Aquatic Thermal Reservoirs of Microbial Life in a Remote and Extreme High Andean Hydrothermal System

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Description of the Sampling Sites
2.2. Sample Collection and Physical–Chemical Analysis
2.3. Enrichment Cultures and Scanning Electron Microscopy (SEM)
2.4. Clonal Analysis of Archaeal and Bacterial 16S rRNA Gene
2.5. Bacterial 16S rRNA Gene Metabarcoding Analysis
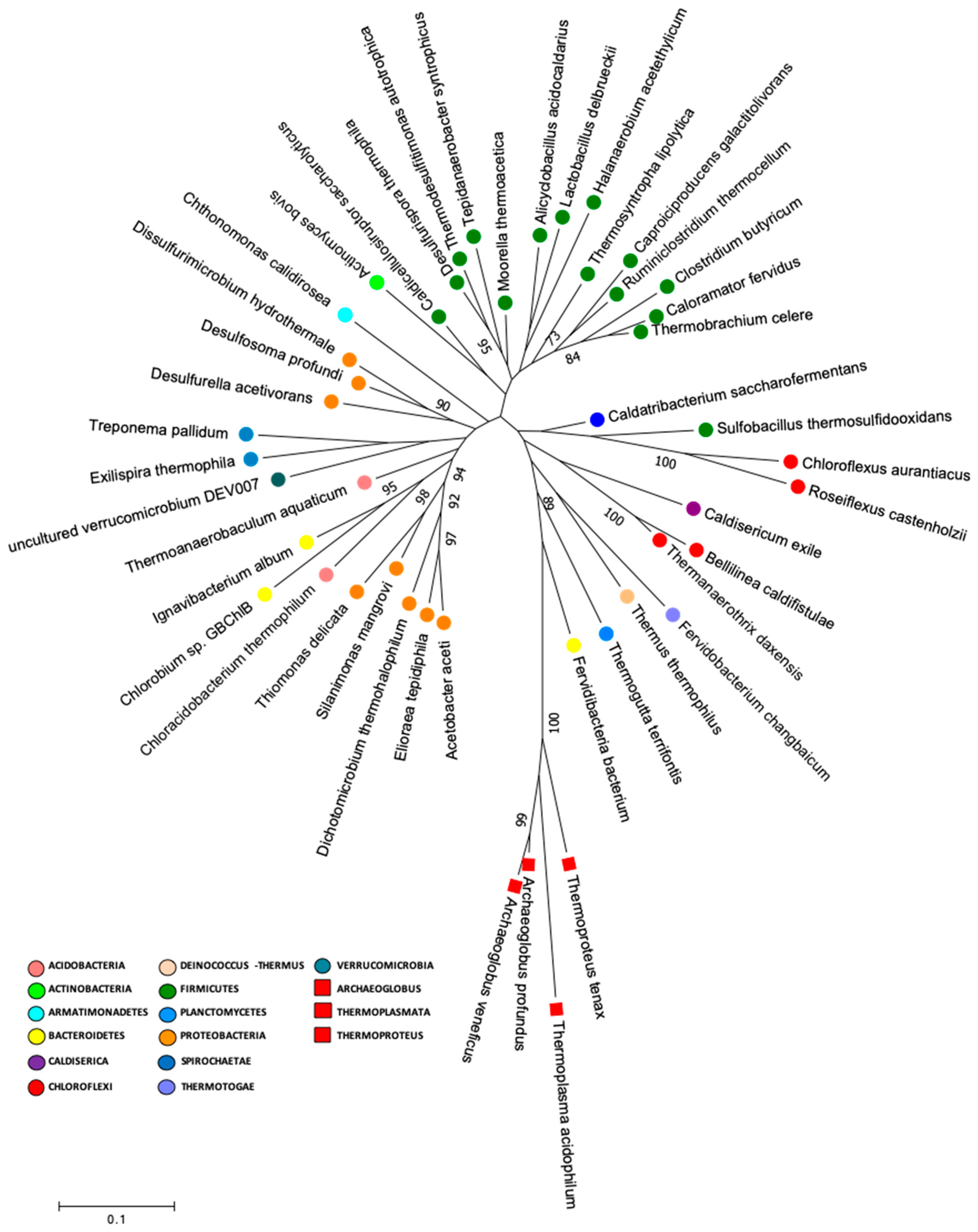
2.6. Early Life Genera and Phylogenetic Analysis
3. Results and Discussion
3.1. Adaptation to Environmental Factors
3.2. Lirima Hot Spring: A Doorway for Early Life
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Woese, C.R.; Kandler, O.; Wheelis, M.L. Towards a natural system of organisms: Proposal for the domains Archaea, Bacteria, and Eucarya. Proc. Natl. Acad. Sci. USA 1990, 87, 4576–4579. [Google Scholar] [CrossRef] [PubMed]
- Farmer, J. Thermophiles, early biosphere evolution, and the origin of life on Earth: Implications for the exobiological exploration of Mars. J. Geophys. Res. 1998, 103, 457–461. [Google Scholar] [CrossRef]
- Stetter, K.O. History of discovery of the first hyperthermophiles. Extremophiles 2006, 10, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Reysenbach, A.L.; Longnecker, K.; Kirshtein, J. Novel bacterial and archaeal lineages from an in situ growth chamber deployed at a Mid-Atlantic Ridge hydrothermal vent. Appl. Environ. Microbiol. 2000, 66, 3798–3806. [Google Scholar] [CrossRef] [PubMed]
- Lane, N.; Allen, J.F.; Martin, W. How did LUCA make a living? Chemiosmosis in the origin of life. BioEssays 2010, 32, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Berg, I.A.; Kockelkorn, D.; Ramos-Vera, W.H.; Say, R.F.; Zarzycki, J.; Hügler, M.; Alber, B.E.; Fuchs, G. Autotrophic carbon fixation in archaea. Nat. Rev. Microbiol. 2010, 8, 447–460. [Google Scholar] [CrossRef]
- Weiss, M.C.; Sousa, F.L.; Mrnjavac, N.; Neukirchen, S.; Roettger, M.; Nelson-Sathi, S.; Martin, W.F. The physiology and habitat of the last universal common ancestor. Nat. Microbiol. 2016, 1, 16116. [Google Scholar] [CrossRef]
- Xiao, X.; Zhang, Y. Life in extreme environments: Approaches to study life-environment co-evolutionary strategies. Sci. China Earth Sci. 2014, 57, 869–877. [Google Scholar] [CrossRef]
- Rasuk, M.C.; Fernández, A.B.; Kurth, D.; Contreras, M.; Novoa, F.; Poiré, D.; Farías, M.E. Bacterial Diversity in Microbial Mats and Sediments from the Atacama Desert. Microb. Ecol. 2016, 71, 44–56. [Google Scholar] [CrossRef]
- Cowan, D.A.; Ramond, J.-B.; Makhalanyane, T.P.; De Maayer, P. Metagenomics of extreme environments. Curr. Opin. Microbiol. 2015, 25, 97–102. [Google Scholar] [CrossRef]
- Wagner, I.D.; Wiegel, J. Diversity of thermophilic anaerobes. Ann. N. Y. Acad. Sci. 2008, 1125, 1–43. [Google Scholar] [CrossRef] [PubMed]
