [Google Scholar] [CrossRef]
- Dassi, E.; Ferretti, P.; Covello, G.; Speccher, A.; Migazzi, A.; Bosco, B.; Rajashekar, B.; Zarbo, C.; Ballabio, C.; Rossetto, D.; et al. The short-term impact of probiotic consumption on the oral cavity microbiome. Sci. Rep. 2018, 8, 10476. [Google Scholar] [CrossRef]
- Saxelin, M. Probiotic formulations and applications, the current probiotics market, and changes in the marketplace: A european perspective. Clin. Infect. Dis. 2008, 46, S76–S79. [Google Scholar] [CrossRef]
- Michaud, D.S.; Izard, J.; Wilhelm-Benartzi, C.S.; You, D.H.; Grote, V.A.; Tjønneland, A.; Dahm, C.C.; Overvad, K.; Jenab, M.; Fedirko, V.; et al. Plasma antibodies to oral bacteria and risk of pancreatic cancer in a large European prospective cohort study. Gut 2013, 62, 1764–1770. [Google Scholar] [CrossRef]
- Zambirinis, C.P.; Levie, E.; Nguy, S.; Avanzi, A.; Barilla, R.; Xu, Y.; Seifert, L.; Daley, D.; Greco, S.H.; Deutsch, M.; et al. TLR9 ligation in pancreatic stellate cells promotes tumorigenesis. J. Exp. Med. 2015, 212, 2077–2094. [Google Scholar] [CrossRef]
- Söder, B.; Jin, L.J.; Klinge, B.; Söder, P.Ö. Periodontitis and premature death: A 16-year longitudinal study in a Swedish urban population. J. Periodontal Res. 2007, 42, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Ali, J.; Pramod, K.; Tahir, M.A.; Ansari, S.H. Autoimmune responses in periodontal diseases. Autoimmun. Rev. 2011, 10, 426–431. [Google Scholar] [CrossRef]
