Genotyping Study of Salmonella 4,[5],12:i:- Monophasic Variant of Serovar Typhimurium and Characterization of the Second-Phase Flagellar Deletion by Whole Genome Sequencing

 , , , , ,
, , , , ,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Isolate Collection
2.2. Whole Genome Sequencing and in Silico Genotyping
2.3. Characterization of the fljAB Operon Deletions by WGS
3. Results
3.1. Characterization of S. 4,[5],12:i:- and S. Typhimurium by WGS and Bioinformatic Tools
3.2. Characterization of the fljAB Operon Deletion Types by WGS
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate Code | Year | Source | Origin | Serovar | Resistance Profile | Phage Type |
|---|---|---|---|---|---|---|
| 692 | 2008 | Human feces | NCM | 4,[5],12:i:- | Susceptible | 104b |
| 693 | 2008 | Human feces | NCM | 4,[5],12:i:- | ASISuT | 193 |
| 694 | 2008 | Human feces | NCM | 4,[5],12:i:- | ASSuT | 195 |
| 695 | 2008 | Human feces | NCM | 4,[5],12:i:- | T | 138 |
| 696 | 2008 | Human feces | NCM | 4,[5],12:i:- | SSuT | NRP |
| 697 | 2008 | Human feces | NCM | 4,[5],12:i:- | T | NRP |
| 698 | 2008 | Human feces | NCM | 4,[5],12:i:- | AT | 104b |
| 699 | 2008 | Human feces | NCM | 4,[5],12:i:- | ASSuT | 193 |
| 701 | 2008 | Human feces | NCM | 4,[5],12:i:- | SIT | 138 |
| 702 | 2008 | Human feces | NCM | 4,[5],12:i:- | SISuT | NRP |
| 703 | 2008 | Human feces | NCM | 4,[5],12:i:- | ASSuT | 7 |
| 704 | 2008 | Human feces | NCM | 4,[5],12:i:- | ASSuT | NRP |
| 705 | 2008 | Human feces | NCM | 4,[5],12:i:- | ASSuT | NRP |
| 711 | 1999 | Chicken sausage | PHL | 4,[5],12:i:- | T | ND |
| 712 | 2000 | Chicken sausage | PHL | 4,[5],12:i:- | SuT | ND |
| 713 | 2000 | Chicken sausage | PHL | 4,[5],12:i:- | ASSuT | ND |
| 714 | 2000 | Chicken sausage | PHL | 4,[5],12:i:- | ASSuT | ND |
| 743 | 2012 | Swine IC | IdAB | 4,[5],12:i:- | ASSu | U311 |
| 744 | 2012 | Swine MLN | IdAB | 4,[5],12:i:- | ASSu | ND |
| 745 | 2012 | Swine MLN | IdAB | 4,[5],12:i:- | ASSuT | ND |
| 746 | 2012 | Swine IC | IdAB | 4,[5],12:i:- | ASSu | ND |
| 747 | 2012 | Swine IC | IdAB | 4,[5],12:i:- | ASSuT | ND |
| 748 | 2015 | Swine IC | IdAB | 4,[5],12:i:- | Susceptible | ND |
| 749 | 2015 | Swine IC | IdAB | 4,[5],12:i:- | ASSu | ND |
| 750 | 2015 | Swine IC | IdAB | 4,[5],12:i:- | A | ND |
| 751 | 2015 | Swine IC | IdAB | 4,[5],12:i:- | ACS | ND |
| 752 | 2015 | Swine IC | IdAB | 4,[5],12:i:- | ACSSu | ND |
| 753 | 2015 | Swine IC | IdAB | 4,[5],12:i:- | ACSISuT | ND |
| 754 | 2015 | Swine IC | IdAB | 4,[5],12:i:- | AS | ND |
| 755 | 2015 | Swine IC | IdAB | 4,[5],12:i:- | ASSuT | ND |
| 757 | 2015 | Swine IC | IdAB | 4,[5],12:i:- | CS | ND |
| 758 | 2015 | Swine IC | IdAB | 4,[5],12:i:- | SSu | ND |
| 739 | 2012 | Swine MLN | IdAB | 4,[5],12:i:1,2 | Susceptible | NRP |
| 756 | 2012 | Swine MLN | IdAB | 4,[5],12:i:1,2 | ACSSuT | ND |
| 759 | 2012 | Swine IC | IdAB | 4,[5],12:i:1,2 | ACSSuT | ND |
| 760 | 2012 | Swine MLN | IdAB | 4,[5],12:i:1,2 | ACSSuTNx | 104b |
| 761 | 2012 | Swine MLN | IdAB | 4,[5],12:i:1,2 | Susceptible | NRP |
| 767 | 2012 | Swine MLN | IdAB | 4,[5],12:i:1,2 | ACSSuT | U302 |
| 773 | 2012 | Swine MLN | IdAB | 4,[5],12:i:1,2 | ACSSuT | 104b |
| 775 | 2012 | Swine MLN | IdAB | 4,[5],12:i:1,2 | ACSSuT | 104b |
| 778 | 2012 | Swine MLN | IdAB | 4,[5],12:i:1,2 | Susceptible | ND |
| 779 | 2012 | Swine MLN | IdAB | 4,[5],12:i:1,2 | ASSuTNxCfx | ND |






References
- Grimont, P.; Weill, F. Antigenic Formulae of the Salmonella Serovars, 9th ed.; WHO Collaborating Center for Reference and Research on Salmonella, Institut Pasteur: Paris, France, 2007. [Google Scholar]
- EFSA; ECDC. The European Union one health 2018 zoonoses report. EFSA J. 2019, 17. [Google Scholar] [CrossRef]
- Echeita, M.-A.; Aladueña, A.; Cruchaga, S.; Usera, M.A. Fast-track communications emergence and spread of an atypical Salmonella enterica subsp. Enterica Soc. 1999, 37, 28220. [Google Scholar]
- Echeita, M.A.; Herrera, S.; Usera, M.A. Atypical, fljB-negative Salmonella enterica subsp. enterica of serovar 4,5,12:i:- appears to be a monophasic variant of serovar Typhimurium. Society 2001, 39, 2981–2983. [Google Scholar] [CrossRef] [PubMed]
- EFSA Panel on Biological Hazards (BIOHAZ). Scientific opinion on monitoring and assessment of the public health risk of “Salmonella Typhimurium-like” strains. EFSA J. 2010, 8. [Google Scholar] [CrossRef]
- Centro Nacional de Epidemiología; CIBER Epidemiología y Salud Pública (CIBERESP); Instituto de Salud Carlos III. Resultados de la Vigilancia Epidemiológica de las Enfermedades Trasmisibles. Informe Anual 2016; Gobierno de España (Ministerio de Ciencias e Innovación y Universidades): Madrid, Spain, 2018.