- Menzel, P.; Gudbergsdóttir, S.R.; Rike, A.G.; Lin, L.; Zhang, Q.; Contursi, P.; Moracci, M.; Kristjansson, J.K.; Bolduc, B.; Gavrilov, S.; et al. Comparative metagenomics of eight geographically remote terrestrial hot springs. Microb. Ecol. 2015, 70, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Garreaud, R.; Vuille, M.; Clement, M.C. The climate of the Altiplano: Observed current conditions and mechanisms of past changes. Palaeogeogr. Palaeoclimatol. Palaeoecol. 2003, 194, 5–22. [Google Scholar] [CrossRef]
- Tassi, F.; Aguilera, F.; Darrah, T.; Vaselli, O.; Capaccioni, B.; Poreda, R.J.; Huertas, A.D. Fluid geochemistry of hydrothermal systems in the Arica-Parinacota, Tarapacá and Antofagasta Regions (Northern Chile). J. Volcanol. Geotherm. Res. 2010, 192, 1–15. [Google Scholar] [CrossRef]
- Procesi, M. Geothermal Potential Evaluation for Northern Chile and Suggestions for New Energy Plans. Energies 2014, 7, 5444–5459. [Google Scholar] [CrossRef]
- Sanchez-Alfaro, P.; Sielfeld, G.; Van Campen, B.; Dobson, P.; Fuentes, V.; Reed, A.; Palma-Behnke, R.; Morata, D. Geothermal barriers, policies and economics in Chile—Lessons for the Andes. Renew. Sustain. Energy Rev. 2015, 51, 1390–1401. [Google Scholar] [CrossRef]
- Filippidou, S.; Junier, T.; Wunderlin, T.; Kooli, W.M.; Palmieri, I.; Al-Dourobi, A.; Molina, V.; Lienhard, R.; Spangenberg, J.E.; Johnson, S.L.; et al. Adaptive strategies in a poly-extreme environment: Differentiation of vegetative cells in Serratia ureilytica and resistance to extreme conditions. Front. Microbiol. 2019, 10, 102. [Google Scholar] [CrossRef]
- Hernández, K.L.; Yannicelli, B.; Olsen, L.M.; Dorador, C.; Menschel, E.J.; Molina, V.; Remonsellez, F.; Hengst, M.B.; Jeffrey, W.H. Microbial activity response to solar radiation across contrasting environmental conditions in salar de huasco, northern chilean altiplano. Front. Microbiol. 2016, 7, 1857. [Google Scholar] [CrossRef]
- Molina, V.; Hernández, K.; Dorador, C.; Eissler, Y.; Hengst, M.; Pérez, V.; Harrod, C. Bacterial active community cycling in response to solar radiation and their influence on nutrient changes in a high-altitude wetland. Front. Microbiol. 2016, 7, 1823. [Google Scholar] [CrossRef]
- Pérez, V.; Hengst, M.; Kurte, L.; Dorador, C.; Jeffrey, W.H.; Wattiez, R.; Molina, V.; Matallana-Surget, S. Bacterial Survival under Extreme UV Radiation: A Comparative Proteomics Study of Rhodobacter sp., Isolated from High Altitude Wetlands in Chile. Front. Microbiol. 2017, 8, 1173. [Google Scholar] [CrossRef]
- Scott, S.; Dorador, C.; Oyanedel, J.P.; Tobar, I.; Hengst, M.; Maya, G.; Harrod, C.; Vila, I. Microbial diversity and trophic components of two high altitude wetlands of the Chilean Altiplano. Gayana 2015, 79, 45–56. [Google Scholar] [CrossRef]