- Rangé, H.; Labreuche, J.; Louedec, L.; Rondeau, P.; Planesse, C.; Sebbag, U.; Bourdon, E.; Michel, J.B.; Bouchard, P.; Meilhac, O. Periodontal bacteria in human carotid atherothrombosis as a potential trigger for neutrophil activation. Atherosclerosis 2014, 236, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, A.; Miura, M.; Fujise, O.; Hamachi, T.; Nishimura, F. Porphyromonas gingivalis gingipain is involved in the detachment and aggregation of Aggregatibacter actinomycetemcomitans biofilm. Mol. Oral Microbiol. 2014, 29, 131–143. [Google Scholar] [CrossRef]
- Jayaprakash, K.; Khalaf, H.; Bengtsson, T. Gingipains from Porphyromonas gingivalis play a significant role in induction and regulation of CXCL8 in THP-1 cells. BMC Microbiol. 2014, 14, 193. [Google Scholar] [CrossRef] [PubMed]
- Dissick, A.; Redman, R.S.; Jones, M.; Rangan, B.V.; Reimold, A.; Griffiths, G.R.; Mikuls, T.R.; Amdur, R.L.; Richards, J.S.; Kerr, G.S. Association of periodontitis with rheumatoid arthritis: A pilot study. J. Periodontol. 2010, 81, 223–230. [Google Scholar] [CrossRef]
- Konig, M.F.; Paracha, A.S.; Moni, M.; Bingham, C.O., III; Andrade, F. Defining the role of Porphyromonas gingivalis peptidylarginine deiminase (PPAD) in rheumatoid arthritis through the study of PPAD biology. Ann. Rheum. Dis. 2015, 74, 2054–2061. [Google Scholar] [CrossRef] [PubMed]
- Mealey, B.L.; Oates, T.W. Diabetes mellitus and periodontal diseases. J. Periodontol. 2006, 77, 1289–1303. [Google Scholar] [CrossRef]
- Taylor, G.W. Bidirectional Interrelationships Between Diabetes and Periodontal Diseases: An Epidemiologic Perspective. Ann. Periodontol. 2001, 6, 99–112. [Google Scholar] [CrossRef]