- Petrovska, L.; Mather, A.E.; Abuoun, M.; Branchu, P.; Harris, S.R.; Connor, T.; Hopkins, K.L.; Underwood, A.; Lettini, A.A.; Page, A.; et al. Microevolution of monophasic Salmonella Typhimurium during epidemic, United Kingdom, 2005–2010. Emerg. Infect. Dis. 2016, 22, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Garaizar, J.; Porwollik, S.; Echeita, A.; Rementeria, A.; Herrera, S.; Wong, R.M.-Y.; Frye, J.; Usera, M.A.; Mc Clelland, M. DNA microarray-based typing of an atypical monophasic Salmonella enterica serovar. J. Clin. Microbiol. 2002, 40, 2074–2078. [Google Scholar] [CrossRef]
- Laorden, L.; Herrera-León, S.; Martínez, I.; Sanchez, A.; Kromidas, L.; Bikandi, J.; Rementeria, A.; Echeita, A.; Garaizar, J. Genetic evolution of the Spanish multidrug-resistant Salmonella enterica 4,5,12:i:- monophasic variant. J. Clin. Microbiol. 2010, 48, 4563–4566. [Google Scholar] [CrossRef]
- Soyer, Y.; Switt, A.M.; Davis, M.A.; Maurer, J.; McDonough, P.L.; Schoonmaker-Bopp, D.J.; Dumas, N.B.; Root, T.; Warnick, L.D.; Gröhn, Y.T.; et al. Salmonella enterica serotype 4,5,12:i:-, an emerging Salmonella serotype that represents multiple distinct clones. J. Clin. Microbiol. 2009, 47, 3546–3556. [Google Scholar] [CrossRef]
- Switt, A.I.M.; Soyer, Y.; Warnick, L.D.; Wiedmann, M. Emergence, distribution, and molecular and phenotypic characteristics of Salmonella enterica serotype 4,5,12:i:-. Foodborne Pathog. Dis. 2009, 6, 407–415. [Google Scholar] [CrossRef]
- Hopkins, K.L.; Kirchner, M.; Guerra, B.; Granier, S.A.; Lucarelli, C.; Porrero, M.C.; Jakubczak, A.; Threlfall, E.J.; Mevius, D.J. Multiresistant Salmonella enterica serovar 4,[5],12:i:- in Europe: A new pandemic strain? Euro Surveill. 2010, 15. [Google Scholar] [CrossRef]
- Cito, F.; Baldinelli, F.; Calistri, P.; Di Giannatale, E.; Scavia, G.; Orsini, M.; Iannetti, S.; Sacchini, L.; Mangone, I.; Candeloro, L.; et al. Outbreak of unusual Salmonella enterica serovar Typhimurium monophasic variant 1,4,[5],12:i:-, Italy, June 2013 to September 2014. Euro Surveill. 2016, 21. [Google Scholar] [CrossRef] [PubMed]
- Mourão, J.; Machado, J.; Novais, C.; Antunes, P.; Peixe, L. Characterization of the emerging clinically-relevant multidrug-resistant Salmonella enterica serotype 4,[5],12:i:- (monophasic variant of S. Typhimurium) clones. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 2249–2257. [Google Scholar] [CrossRef] [PubMed]
- Dionisi, A.M.; Graziani, C.; Lucarelli, C.; Filetici, E.; Villa, L.; Owczarek, S.; Caprioli, A.; Luzzi, I. Molecular characterization of multidrug-resistant strains of Salmonella enterica serotype Typhimurium and monophasic variant (S. 4,[5],12:i:-) isolated from human infections in Italy. Foodborne Pathog. Dis. 2009, 6, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Lucarelli, C.; Dionisi, A.M.; Filetici, E.; Owczarek, S.; Luzzi, I.; Villa, L. Nucleotide sequence of the chromosomal region conferring multidrug resistance (R-type ASSuT) in Salmonella Typhimurium and monophasic Salmonella Typhimurium strains. J. Antimicrob. Chemother. 2012, 67, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Garcia, P.; Hopkins, K.L.; Garcia, V.; Beutlich, J.; Mendoza, M.C.; Threlfall, J.; Mevius, D.; Helmuth, R.; Rodicio, M.R.; Guerra, B. Diversity of plasmids encoding virulence and resistance functions in Salmonella enterica subsp. enterica serovar Typhimurium monophasic variant 4,[5],12:i:- strains circulating in Europe. PLoS ONE 2014, 9, e89635. [Google Scholar] [CrossRef]