- McAullife, C. GC determination of solutes by multiple phase equilibration. Chem. Technol. 1971, 1, 46–51. [Google Scholar]
- Jurgens, G.; Glockner, F.O.; Amann, R.; Saano, A.; Montonen, L.; Likolammi, M.; Munster, U. Identification of novel Archaea in bacterioplankton of a boreal forest lake by phylogenetic analysis and fluorescent in situ hybridization. FEMS Microbiol. Ecol. 2000, 34, 45–56. [Google Scholar] [CrossRef]
- Stackebrandt, E.; Liesack, W. Nucleic acids and classification. In Handbook of New Bacterial Systematics; Goodfellow, M., O’Donnell, A.G., Eds.; Academic Press: London, UK, 1993; pp. 152–189. [Google Scholar]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning—A Laboratory Manual; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1989. [Google Scholar]
- Wright, E.S.; Yilmaz, L.S.; Noguera, D.R. DECIPHER, a search-based approach to chimera identification for 16S rRNA sequences. Appl. Environ. Microbiol. 2012, 78, 717–725. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucl. Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-High-Throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina paired-end read merger. Bioinformatics 2014, 30, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Schloss, P.; Westcott, S. Assessing and Improving Methods Used in Operational Taxonomic Unit-Based Approaches for 16S rRNA Gene Sequence Analysis. Appl. Environ. Microbiol. 2011, 77, 3219–3226. [Google Scholar] [CrossRef]
- Oksanen, F.; Blanchet, G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. R package Version 2.5-4; R Development Core Team: Vienna, Austria, 2019. [Google Scholar]
- Good, I. The population frequencies of species and the estimation of population parameters. Biometrika 1953, 40, 237–264. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M. Estimation of the number of nucleo-tide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef]
- Kumar, S.; Tamura, K.; Nei, M. MEGA3: An integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief. Bioinform. 1954, 5, 150–163. [Google Scholar] [CrossRef] [PubMed]
- Arcos, R.; Clavero, J.; Giavelli, A.; Simmons, S.; Aguirre, I.; Martini, S.; Mayorga, C.; Pineda, G.; Parra, J.; Soffia, J. Surface exploration at Pampa Lirima geothermal project, central Andes of northern Chile. Geotherm. Resour. Counc. Trans. 2011, 35, 689–693. [Google Scholar]
- Mardanov, A.V.; Gumerov, V.M.; Beletsky, A.V.; Perevalova, A.N.; KArpov, G.A.; Bonch-Osmolovskaya, A.B.; Ravin, N.V. Uncultured archaea dominate in the thermal groundwater of Uzon Caldera, Kamchatka. Extremophiles 2011, 15, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Kambura, A.K.; Mwirichia, R.K.; Kasili, R.W.; Karanja, E.N.; Makonde, H.M.; Boga, H.I. Bacteria and Archaea diversity within the hot springs of Lake Magadi and Little Magadi in Kenya. BMC Microbiol. 2016, 16, 136. [Google Scholar] [CrossRef] [PubMed]