- Scannapieco, F.A.; Dasanayake, A.P.; Chhun, N. Does periodontal therapy reduce the risk for systemic diseases? Dent. Clin. N. Am. 2010, 54, 163–181. [Google Scholar] [CrossRef]
- Devine, D.A.; Marsh, P.D.; Meade, J. Modulation of host responses by oral commensal bacteria. J. Oral Microbiol. 2015, 7, 26941. [Google Scholar] [CrossRef] [PubMed]
- López-López, A.; Camelo-Castillo, A.; Ferrer, M.D.; Simon-Soro, Á.; Mira, A. Health-associated niche inhabitants as oral probiotics: The case of Streptococcus dentisani. Front. Microbiol. 2017, 8, 379. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Palmer, S.R.; Ahn, S.J.; Richards, V.P.; Williams, M.L.; Nascimento, M.M.; Burne, R.A. A highly arginolytic Streptococcus species that potently antagonizes Streptococcus mutans. Appl. Environ. Microbiol. 2016, 82, 2187–2201. [Google Scholar] [CrossRef] [PubMed]
- Loozen, G.; Boon, N.; Pauwels, M.; Slomka, V.; Rodrigues Herrero, E.; Quirynen, M.; Teughels, W. Effect of Bdellovibrio bacteriovorus HD100 on multispecies oral communities. Anaerobe 2015, 35, 45–53. [Google Scholar] [CrossRef]
- Xiao, E.; Mattos, M.; Vieira, G.H.A.; Chen, S.; Corrêa, J.D.; Wu, Y.; Albiero, M.L.; Bittinger, K.; Graves, D.T. Diabetes enhances IL-17 expression and alters the oral microbiome to increase its pathogenicity. Cell Host Microbe 2017, 22, 120–128. [Google Scholar] [CrossRef]
- Knapp, J.S. Historical perspectives and identification of Neisseria and related species. Clin. Microbiol. Rev. 1988, 1, 415–431. [Google Scholar] [CrossRef]