- García, P.; Malorny, B.; Rodicio, M.R.; Stephan, R.; Hächler, H.; Guerra, B.; Lucarelli, C. Horizontal acquisition of a multidrug-resistance module (R-type ASSuT) is responsible for the monophasic phenotype in a widespread clone of Salmonella serovar 4,[5],12:i:-. Front. Microbiol. 2016, 7, 1–7. [Google Scholar] [CrossRef]
- Wasyl, D.; Hoszowski, A. Occurrence and characterization of monophasic Salmonella enterica serovar Typhimurium (1,4,[5],12:i:-) of non-human origin in Poland. Foodborne Pathog. Dis. 2012, 9, 1037–1043. [Google Scholar] [CrossRef]
- Barco, L.; Longo, A.; Lettini, A.A.; Cortini, E.; Saccardin, C.; Minorello, C.; Olsen, J.E.; Ricci, A. Molecular characterization of “inconsistent” variants of Salmonella Typhimurium isolated in Italy. Foodborne Pathog. Dis. 2014, 11, 497–499. [Google Scholar] [CrossRef]
- Boland, C.; Bertrand, S.; Mattheus, W.; Dierick, K.; Wattiau, P. Molecular typing of monophasic Salmonella 4,[5]:i:- strains isolated in Belgium (2008–2011). Vet. Microbiol. 2014, 168, 447–450. [Google Scholar] [CrossRef]
- CLSI (Clinical and Laboratory Standards Institute). Performance Standards for Antimicrobial Disk and Dilution Susceptibility Tests for Bacteria Isolated from Animals, 4th ed.; CLSI: Wayne, PA, USA, 2018. [Google Scholar]
- Anderson, E.S.; Ward, L.R.; Saxe, M.J.; de Sa, J.D. Bacteriophage-typing designations of Salmonella Typhimurium. J. Hyg. (Lond.) 1977, 78, 297–300. [Google Scholar] [CrossRef]
- Machado, M.; Halkilahti, J.; Jaakkonen, A.; Silva, D.; Mendes, I.; Nalbantoglu, Y.; Borges, V.; Ramirez, M.; Rossi, M.; Carriço, J. GitHub-B-UMMI/INNUca: INNUENDO Quality Control of Reads, De Novo Assembly and Contigs Quality Assessment, and Possible Contamination Search. Available online: https://github.com/B-UMMI/INNUca (accessed on 10 November 2020).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; den Bakker, H.C.; Li, S.; Chen, J.; Dinsmore, B.A.; Lane, C.; Lauer, A.C.; Fields, P.I.; Deng, X. SeqSero2: Rapid and improved Salmonella serotype determination using Whole-Genome Sequencing Data. Appl. Environ. Microbiol. 2019, 85. [Google Scholar] [CrossRef] [PubMed]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020. [Google Scholar] [CrossRef] [PubMed]
- Carattoli, A. Plasmids and the spread of resistance. Int. J. Med. Microbiol. 2013, 303, 298–304. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef]
- San Millán, R.M.; Martínez-Ballesteros, I.; Rementeria, A.; Garaizar, J.; Bikandi, J. Online exercise for the design and simulation of PCR and PCR-RFLP experiments. BMC Res. Notes 2013, 6, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Alikhan, N.-F.; Zhou, Z.; Sergeant, M.J.; Achtman, M. A genomic overview of the population structure of Salmonella. PLoS Genet. 2018, 14, e1007261. [Google Scholar] [CrossRef]
- EFSA; ECDC. The European Union summary report on trends and sources of zoonoses, zoonotic agents and food-borne outbreaks in 2017. EFSA J. 2018, 16. [Google Scholar] [CrossRef]