- Panda, A.K.; Bisht, S.S.; De Mandal, S.; Kumar, N.S. Bacterial and archeal community composition in hot springs from Indo-Burma region, North-east India. AMB Express 2016, 6, 111. [Google Scholar] [CrossRef] [PubMed]
- Hohn, M.J.; Hedlund, B.P.; Huber, H. Detection of 16S rDNA sequences representing the novel phylum “Nanoarchaeota”: Indication for a broad distribution in high temperature. Syst. Appl. Microbiol. 2002, 25, 551–554. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Casanueva, A.; Galada, N.; Baker, G.C.; Grant, W.D.; Heaphy, S.; Jones, B.; Yanhe, M.; Ventose, A.; Blamey, J.; Cowan, D.A. Nanoarchaeal 16S rRNA gene sequences are widely dispersed in hyperthermophilic and mesophilic halophilic environments. Extremophiles 2008, 12, 651–656. [Google Scholar] [CrossRef]
- Li, H.; Yang, Q.; Li, J.; Gao, H.; Li, P.; Zhou, H. The impact of temperature on microbial diversity and AOA activity in the Tengchong Geothermal Field, China. Sci. Rep. 2015, 5, 17056. [Google Scholar] [CrossRef]
- Salonen, A.; Nikkilä, J.; Jalanka-Tuovinen, J.; Immonen, O.; Rajilić-Stojanović, M.; Kekkonen, R.A.; Palva, A.; de Vos, W.M. Comparative analysis of fecal DNA extraction methods with phylogenetic microarray: Effective recovery of bacterial and archaeal DNA using mechanical cell lysis. J. Microbiol. Methods 2010, 81, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Bryant, D.A.; Frigaard, N.U. Prokaryotic photosynthesis and phototrophy illuminated. Trends Microbiol. 2006, 14, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Bryant, D.A.; Liu, Z.; Li, T.; Zhao, F.; Garcia, A.M.; Klatt, C.G.; Ward, D.M.; Frigaard, N.-U.; Overmann, J. Comparative and functional genomics of anoxygenic green Bacteria from the taxa Chlorobi, Chloroflexi, and Acidobacteria. In Functional Genomics and Evolution of Photosynthetic Systems; Burnap, R., Vermaas, W., Eds.; Advances in Photosynthesis and Respiration; Springer: Dordrecht, The Netherlands, 2012; Volume 33. [Google Scholar]
- Zeng, Y.; Feng, F.; Medová, H.; Dean, J.; Koblížek, M. Functional type 2 photosynthetic reaction centers found in the rare Bacterial phylum Gemmatimonadetes. Proc. Natl. Acad. Sci. USA 2014, 111, 7795–7800. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.S.; Chan, K.G.; Tay, Y.L.; Chua, Y.H.; Goh, K.M. Diversity of thermophiles in a Malaysian hot spring determined using 16S rRNA and shotgun metagenome sequencing. Front. Microbiol. 2015, 6, 177. [Google Scholar] [CrossRef] [PubMed]
- Boyd, E.S.; Fecteau, K.M.; Havig, J.R.; Shock, E.L.; Peters, J.W. Modeling the habitat range of phototrophs in Yellowstone NationalPark: Toward the development of a comprehensive fitness landscape. Front. Microbiol. 2012, 3, 221. [Google Scholar] [CrossRef] [PubMed]
- Frigaard, N.U.; Dahl, C. Sulfur metabolism in phototrophic sulfur bacteria. Adv. Microb. Physiol. 2008, 54, 103–200. [Google Scholar] [CrossRef]