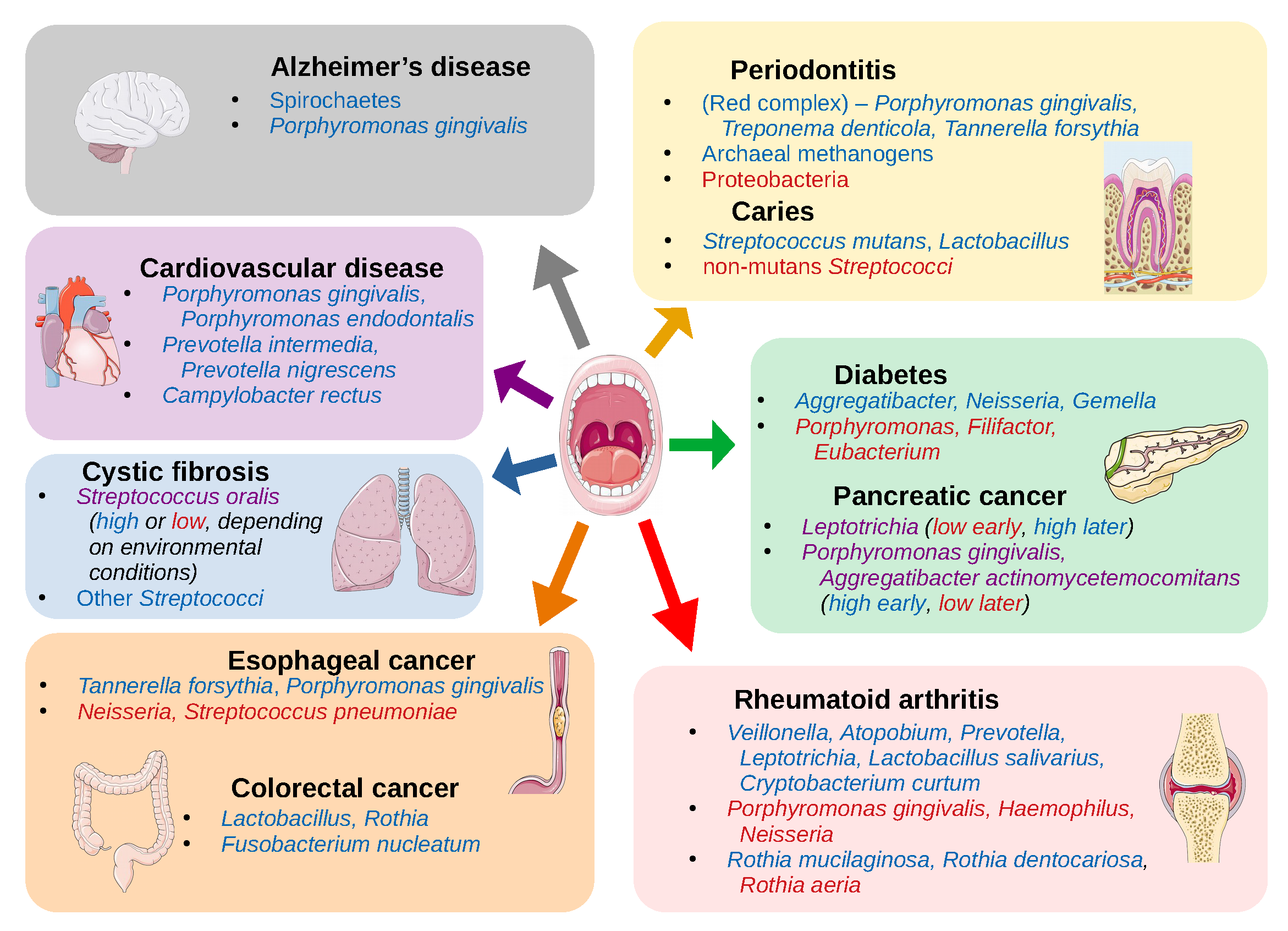{kind=link}
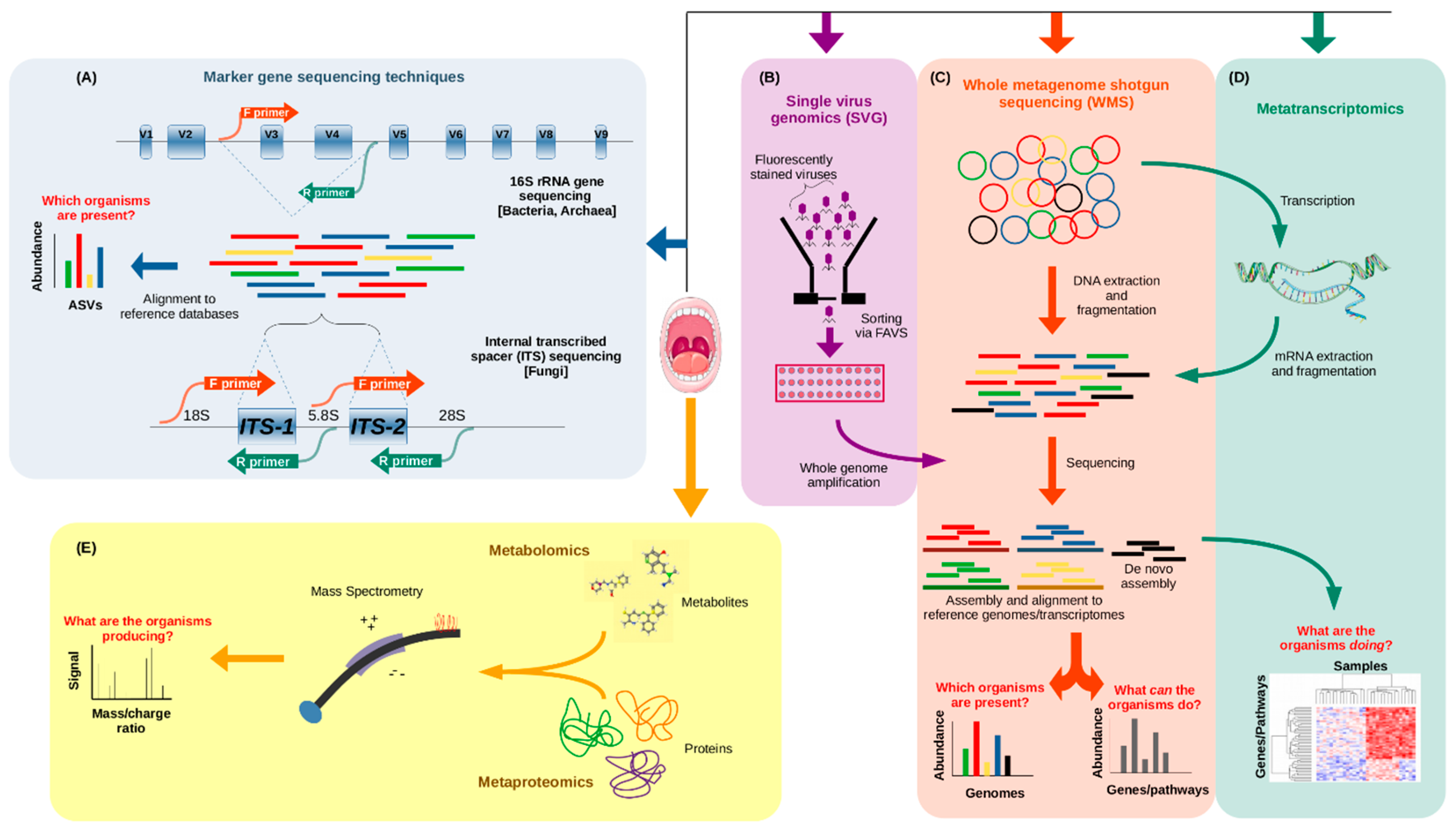{kind=link}
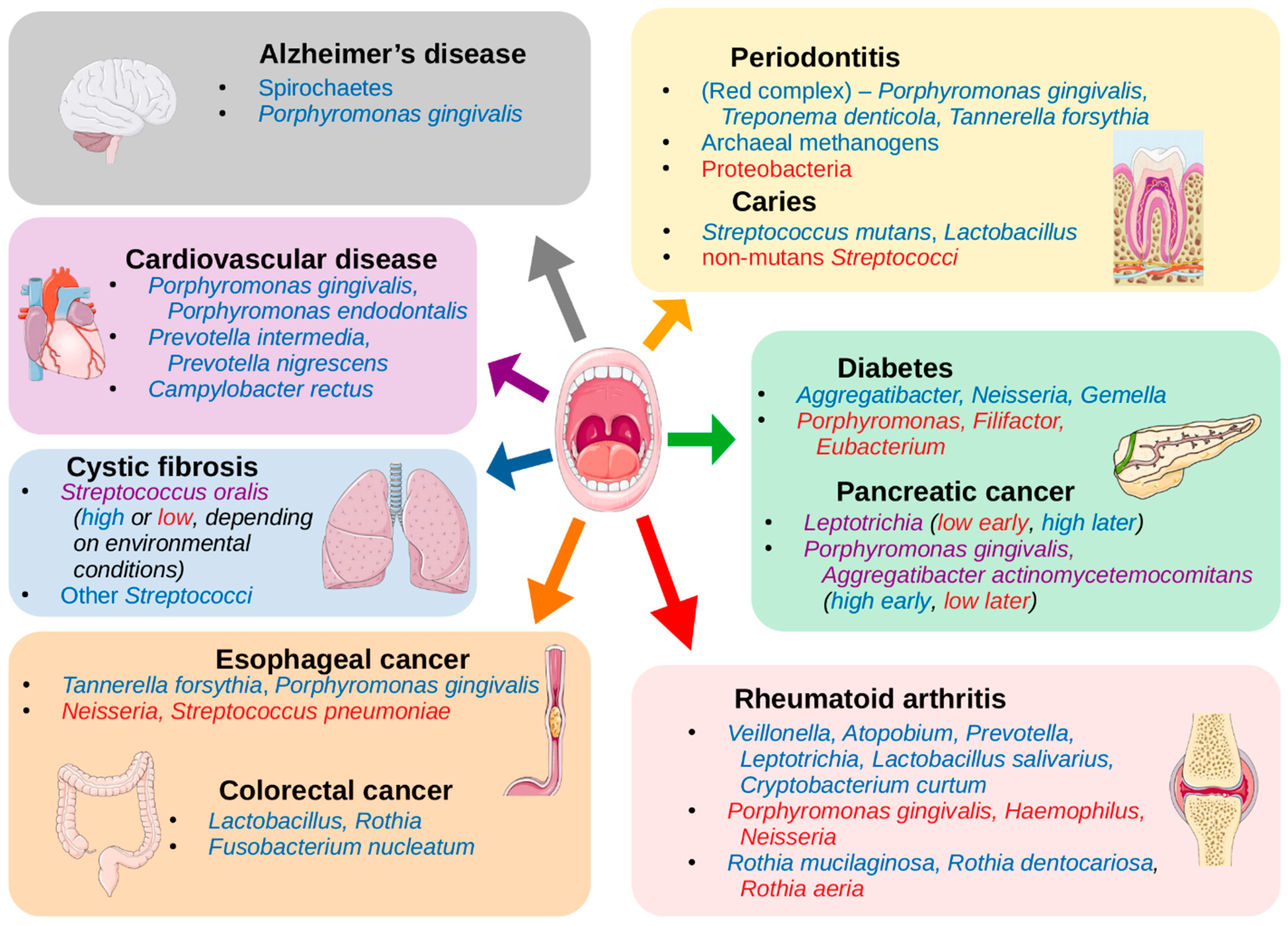{kind=link}
{kind=link}
| Disease | Associated Organisms | Inhibited Organisms | Reference |
|---|---|---|---|
| Periodontitis | Phyla: Spirochaetes, Synergistetes and Bacteroidetes Classes: Clostridia, Negativicutes and Erysipelotrichia Genera: Prevotella, Fusobacterium Species: Porphyromonas gingivalis, Treponema denticola, Tannerella forsythia, Filifactor alocis, Parvimonas micra, Aggregatibacter actinomycetemcomitans Archaea: Methanobrevibacter oralis, Methanobacterium curvum/congolense, and Methanosarcina mazeii | Phyla: Proteobacteria Classes: Bacilli Genera: Streptococcus, Actinomyces, Granulicatella | [14,15,16,17,18,19,20,21] |
| Dental caries | Genera: Neisseria, Selenomonas, Propionibacterium Species: Streptococcus mutans, Lactobacillus spp. Fungi: Candida albicans | Species: non-mutans Streptococci, Corynebacterium matruchotii, Capnocytophaga gingivalis, Eubacterium IR009, Campylobacter rectus, Lachnospiraceae sp. C1 | [22,23] |
| Oral cancer | Species: Capnocytophaga gingivalis, Prevotella melaninogenica and Streptococcus mitis, Peptostreptococcus stomatis*, Streptococcus salivarius*, Streptococcus gordonii*, Gemella haemolysans*, Gemella morbillorum*, Johnsonella ignava* and Streptococcus parasanguinis I* | Species: Granulicatella adiacens* | [24,25,26] |
| Esophageal cancer | Species: Tannerella forsythia, Porphyromonas gingivalis | Genera: Neisseria Species: Streptococcus pneumoniae | [27] |