- Lockman, H.A.; Curtiss, R. Salmonella Typhimurium mutants lacking flagella or motility remain virulent in BALB/c mice. Infect. Immun. 1990, 58, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Boland, C.; Van Hessche, M.; Mahillon, J.; Wattiau, P. A liquid bead array for the identification and characterization of fljB-positive and fljB-negative monophasic variants of Salmonella Typhimurium. Food Microbiol. 2018, 71, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Llarena, A.; Ribeiro-Gonçalves, B.F.; Nuno Silva, D.; Halkilahti, J.; Machado, M.P.; Da Silva, M.S.; Jaakkonen, A.; Isidro, J.; Hämäläinen, C.; Joenperä, J.; et al. INNUENDO: A cross-sectoral platform for the integration of genomics in the surveillance of food-borne pathogens. EFSA Support. Publ. 2018, 15. [Google Scholar] [CrossRef]
- Uelze, L.; Borowiak, M.; Deneke, C.; Szabo, I.; Fischer, J.; Tausch, S.H.; Malorny, B. Performance and accuracy of four oppen-source tools for in silico serotyping of Salmonella spp. based on Whole-Genome Short-Read Sequencing Data. Appl. Environ. Microbiol. 2020, 86. [Google Scholar] [CrossRef]
- Banerji, S.; Simon, S.; Tille, A.; Fruth, A.; Flieger, A. Genome-based Salmonella serotyping as the new gold standard. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Diep, B.; Barretto, C.; Portmann, A.C.; Fournier, C.; Karczmarek, A.; Voets, G.; Li, S.; Deng, X.; Klijn, A. Salmonella serotyping; comparison of the traditional method to a Microarray-based method and an in silico platform using Whole Genome Sequencing Data. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Boland, C.; Bertrand, S.; Mattheus, W.; Dierick, K.; Jasson, V.; Rosseel, T.; Van Borm, S.; Mahillon, J.; Wattiau, P. Extensive genetic variability linked to IS26 insertions in the fljB promoter region of atypical monophasic variants of Salmonella enterica serovar Typhimurium. Appl. Environ. Microbiol. 2015, 81, 3169–3175. [Google Scholar] [CrossRef]
- Bugarel, M.; Vignaud, M.-L.; Moury, F.; Fach, P.; Brisabois, A. Molecular identification in monophasic and nonmotile variants of Salmonella enterica serovar Typhimurium. Microbiologyopen 2012, 1, 481–489. [Google Scholar] [CrossRef]
- San Román, B.; Garrido, V.; Sanchez, S.; Martinez-Ballesteros, I.; Garaizar, J.; Mainar-Jaime, R.C.; Migura-Garcia, L.; Grilló, M.J. Relationship between Salmonella infection, shedding and serology in fattening pigs in low-moderate prevalence areas. Zoonoses Public Health 2018, 65, 481–489. [Google Scholar] [CrossRef]
| Isolate Code | SeqSero2 | INNUca MLST | cgMLSTFinder | ResFinder profile | SPIFinder | PlasmidFinder | PHASTER | In Silico IS26 PCR |
|---|---|---|---|---|---|---|---|---|
| 692 | 4,[5],12:i:- | 19 | 123420 | S | SPI-1, SPI-3, SPI-5, SPI-13, C63PI | IncFIB(S), IncFII(S) | G2, EGF2, F2 | − |
| 693 | 4,[5],12:i:1,2 | 34 | 84985 | ASSuT | SPI-1, SPI-2, SPI-3, SPI-5, SPI-13, SPI-14, C63PI | IncQ1 | S3, G1, HP2, Ephi20 | + |
| 694 | 4,[5],12:i:1,2 | 34 | 78574 | ASSuT | SPI-1, SPI-2, SPI-3, SPI-5, SPI-13, SPI-14, C63PI | IncQ1 | S3, G1 | + |
| 695 | 4,[5],12:i:- | 34 | 141108 | ST | SPI-1, SPI-2, SPI-3, SPI-4, SPI-5, SPI-13, SPI-14, C63PI | S3, G1 | + | |
| 696 | 4,[5],12:i:1,2 | 34 | 21377 | SSuT | SPI-1, SPI-2, SPI-3, SPI-5, SPI-13, SPI-14, C63PI | IncQ1, IncFII | S3, G1, HP2, SfI | + |