- Elshahed, M.S.; Senko, J.M.; Najar, F.Z.; Kenton, S.M.; Roe, B.A.; Dewer, T.A.; Spear, J.R.; Krumholz, L.R. Bacterial diversity and sulfur cycling in a mesophilic sulfide-rich spring. Appl. Environ. Microbiol. 2003, 69, 5609–5621. [Google Scholar] [CrossRef]
- Wahlund, T.M.; Woese, C.R.; Castenholz, R.W.; Madigan, M.T. A thermophilic green sulfur bacterium from New Zealand hot springs, Chlorobium tepidum sp.nov. Arch. Microbiol. 1991, 156, 81–90. [Google Scholar] [CrossRef]
- Dahl, C. Sulfur metabolism in phototrophic bacteria. In Modern Topics in the Phototrophic Prokaryotes; Hallenbeck, P., Ed.; Springer: Cham, Switzerland, 2017; pp. 27–66. [Google Scholar] [CrossRef]
- Thiel, V.; Wood, J.M.; Olsen, M.T.; Tank, M.; Klatt, C.G.; Ward, D.M.; Bryant, D.A. The dark side of the Mushroom Spring microbial mat: Life in the shadow of Chlorophototrophs. I. Microbial diversity based on 16S rRNA gene amplicons and metagenomic sequencing. Front. Microbiol. 2016, 7, 919. [Google Scholar] [CrossRef]
- Cuecas, A.; Portillo, M.C.; Kanoksilapatham, W.; Gonzalez, J.M. Bacterial distribution along a 50 °C temperature gradient reveals a parceled out hot spring environment. Microb. Ecol. 2014, 68, 729–739. [Google Scholar] [CrossRef]
- Hou, W.; Wang, S.; Dong, H.; Jiang, H.; Briggs, B.R.; Peacock, J.P.; Huang, Q.; Huang, L.; Wu, G.; Zhi, X.; et al. A comprehensive census of microbial diversity in hot springs of Tengchong, Yunnan Province China using 16S rRNA gene pyrosequencing. PLoS ONE 2013, 8, e53350. [Google Scholar] [CrossRef] [PubMed]
- Nobu, M.K.; Dodsworth, J.A.; Murugapiran, S.K.; Rinke, C.; Gies, E.A.; Webster, G.; Schwientek, P.; Kille, P.; Parkes, R.J.; Sass, H.; et al. Phylogeny and physiology of candidate phylum “Atribacteria” (OP9/JS1) inferred from cultivation-independent genomics. ISME J. 2016, 10, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Inskeep, W.P.; Rusch, D.B.; Jay, Z.J.; Herrgard, M.J.; Kozubal, M.A.; Richardson, T.H.; Macur, R.E.; Hamamura, N.; Jennings, R.; Fouke, B.W.; et al. Metagenomes from high-temperature chemotrophic systems reveal geochemical controls on microbial community structure and function. PLoS ONE 2010, 5, e9773. [Google Scholar] [CrossRef] [PubMed]
- Swingley, W.D.; Meyer-Dombard, D.A.-R.; Shock, E.L.; Alsop, E.B.; Falenski, H.D.; Havig, J.R.; Raymond, J. Coordinating environmental genomics and geochemistry reveals metabolic transitions in a hot spring ecosystem. PLoS ONE 2012, 7, e38108. [Google Scholar] [CrossRef]
- Delgado-Serrano, L.; López, G.; Bohorquez, L.C.; Bustos, J.R.; Rubiano, C.; Osorio-Forero, C.; Junca, H.; Baena, S.; Zambrano, M.M. Neotropical Andes hot springs harbor diverse and distinct planktonic microbial communities. FEMS Microbiol. Ecol. 2014, 89, 56–66. [Google Scholar] [CrossRef]