| Disease | Associated Organisms | Inhibited Organisms | Reference |
|---|---|---|---|
| Colorectal cancer | Genera: Lactobacillus, Rothia Species: Fusobacterium nucleatum | [12,28,29,30] | |
| Pancreatic cancer | Genera: Leptotrichia (later in progression of disease) Species: Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans (at onset of disease) | Genera: Leptotrichia (at onset of disease) Species: Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans (later in progression of disease) | [31,32] |
| Cystic fibrosis | Species: Streptococcus oralis (depends on environmental conditions), S. mitis, S. gordonii and S. sanguinis | Species: Streptococcus oralis (depends on environmental conditions) | [33] |
| Cardiovascular disease | Species: Campylobacter rectus, Porphyromonas gingivalis, Porphyromonas endodontalis, Prevotella intermedia, Prevotella nigrescens, (oral commensals that were found on athersclerotic plaques - not necessarily at high abundance in oral cavity) | [34,35] | |
| Rheumatoid arthritis | Genera: Veillonella, Atopobium, Prevotella, Leptotrichia Species: Rothia mucilaginosa, Rothia dentocariosa, Lactobacillus salivarius, Cryptobacterium curtum | Genera: Haemophilus, Neisseria Species: Porphyromonas gingivalis, Rothia aeria | [36,37,38,39] |
| Alzheimer’s disease | Phyla: Spirochaetes Species: Porphyromonas gingivalis | [40,41,42] | |
| Diabetes | Genera: Aggregatibacter, Neisseria, Gemella, Eikenella, Selenomonas, Actinomyces, Capnocytophaga, Fusobacterium, Veillonella, Streptococcus | Genera: Porphyromonas, Filifactor, Eubacterium, Synergistetes, Tannerella, Treponema | [43] |