| 697 | 4,[5],12:i:- | 19 | 85377 | CSSuTTm | SPI-1, SPI-2, SPI-3, SPI-4, SPI-5, SPI-13, SPI-14, C63PI | IncR | G2, EGF2 | + |
| 698 | 4,[5],12:i:- | 34 | 31310 | AST | SPI-1, SPI-2, SPI-3, SPI-5, SPI-13, SPI-14, C63PI | S3, G1, HP2, SfI | + | |
| 699 | 4,[5],12:i:- | 34 | 3719 | ASSuT | SPI-1, SPI-2, SPI-3, SPI-5, SPI-13, SPI-14, C63PI | IncQ1, Col156 | S3, G1, HP2, SfI | + |
| 701 | 4,[5],12:i:- | 34 | 141108 | ST | SPI-1, SPI-2, SPI-3, SPI-5, SPI-13, C63PI | S3, G1 | + | |
| 702 | 4,[5],12:i:- | 19 | 85377 | CSSuTTm | SPI-1, SPI-2, SPI-3, SPI-4, SPI-5, SPI-13, SPI-14, C63PI | IncR | G2, EGF2 | + |
| 703 | 4,[5],12:i:- | 34 | 132646 | ASSuT | SPI-1, SPI-2, SPI-3, SPI-5, SPI-13, SPI-14, C63PI | G2, HP2 | + | |
| 704 | 4,[5],12:i:- | 34 | 132646 | ASSuT | SPI-1, SPI-2, SPI-3, SPI-4, SPI-5, SPI-13, SPI-14, C63PI | IncQ1 | G1, HP2 | + |
| 705 | 4,[5],12:i:- | 34 | 6912 | ST | SPI-1, SPI-2, SPI-3, SPI-5, SPI-13, SPI-14, C63PI | G1 | + | |
| 711 | 4,[5],12:i:- | 19 | 156249 | CSSuT | SPI-1, SPI-2, SPI-3, SPI-5, SPI-13, SPI-14, C63PI | IncI1, IncA/C2 | G1, HP2 | + |
| 712 | 4,[5],12:i:- | 19 | 156249 | CSSuTTmNxCip | SPI-1, SPI-2, SPI-3, SPI-5, SPI-13, SPI-14, C63PI | Col(BS512), IncA/C2 | G1, G2, HP2, EST104 | + |
| 713 | 4,[5],12:i:- | 19 | 156249 | ACSSuTTm | SPI-1, SPI-3, SPI-5, SPI-13, SPI-14, C63PI | Col(BS512), IncA/C2 | G1, EST104 | + |
| 714 | 4,[5],12:i:- | 19 | 156249 | ACSSuTTm | SPI-1, SPI-2, SPI-3, SPI-5, SPI-14, C63PI | Col(BS512), IncA/C2 | G1, G2, HP2, EST104 | + |
| 743 | 4,[5],12:i:- | 34 | 132646 | ASSu | SPI-1, SPI-3, SPI-4, SPI-5, SPI-13, SPI-14, C63PI | IncQ1 | S3, G1, G2, HP2 | + |
| 744 | 4,[5],12:i:- | 34 | 43443 | ASSu | SPI-5, SPI-13, SPI-14, C63PI | IncQ1 | S3, G1, G2, HP2 | + |
| 745 | 4,[5],12:i:- | 34 | 165159 | ASSuT | SPI-5, SPI-13, SPI-14, C63PI | IncQ1 | S3, G1, G2, HP2, SfI | + |
| 746 | 4,[5],12:i:- | 34 | 132646 | ASSu | SPI-1, SPI-2, SPI-3, SPI-4, SPI-5, SPI-13, SPI-14 | IncQ1 | G1 | + |
| 747 | 4,[5],12:i:- | 34 | 100540 | ASSuT | SPI-1, SPI-3, SPI-5, SPI-13, SPI-14, C63PI | IncQ1, Col440l | G1, Ep460 | + |
| 748 | 4,[5],12:i:- | 34 | 26728 | S | SPI-1, SPI-2, SPI-3, SPI-5, SPI-13, SPI-14, C63PI | G1, EfiAA91, SfII | + | |
| 749 | 4,[5],12:i:- | 34 | 26728 | ASSu | SPI-1, SPI-2, SPI-3, SPI-5, SPI-9, SPI-13, SPI-14, C63PI | IncQ1 | S3, G1, EfiAA91, SfI | + |
| 750 | 4,[5],12:i:- | 34 | 26728 | ASSu | SPI-1, SPI-2, SPI-3, SPI-5, SPI-13, SPI-14, C63PI | IncQ1 | G1, EfiAA91, E186, SfI | + |
| 751 | 4,[5],12:i:- | 34 | 29699 | CS | SPI-1, SPI-3, SPI-5, SPI-13, SPI-14, C63PI | IncI1 | G1, E186, EfiAA91, SfII | + |
| 752 | 4,[5],12:i:- | 34 | 89891 | CS | SPI-3, SPI-5, SPI-13, SPI-14, C63PI | IncI1 | S3, G1, G2, E186, EfiAA91, ESfV | + |
| 753 | 4,[5],12:i:- | 34 | 144738 | SSu | SPI-1, SPI-2, SPI-3, SPI-4, SPI-5, SPI-13, SPI-14, C63PI | IncQ1 | S3, G1, G2, EST104, Epro147 | + |
| 754 | 4,[5],12:i:- | 34 | 89891 | S | SPI-1, SPI-3, SPI-5, SPI-13, SPI-14 | IncI1 | S3, G1, E186, EfiAA91, Ep460 | + |
| 755 | 4,[5],12:i:- | 34 | 84985 | ASSuT | SPI-1, SPI-2, SPI-3, SPI-5, SPI-13, SPI-14, C63PI | IncQ1 | S3, G1, G2, HP2 | + |
| 757 | 4,[5],12:i:- | 34 | 29699 | CS | SPI-1, SPI-2, SPI-3, SPI-5, SPI-13, SPI-14, C63PI | IncI1 | G1, G2 | + |