- Cole, J.K.; Peacock, J.P.; Dodsworth, J.A.; Williams, A.J.; Thompson, D.B.; Dong, H.; Wu, G.; Hedlund, B.P. Sediment microbial communities in Great Boiling Spring are controlled by temperature and distinct from water communities. ISME J. 2013, 7, 718–729. [Google Scholar] [CrossRef]
- Nordstrom, D.K.; Ball, J.W.; McCleskey, R.B. Ground Water to Surface Water: Chemistry of Thermal Outflows in Yellowstone National Park Geothermal Biology and Geochemistry in Yellowstone National Park; Skeep, W.P., Varley, J., Eds.; Thermal Biology Institute, Montana State University: Bozeman, MT, USA, 2005. [Google Scholar]
- Fouke, B.W. Hot-spring systems geobiology: Abiotic and biotic influences on travertine formation at Mammoth Hot Springs, Yellowstone National Park, USA. Sedimentology 2011, 58, 170–219. [Google Scholar] [CrossRef]
- Urbieta, M.S.; Donati, E.R.; Chan, K.G.; Shahar, S.; Sin, L.L.; Goh, K.M. Thermophiles in the genomic era: Biodiversity, science, and applications. Biotechnol. Adv. 2015, 33, 633–647. [Google Scholar] [CrossRef]
- Pérez, V.; Dorador, C.; Molina, V.; Yáñez, C.; Hengst, M. Rhodobacter sp. Rb3, an aerobic anoxygenic phototroph which thrives in the polyextreme ecosystem of the Salar de Huasco, in the Chilean Altiplano. Antonie Van Leeuwenhoek 2018, 111, 1449–1465. [Google Scholar] [CrossRef]
- Roberts, M.S.; Cohan, F.M. Recombination and migration rates in natural populations of Bacillus subtilis and Bacillus mojavensis. Evolution 1995, 49, 1081–1094. [Google Scholar] [CrossRef]
- Filippidou, S.; Wunderlin, T.; Junier, T.; Jeanneret, N.; Dorador, C.; Molina, V.; Johnson, D.R.; Junier, P. A Combination of Extreme Environmental Conditions Favor the Prevalence of Endospore-Forming Firmicutes. Front. Microbiol. 2016, 7, 1707. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Fukui, M. Molecular characterization of community structures and sulfur metabolism within microbial streamers in Japanese hot springs. Appl. Environ. Microbiol. 2003, 69, 7044–7057. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.; Eder, W.; Heldwein, S.; Wanner, G.; Huber, H.; Rachel, R.; Stetter, K.O. Thermocrinis ruber gen. nov., sp. nov., a pink-filament-forming hyperthermophilic bacterium isolated from Yellowstone National Park. Appl. Environ. Microbiol. 1998, 64, 3576–3583. [Google Scholar] [CrossRef] [PubMed]
- Strazzulli, A.; Iacono, R.; Giglio, R.; Moracci, M.; Cobucci-Ponzano, B. Metagenomics of Hyperthermophilic Environments: Biodiversity and Biotechnology. In Microbial Ecology of Extreme Environments; Chénard, C., Lauro, F., Eds.; Springer: Cham, Switzerland, 2017. [Google Scholar]
- Fru, C. Microbial evolution of sulphate reduction when lateral gene transfer is geographically restricted. Int. J. Syst. Evol. Microbiol. 2011, 61, 1725–1735. [Google Scholar] [CrossRef] [PubMed]