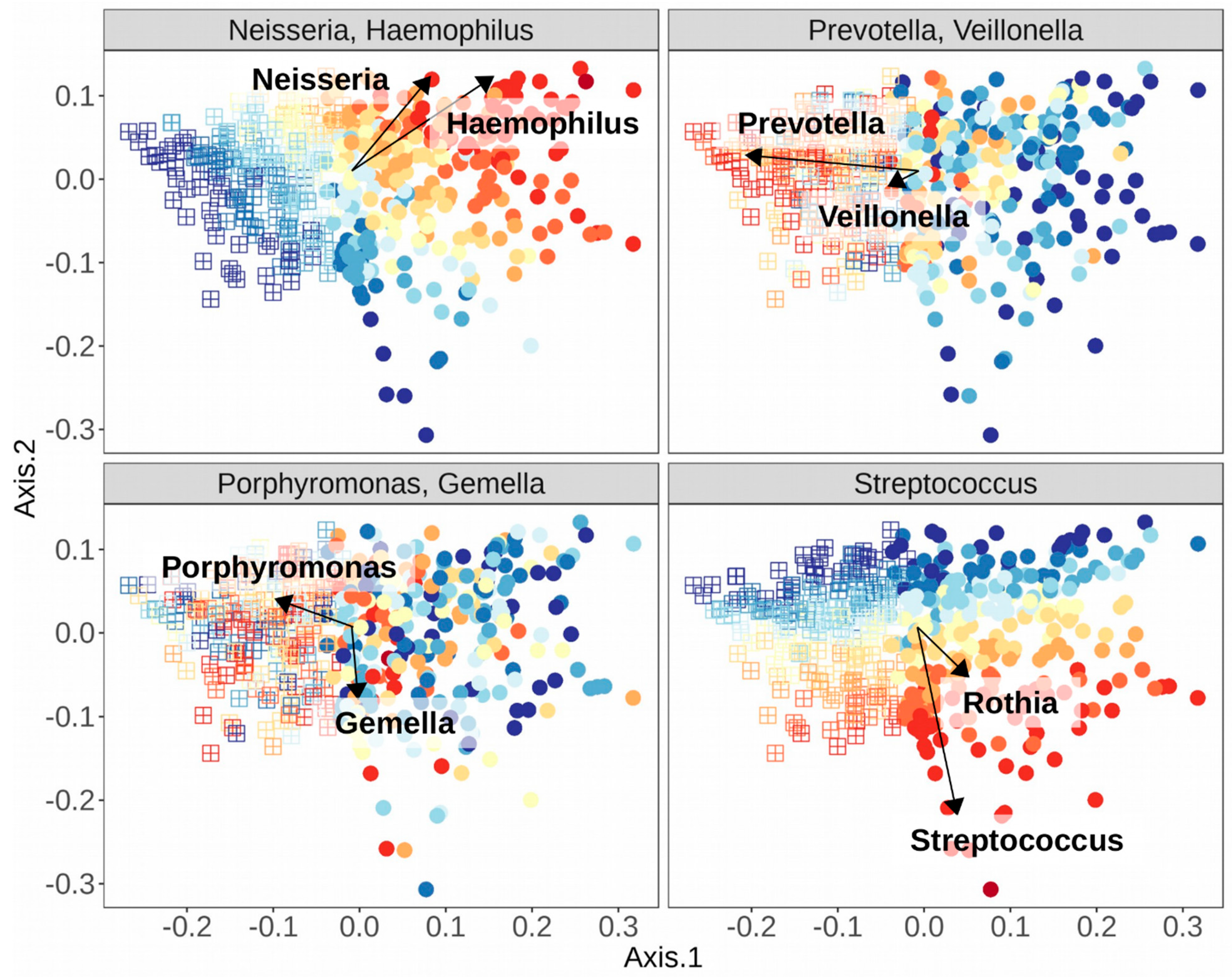
| Genus | References | |
|---|---|---|
| Stomatotype 1 | ||
| Neisseria | [10,48,49,51] | |
| Haemophilus | [10,48,51] | |
| Stomatotype 2 | ||
| Prevotella | [10,48,49,51] | |
| Veillonella | [10,48,51] | |
| Variable Stomatotypes | ||
| Streptococcus—varies depending on study and species | [10,48,49,51] | |
| Gemella—co-occurs with Streptococcus and Porphyromonas | [49,51] | |
| Porphyromonas—may co-occur with Streptococcus, Gemella, or Neisseria | [48,51] | |
| Rothia—co-occurs with varying species of Streptococcus, depending on study | [49,51] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Willis, J.R.; Gabaldón, T. The Human Oral Microbiome in Health and Disease: From Sequences to Ecosystems. Microorganisms 2020, 8, 308. https://doi.org/10.3390/microorganisms8020308
Willis JR, Gabaldón T. The Human Oral Microbiome in Health and Disease: From Sequences to Ecosystems. Microorganisms. 2020; 8(2):308. https://doi.org/10.3390/microorganisms8020308
Chicago/Turabian StyleWillis, Jesse R., and Toni Gabaldón. 2020. "The Human Oral Microbiome in Health and Disease: From Sequences to Ecosystems" Microorganisms 8, no. 2: 308. https://doi.org/10.3390/microorganisms8020308
APA StyleWillis, J. R., & Gabaldón, T. (2020). The Human Oral Microbiome in Health and Disease: From Sequences to Ecosystems. Microorganisms, 8(2), 308. https://doi.org/10.3390/microorganisms8020308