| 758 | 4,[5],12:i:- | 34 | 144738 | SSu | SPI-1, SPI-2, SPI-3, SPI-4, SPI-5, SPI-13, SPI-14, C63PI | IncQ1 | G1, EST104, Epro147 | + |
| 739 | 4,[5],12:i:1,2 | 19 | 35732 | S | SPI-1, SPI-2, SPI-3, SPI-5, SPI-13, SPI-14, C63PI | ColpVC, IncFIB(S), IncFII(S), IncX1 | S3, G1, G2 | − |
| 756 | 4,[5],12:i:1,2 | 19 | 45281 | ACSSuT | SPI-1, SPI-2, SPI-3, SPI-5, SPI-13, SPI-14, C63PI | IncFIB(S), IncFII(S) | EST104 | − |
| 759 | 4,[5],12:i:1,2 | 19 | 78568 | ACSSuT | SPI-1, SPI-5, SPI-13, SPI-14, C63PI | IncX1, IncFII(S) | G1, G2, EST104 | + |
| 760 | 4,[5],12:i:1,2 | 19 | 20179 | ACSSuTNxCip | SPI-1, SPI-2, SPI-3, SPI-5, SPI-13, SPI-14, C63PI | IncFII(S) | G1, G2, EST104, HP2 | + |
| 761 | 4,[5],12:i:1,2 | 19 | 35732 | S | SPI-1, SPI-2, SPI-3, SPI-4, SPI-5, SPI-13, SPI-14, C63PI | ColpVC | S3, G1, G2 | − |
| 767 | 4,[5],12:i:1,2 | 19 | 45281 | ACSSuT | SPI-1, SPI-2, SPI-3, SPI-4, SPI-5, SPI-13, SPI-14, C63PI | IncFIB(S), IncFII(S) | G1, G2, EST104 | − |
| 773 | 4,[5],12:i:1,2 | 19 | 45281 | ACSSuT | SPI-1, SPI-2, SPI-3, SPI-4, SPI-5, SPI-13, SPI-14, C63PI | IncFIB(S), IncFII(S) | S3, G1, G2, EST104 | − |
| 775 | 4,[5],12:i:1,2 | 19 | 45281 | ACSSuT | SPI-1, SPI-2, SPI-3, SPI-4, SPI-5, SPI-13, SPI-14, C63PI | IncFIB(S), IncFII(S) | S3, G1, G2, EST104 | − |
| 778 | 4,[5],12:i:1,2 | 19 | 35732 | S | SPI-1, SPI-2, SPI-3, SPI-4, SPI-5, SPI-13, SPI-14, C63PI | ColpVC, IncFIB(S), IncFII(S), IncX1 | G1, G2 | − |
| 779 | 4,[5],12:i:1,2 | 34 | 95263 | ASSuTCol | SPI-1, SPI-3, SPI-5, SPI-13, SPI-14, C63PI | IncQ1, IncHI2, IncHI2A | S3, G1 | + |
| Isolate Code | SeqSero2 | Serotyping | ResFinder | Antibiogram Resistance Profile | |
|---|---|---|---|---|---|
| Antibiotic Resistance Genes and Mutations | Profile | ||||
| 692 | 4,[5],12:i:- | 4,[5],12:i:- | aac(6′)-laa | S | Susceptible |
| 693 | 4,[5],12:i:1,2 | 4,[5],12:i:- | blaTEM-1B; aac(6’)-laa; aph(3″)-lb; aph(6)-ld; Sul2; tet(B) | ASSuT | ASISuT |
| 694 | 4,[5],12:i:1,2 | 4,[5],12:i:- | blaTEM-1B; aac(6´)-laa; aph(3″)-lb; aph(6)-ld; Sul2; tet(B) | ASSuT | ASSuT |
| 695 | 4,[5],12:i:- | 4,[5],12:i:- | aac(6´)-laa; tet(B) | ST | T |
| 696 | 4,[5],12:i:1,2 | 4,[5],12:i:- | aac(6´)-laa; aph(3″)-lb; Sul2; tet(B) | SSuT | SSuT |
| 697 | 4,[5],12:i:- | 4,[5],12:i:- | cmlA1; aac(6´)-laa; aph(3″)-lb; aph(6)-ld; aadA1; aadA2; Sul3; dfrA12; tet(B) | CSSuTTm | T |
| 698 | 4,[5],12:i:- | 4,[5],12:i:- | blaTEM-1B; aac(6´)-laa; tet(B) | AST | AT |
| 699 | 4,[5],12:i:- | 4,[5],12:i:- | blaTEM-1B; aac(6´)-laa; aph(3″)-lb; aph(6)-ld; Sul2; tet(B) | ASSuT | ASSuT |
| 701 | 4,[5],12:i:- | 4,[5],12:i:- | aac(6´)-laa; tet(B) | ST | SIT |
| 702 | 4,[5],12:i:- | 4,[5],12:i:- | cmlA1; aac(6´)-laa; aph(3″)-lb; aph(6)-ld; aadA1; aadA2; Sul3; dfrA12; tet(B) | CSSuTTm | SISuT |
| 703 | 4,[5],12:i:- | 4,[5],12:i:- | blaTEM-1B; aac(6´)-laa; aph(3″)-lb; aph(6)-ld; Sul2; tet(B) | ASSuT | ASSuT |
| 704 | 4,[5],12:i:- | 4,[5],12:i:- | blaTEM-1B; aac(6´)-laa; aph(3″)-lb; aph(6)-ld; Sul2; tet(B) | ASSuT | ASSuT |
| 705 | 4,[5],12:i:- | 4,[5],12:i:- | aac(6´)-laa; tet(B) | ST | ASSuT |
| 711 | 4,[5],12:i:- | 4,[5],12:i:- | cmlA1; aac(6′)-Iaa; aadA8b; Sul1; Sul2; Sul3; tet(A) | CSSuT | T |
| 712 | 4,[5],12:i:- | 4,[5],12:i:- | cmlA1; aac(6′)-Iaa; Sul1; Sul2; Sul3; dfrA12; tet(A); gyrB p.E466D mutation | CSSuTTmNxCip | SuT |