- Schuler, C.G.; Havig, J.R.; Hamilton, T.L. Hot spring microbial community composition, morphology, and carbon fixation: Implications for interpreting the ancient rock record. Front. Earth Sci. 2017, 5, 97. [Google Scholar] [CrossRef]
- Des Marais, D.J. Stable light isotope biogeochemistry of hydrothermal systems. In Evolution of Hydrothermal Ecosystems on Earth and Mars? Bock, G., Goode, J., Eds.; John Wiley & Sons: New York, NY, USA, 1996; pp. 83–93, 273–299. [Google Scholar]
- Farmer, J.D. Hydrothermal systems: Doorways to early biosphere evolution. GSA Today 2000, 10, 1–9. [Google Scholar]
- Garrity, G.; Boone, D.R.; Castenholtz, R.W. The Archaea and the Deeply Branching and Phototrophic Bacteria. In Bergey’s Manual of Systematic Bacteriology, 2nd ed.; Springer: Heidelberg, Germany, 2001; Volume 1. [Google Scholar]
- Hug, L.A.; Brett, B.J.; Anantharaman, K.; Brown, C.T.; Probst, A.J.; Castelle, C.J.; Butterfield, C.N.; Hernsdorf, A.W.; Amano, Y.; Ise, K.; et al. A new view of the tree of life. Nat. Microbiol. 2016, 1, 16048. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Physicochemical Parameters | Sampling Year | P42 | P53A | P53B | P53C | P72 |
|---|---|---|---|---|---|---|
| Temperature (°C) | 2011 | 45 | 51 | 51 | 51 | ND |
| 2013 | 42 | 48 | 48 | 53 | 72 | |
| pH | 2011 | 7.92 | 7.6 | 7.6 | 7.6 | ND |
| 2013 | 7.8 | 7.2 | 7.2 | 7.2 | 5.2 | |
| Conductivity (µS/cm) | 2011 | 2520 | 2100 | 2100 | 2100 | ND |
| 2013 | 1837 | 1875 | 1875 | 1875 | 1848 | |
| Salinity (psu) | 2011 | 1 | 1 | 1 | 1 | ND |
| 2013 | 0.89 | 0.71 | 0.71 | 0.71 | 0.86 | |
| Redox potential (mV) | 2013’ | −152.4 | −246.7 | −246.7 | −246.7 | −316.9 |
| Type of Soil | Mat | Mat | Sediment | Sediment | Sediment | |
| Altitude (m a.s.l.) | 4016 | 4016 | 4016 | 4016 | 4,016 | |
| GPS coordinates | 19°51′7.75″S; 68°54′24.20″W | 19°51′7.75″S; 68°54′24.20″W | 19°51′7.75″S; 68°54′24.20″W | 19°51′7.75″S; 68°54′24.20″W | 19°51′5.69″S; 68°54′23.57″W |
| Characteristics1 | Genus | Phylum | Metabarcoding2 | Clones2 | |||||
|---|---|---|---|---|---|---|---|---|---|
| P42 | P53A | P53B | P53C | P72 | P42 | P53 | |||
| ◯■❖ | Chloracidobacterium | Acidobacteria | |||||||
| ◯■♦ | Thermoanaerobaculum | Acidobacteria | |||||||
 ☐ ☐ | Actinomyces | Actinobacteria | |||||||
| ◯■ | Chthonomonadales | Armatimonadetes | |||||||
| ■ | Candidatus Caldatribacterium | Atribacteria | |||||||
| ☑ | Fervidibacteria | Bacteroidetes | |||||||
| ■ | GBChlB | Bacteroidetes | |||||||
| ■ | Ignavibacterium | Bacteroidetes | |||||||
| ■ | Caldisericum | Caldiserica | |||||||
| ◯☐ | Bellilinea | Chloroflexi | |||||||
| ■ | Chloroflexus | Chloroflexi | |||||||
| ◯■ | Roseiflexus | Chloroflexi | |||||||
| ◯■ | Thermanaerothrix | Chloroflexi | |||||||
| ◯■ | Thermus | Deinococcus–Thermus | |||||||
◯♦■ | Alicyclobacillus | Firmicutes | |||||||