| 713 | 4,[5],12:i:- | 4,[5],12:i:- | blaTEM-1B; cmlA1; aac(3)-IV; aac(6′)-Iaa; Sul1; Sul2; Sul3; dfrA12; tet(A) | ACSSuTTm | ASSuT |
| 714 | 4,[5],12:i:- | 4,[5],12:i:- | blaTEM-1B; cmlA1; aac(3)-IV; aac(6′)-Iaa; Sul1; Sul2; Sul3; dfrA12; tet(A) | ACSSuTTm | ASSuT |
| 743 | 4,[5],12:i:- | 4,[5],12:i:- | blaTEM-1B; aac(6′)-laa; aph(3″)-lb; aph(6)-ld; Sul2 | ASSu | ASSu |
| 744 | 4,[5],12:i:- | 4,[5],12:i:- | blaTEM-1B; aac(6′)-laa; aph(3″)-lb; aph(6)-ld; Sul2 | ASSu | ASSu |
| 745 | 4,[5],12:i:- | 4,[5],12:i:- | blaTEM-1B; aac(6′)-laa; aph(3″)-lb; aph(6)-ld; Sul2; tet(B) | ASSuT | ASSuT |
| 746 | 4,[5],12:i:- | 4,[5],12:i:- | blaTEM-1B; aac(6′)-laa; aph(3″)-lb; aph(6)-ld; Sul2 | ASSu | ASSu |
| 747 | 4,[5],12:i:- | 4,[5],12:i:- | blaTEM-1B; aac(6′)-laa; aph(3″)-lb; aph(6)-ld; Sul2; tet(B) | ASSuT | ASSuT |
| 748 | 4,[5],12:i:- | 4,[5],12:i:- | aac(6′)-laa | S | Susceptible |
| 749 | 4,[5],12:i:- | 4,[5],12:i:- | blaTEM-1B; aac(6′)-laa; aph(3″)-lb; aph(6)-ld; Sul2 | ASSu | ASSu |
| 750 | 4,[5],12:i:- | 4,[5],12:i:- | blaTEM-1B; aac(6′)-laa; aph(3″)-lb; aph(6)-ld; Sul2 | ASSu | A |
| 751 | 4,[5],12:i:- | 4,[5],12:i:- | floR; aac(3)-IV; aac(6′)-laa; aph(3″)-lb; aph(4)-la; aph(6)-ld | CS | ACS |
| 752 | 4,[5],12:i:- | 4,[5],12:i:- | floR; aac(3)-IV; aac(6′)-laa; aph(3″)-lb; aph(4)-la; aph(6)-ld | CS | ACSSu |
| 753 | 4,[5],12:i:- | 4,[5],12:i:- | aac(6′)-laa; aph(3″)-lb; aph(6)-ld; Sul2 | SSu | ACSISuT |
| 754 | 4,[5],12:i:- | 4,[5],12:i:- | aac(3)-IV; aac(6′)-laa; aph(3″)-lb; aph(4)-la; aph(6)-ld | S | AS |
| 755 | 4,[5],12:i:- | 4,[5],12:i:- | blaTEM-1B; aac(6′)-laa; aph(3″)-lb; aph(6)-ld; Sul2; tet(B) | ASSuT | ASSuT |
| 757 | 4,[5],12:i:- | 4,[5],12:i:- | floR; aac(3)-IV; aac(6′)-laa; aph(3″)-lb; aph(4)-la; aph(6)-ld | CS | CS |
| 758 | 4,[5],12:i:- | 4,[5],12:i:- | aac(6′)-laa; aph(3″)-lb; aph(6)-ld; Sul2 | SSu | SSu |
| 739 | 4,[5],12:i:1,2 | 4,[5],12:i:1,2 | aac(6′)-laa | S | Susceptible |
| 756 | 4,[5],12:i:1,2 | 4,[5],12:i:1,2 | blaCARB-2; floR; aac(6′)-laa; aadA2; Sul1; tet(G) | ACSSuT | ACSSuT |
| 759 | 4,[5],12:i:1,2 | 4,[5],12:i:1,2 | blaOXA-1; catA1; aac(6′)-laa; aadA1; Sul1; tet(B) | ACSSuT | ACSSuT |
| 760 | 4,[5],12:i:1,2 | 4,[5],12:i:1,2 | blaOXA-1; catA1; aac(6′)-laa; aadA1; Sul1; tet(B); gyrA p. D87N mutation | ACSSuTNxCip | ACSSuTNx |
| 761 | 4,[5],12:i:1,2 | 4,[5],12:i:1,2 | aac(6′)-laa | S | Susceptible |
| 767 | 4,[5],12:i:1,2 | 4,[5],12:i:1,2 | blaCARB-2; floR; aac(6′)-laa; aadA2; Sul1; tet(G) | ACSSuT | ACSSuT |
| 773 | 4,[5],12:i:1,2 | 4,[5],12:i:1,2 | blaCARB-2; floR; aac(6′)-laa; aadA2; Sul1; tet(G) | ACSSuT | ACSSuT |
| 775 | 4,[5],12:i:1,2 | 4,[5],12:i:1,2 | blaCARB-2; floR; aac(6′)-laa; aadA2; Sul1; tet(G) | ACSSuT | ACSSuT |
| 778 | 4,[5],12:i:1,2 | 4,[5],12:i:1,2 | aac(6′)-laa | S | Susceptible |
| 779 | 4,[5],12:i:1,2 | 4,[5],12:i:1,2 | blaTEM-1B; blaCTX-M-9; aac(6′)-laa; aadA2; aph(3″)-lb; aph(6)-ld; Sul1; Sul2; dfrA16; tet(A); mcr-9 | ASSuTCol | ASSuTNxCfx |
| Deletion Type | Subtype | No. of Strains | Starting Point | Ending Point | Inserted Fragment † |
|---|---|---|---|---|---|
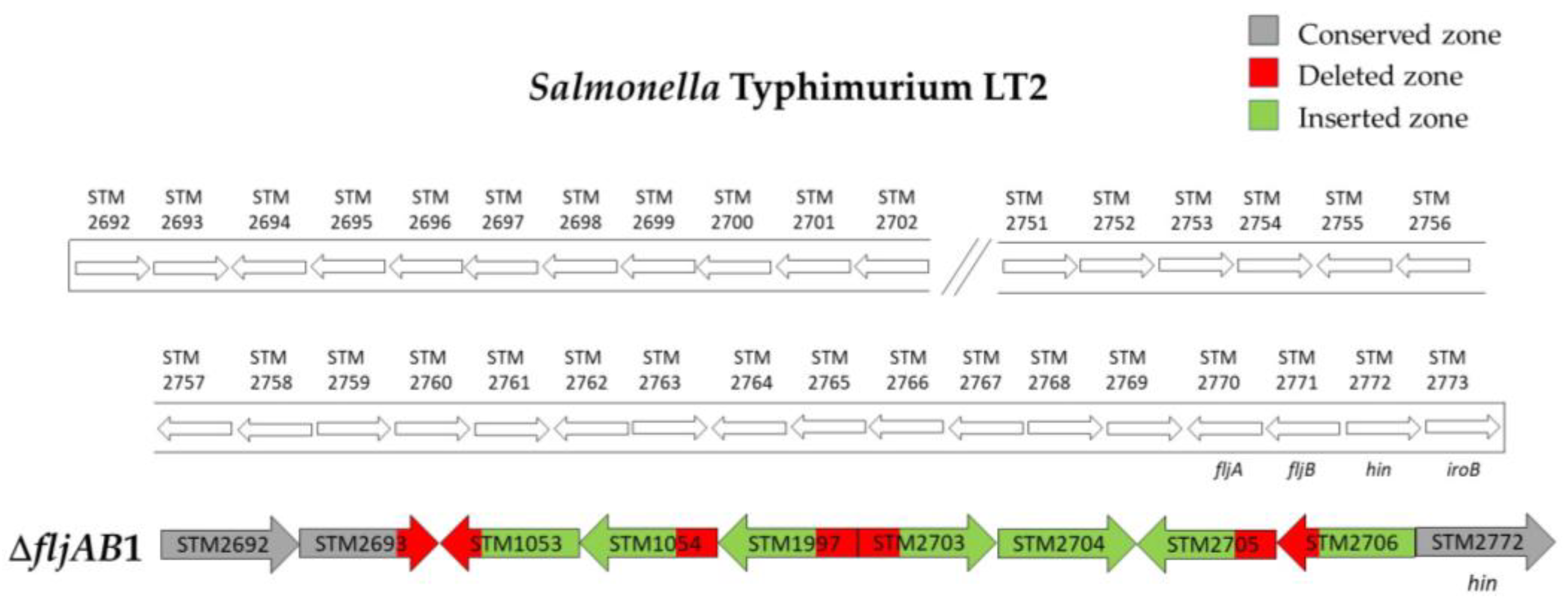
| ΔfljAB1 | 4 | 98 bp of STM2693 | 10 bp downstream of STM2771 (fljB) | 5654 bp (see Figure A1) | |
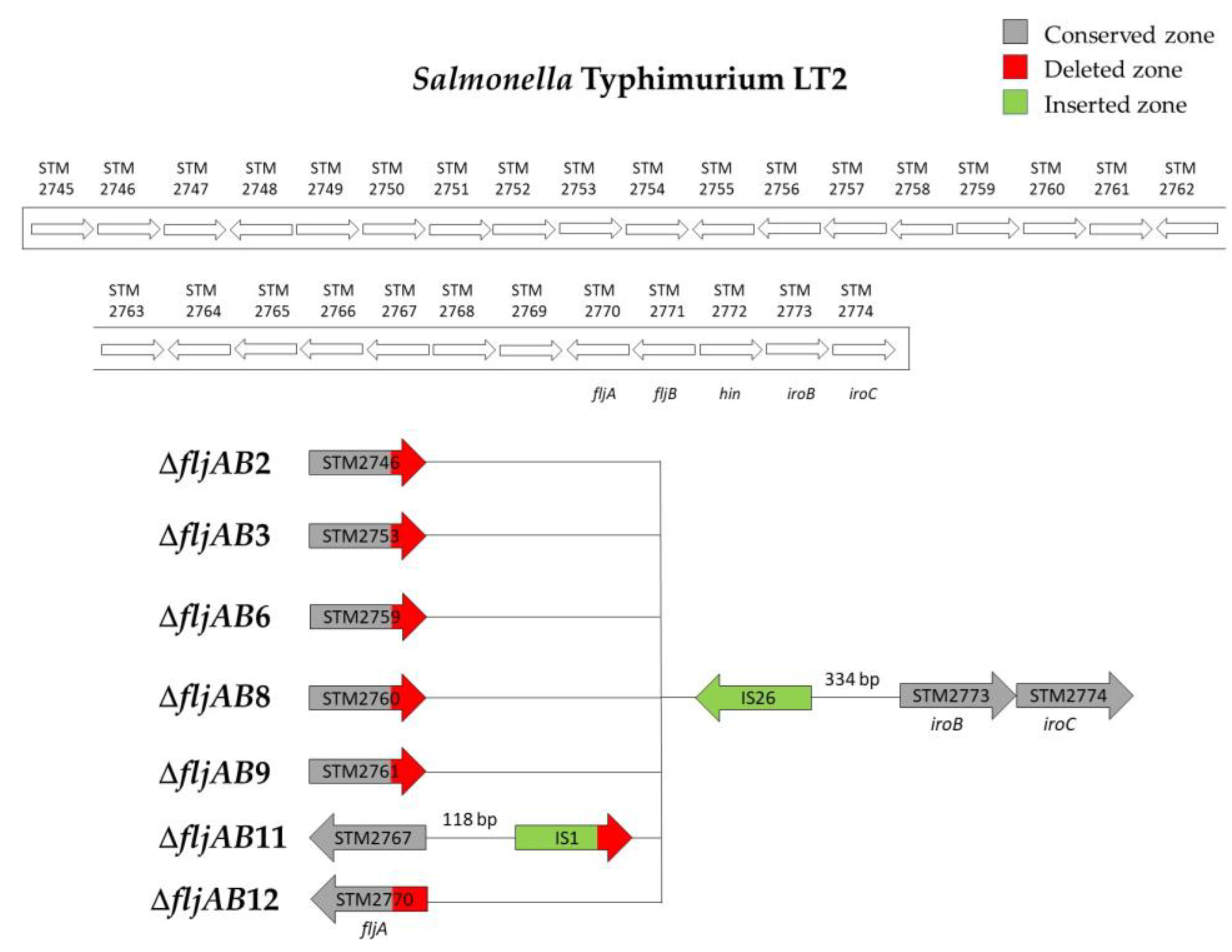
| ΔfljAB2 | ΔfljAB2-A | 1 | 1201 bp of STM2746 | 334 bp upstream from STM2773 (iroB) | 820 bp (one IS26) |
| ΔfljAB2-B | 1 | 1263 bp of STM2746 | |||
| ΔfljAB3 | ΔfljAB3-A | 1 | 177 of STM2753 | 334 bp upstream from STM2773 (iroB) | 820 bp (one IS26) |
| ΔfljAB3-B | 1 | 207 bp of STM2753 | |||
| ΔfljAB3-C | 1 | 353 bp of STM2753 | |||
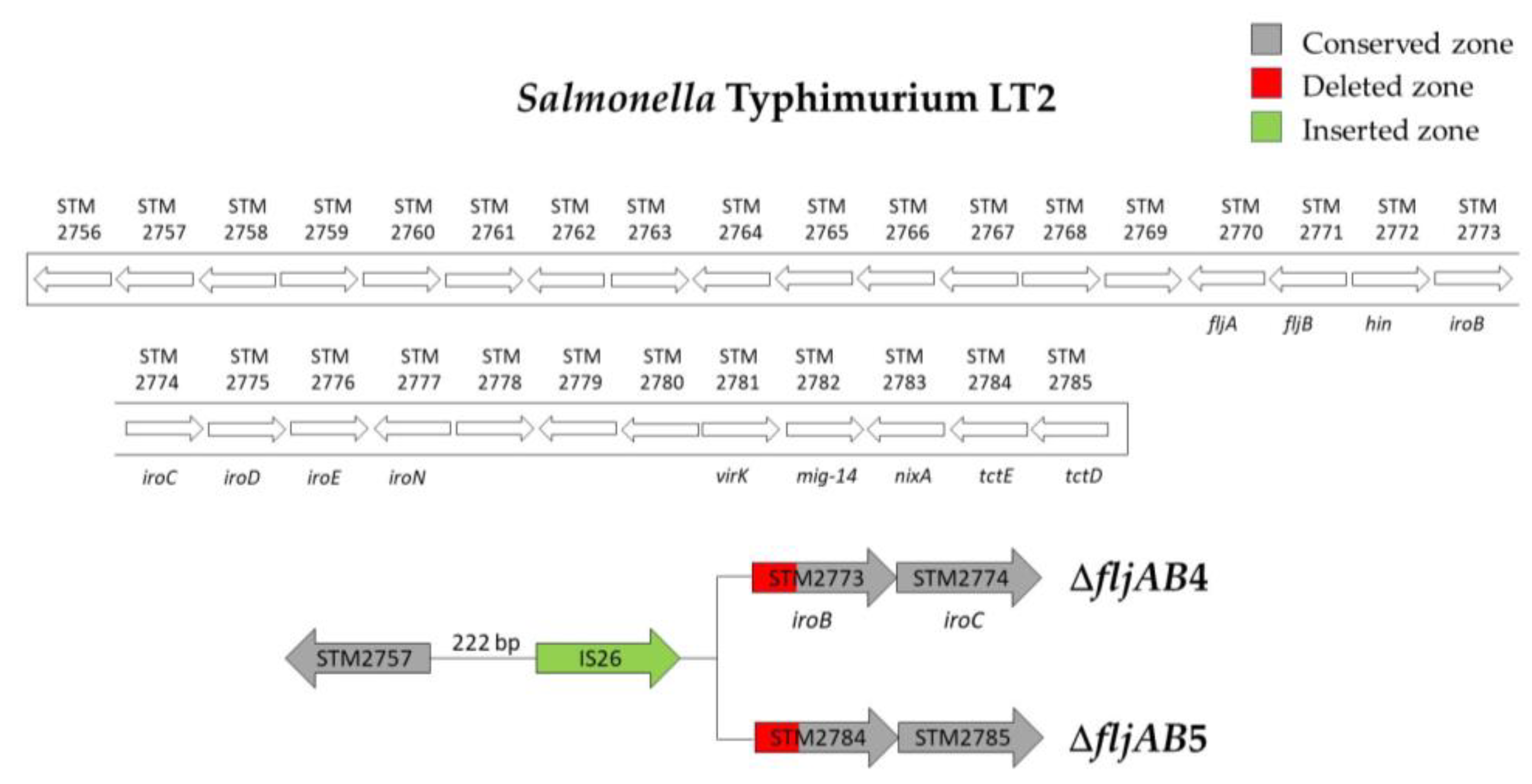
| ΔfljAB4 | 3 | 222 bp downstream of STM2757 | 571 bp of STM2773 (iroB) | 820 bp (one IS26) | |
| ΔfljAB5 | 1 | 222 bp downstream of STM2757 | 848 bp of the STM2784 | 820 bp (one IS26) | |
| ΔfljAB6 | ΔfljAB6-A | 1 | 1079 bp of STM2759 | 334 bp upstream from STM2773 (iroB) | 820 bp (one IS26) |
| ΔfljAB6-B | 2 | 142 bp downstream of the STM2759 | |||
| ΔfljAB7 | 1 | 998 bp downstream of STM2759 | 475 bp of STM2774 (iroC) | 1640 bp (two IS26) | |
| ΔfljAB8 | 7 | 88 bp of STM2760 | 334 bp upstream from STM2773 (iroB) | 820 bp (one IS26) | |
| ΔfljAB9 | 1 | 175 bp of the STM2761 | 334 bp upstream from STM2773 (iroB) | 820 bp (one IS26) | |
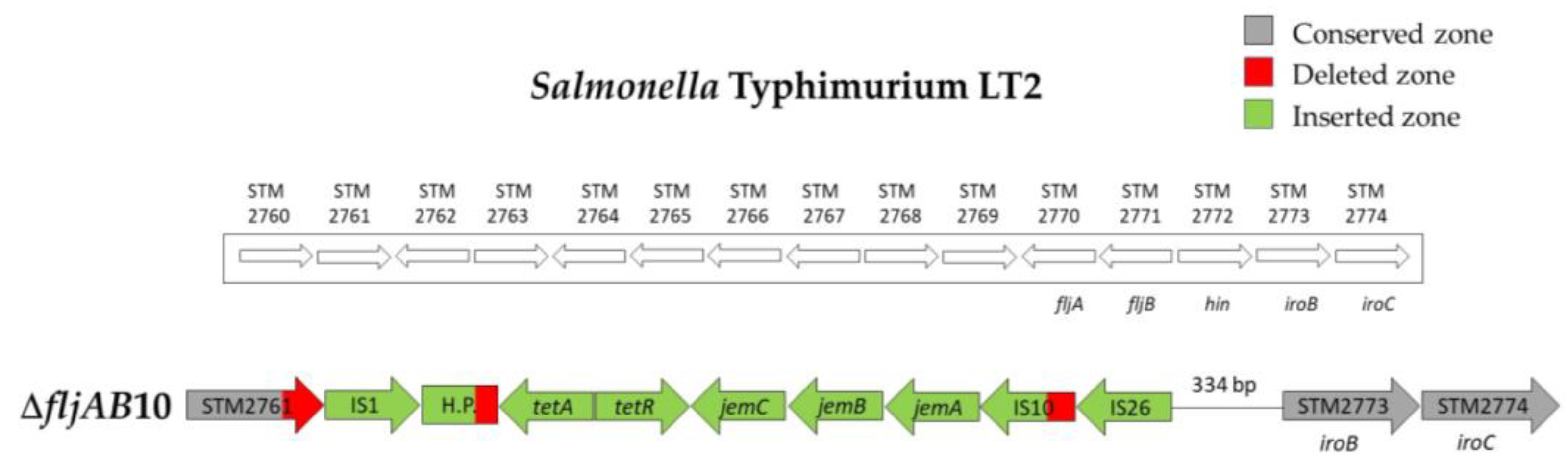
| ΔfljAB10 | 2 | 1125 bp of the STM2761 | 334 bp upstream from STM2773 (iroB) | 7648 bp (see Figure A5) | |
| ΔfljAB11 | 1 | 118 bp downstream of STM2767 | 334 bp upstream from STM2773 (iroB) | 1455 bp (see Figure A2) | |
| ΔfljAB12 | 1 | 155 bp of the fljA gene | 334 bp upstream from STM2773 (iroB) | 820 bp (one IS26) | |
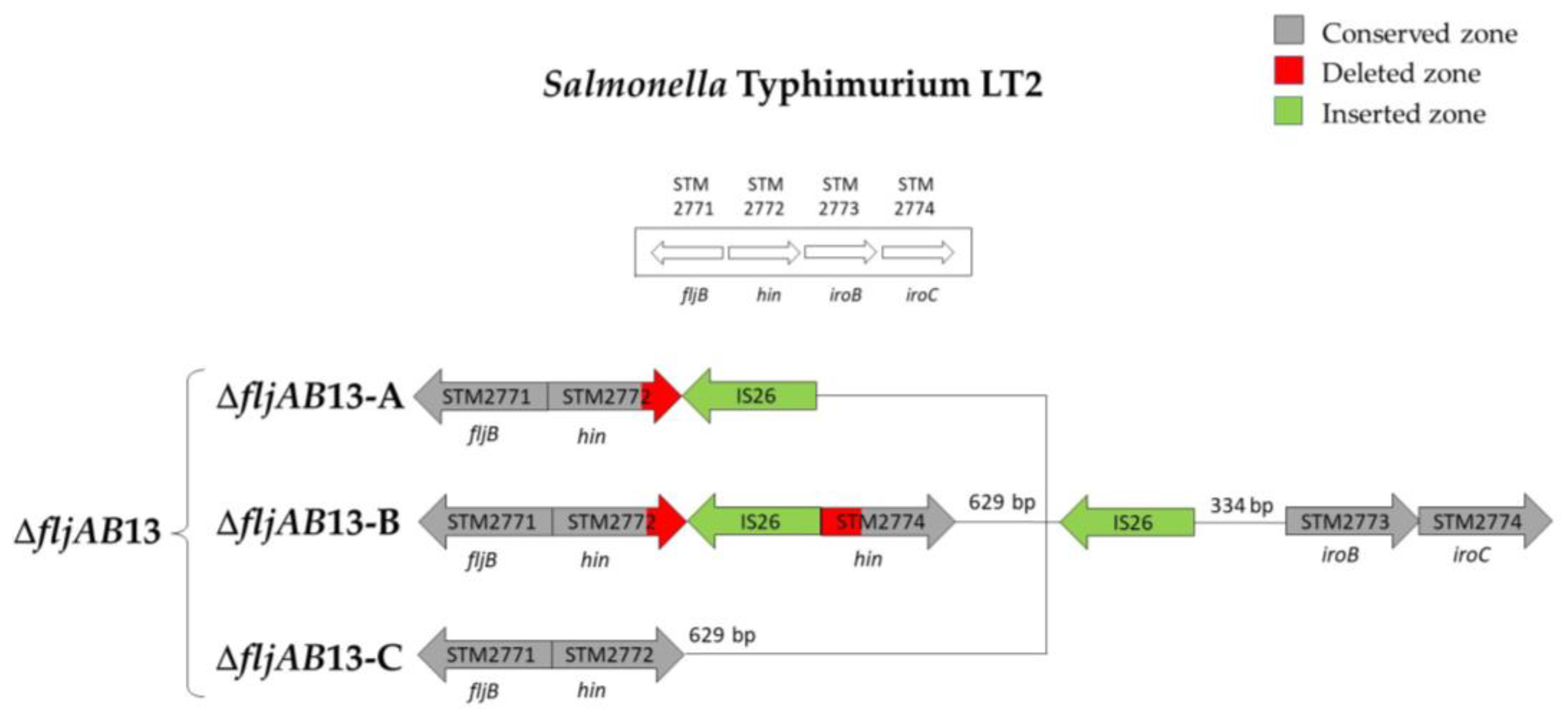
| ΔfljAB13 | ΔfljAB13-A | 1 | fljB-positive | 1640 bp (two IS26) | |
| ΔfljAB13-B | 1 | ||||
| ΔfljAB13-C | 1 | 820 bp (one IS26) | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arrieta-Gisasola, A.; Atxaerandio-Landa, A.; Garrido, V.; Grilló, M.J.; Martínez-Ballesteros, I.; Laorden, L.; Garaizar, J.; Bikandi, J. Genotyping Study of Salmonella 4,[5],12:i:- Monophasic Variant of Serovar Typhimurium and Characterization of the Second-Phase Flagellar Deletion by Whole Genome Sequencing. Microorganisms 2020, 8, 2049. https://doi.org/10.3390/microorganisms8122049
Arrieta-Gisasola A, Atxaerandio-Landa A, Garrido V, Grilló MJ, Martínez-Ballesteros I, Laorden L, Garaizar J, Bikandi J. Genotyping Study of Salmonella 4,[5],12:i:- Monophasic Variant of Serovar Typhimurium and Characterization of the Second-Phase Flagellar Deletion by Whole Genome Sequencing. Microorganisms. 2020; 8(12):2049. https://doi.org/10.3390/microorganisms8122049
Chicago/Turabian StyleArrieta-Gisasola, Ainhoa, Aitor Atxaerandio-Landa, Victoria Garrido, María Jesús Grilló, Ilargi Martínez-Ballesteros, Lorena Laorden, Javier Garaizar, and Joseba Bikandi. 2020. "Genotyping Study of Salmonella 4,[5],12:i:- Monophasic Variant of Serovar Typhimurium and Characterization of the Second-Phase Flagellar Deletion by Whole Genome Sequencing" Microorganisms 8, no. 12: 2049. https://doi.org/10.3390/microorganisms8122049
APA StyleArrieta-Gisasola, A., Atxaerandio-Landa, A., Garrido, V., Grilló, M. J., Martínez-Ballesteros, I., Laorden, L., Garaizar, J., & Bikandi, J. (2020). Genotyping Study of Salmonella 4,[5],12:i:- Monophasic Variant of Serovar Typhimurium and Characterization of the Second-Phase Flagellar Deletion by Whole Genome Sequencing. Microorganisms, 8(12), 2049. https://doi.org/10.3390/microorganisms8122049