| ■ | Caldicellulosiruptor | Firmicutes | |||||||
| ■ | Caloramator | Firmicutes | |||||||
| ☐ | Caproiciproducens | Firmicutes | |||||||
| ☐ | Clostridium sensu stricto | Firmicutes | |||||||
| ■ | Desulfurispora | Firmicutes | |||||||
| ☐ | Halanaerobium | Firmicutes | |||||||
| ❖ | Lactobacillus | Firmicutes | |||||||
| ◯■ | Moorella | Firmicutes | |||||||
| ◯■ | Ruminiclostridium | Firmicutes | |||||||
| ◯☐♦ | Sulfobacillus | Firmicutes | |||||||
| ■ | Tepidanaerobacter | Firmicutes | |||||||
| ◯■ | Thermobrachium | Firmicutes | |||||||
| ■ | Thermodesulfitimonas | Firmicutes | |||||||
| ■ | Thermosyntropha | Firmicutes | |||||||
| ◯■ | Thermogutta | Planctomycetes | |||||||
| ◯❖■ | Acetobacter | Proteobacteria | |||||||
| ■ | Desulfosoma | Proteobacteria | |||||||
| ■ | Desulfurella | Proteobacteria | |||||||
| ◯■ | Dichotomicrobium | Proteobacteria | |||||||
| ■ | Dissulfurimicrobium | Proteobacteria | |||||||
| ◯■ | Elioraea | Proteobacteria | |||||||
| ■ | Silanimonas | Proteobacteria | |||||||
| ◯❖ | Thiomonas | Proteobacteria | |||||||
| ■ | Exilispira | Spirochaetae | |||||||
| ■ | Treponema | Spirochaetae | |||||||
| ■ | Fervidobacterium | Thermotogae | |||||||
| ■ | DEV007 | Verrucomicrobia | |||||||
Anaerobic; ◯ Aerobic; ◯ Facultative anaerobic; ■ Thermophilic; ☐ Thermotolerant; ☑ Hyperthermophilic; ♦ Acidophilic; ❖ Acidotolerant; and Spore former; Highlight in blue: <0.1%; Highlight in yellow: 0.1–1%; Highlight in bright green: >0.1%.© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez, V.; Cortés, J.; Marchant, F.; Dorador, C.; Molina, V.; Cornejo-D’Ottone, M.; Hernández, K.; Jeffrey, W.; Barahona, S.; Hengst, M.B. Aquatic Thermal Reservoirs of Microbial Life in a Remote and Extreme High Andean Hydrothermal System. Microorganisms 2020, 8, 208. https://doi.org/10.3390/microorganisms8020208
Pérez V, Cortés J, Marchant F, Dorador C, Molina V, Cornejo-D’Ottone M, Hernández K, Jeffrey W, Barahona S, Hengst MB. Aquatic Thermal Reservoirs of Microbial Life in a Remote and Extreme High Andean Hydrothermal System. Microorganisms. 2020; 8(2):208. https://doi.org/10.3390/microorganisms8020208
Chicago/Turabian StylePérez, Vilma, Johanna Cortés, Francisca Marchant, Cristina Dorador, Verónica Molina, Marcela Cornejo-D’Ottone, Klaudia Hernández, Wade Jeffrey, Sergio Barahona, and Martha B. Hengst. 2020. "Aquatic Thermal Reservoirs of Microbial Life in a Remote and Extreme High Andean Hydrothermal System" Microorganisms 8, no. 2: 208. https://doi.org/10.3390/microorganisms8020208
APA StylePérez, V., Cortés, J., Marchant, F., Dorador, C., Molina, V., Cornejo-D’Ottone, M., Hernández, K., Jeffrey, W., Barahona, S., & Hengst, M. B. (2020). Aquatic Thermal Reservoirs of Microbial Life in a Remote and Extreme High Andean Hydrothermal System. Microorganisms, 8(2), 208. https://doi.org/10.3390/microorganisms8020208