Eukaryotic and Prokaryotic Microbiota Interactions



Abstract
1. Introduction
2. Methods
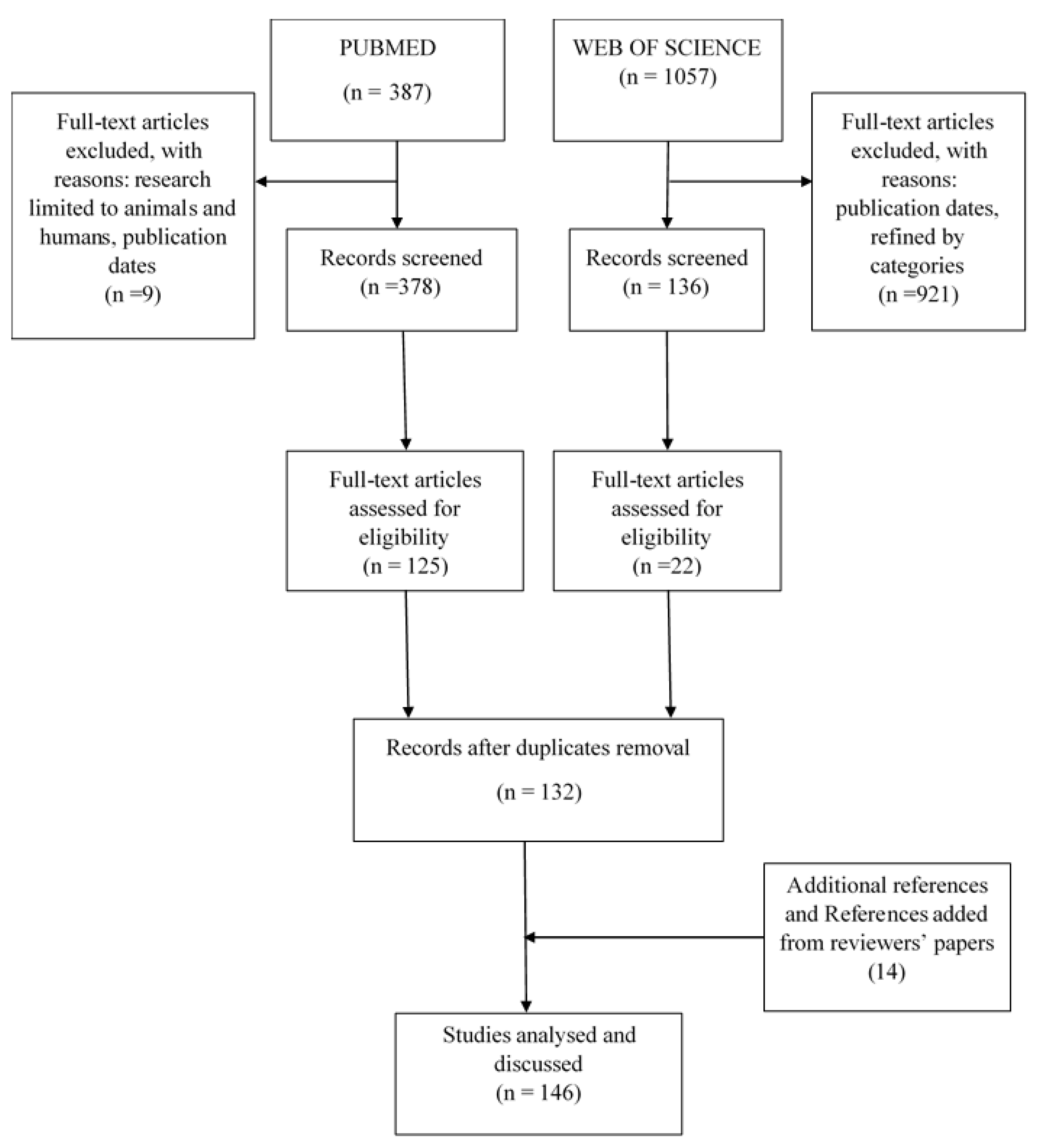
2.1. Literature Search Strategy
2.2. Data Extraction
3. Results
3.1. Different Methods Characterizing Microbiota
3.1.1. Culturomics and Culture-Based Methods
3.1.2. Polymerase Chain Reaction
3.1.3. Denaturing and Temperature Gradient Gel Electrophoresis (DGGE)
3.1.4. Sequencing of 16S rRNA Amplicons or 16S Metabarcoding
3.1.5. Whole Genome Shotgun (WGS)
3.2. Impact of Eukaryotes on Bacterial Community
3.2.1. Protozoa—Bacterial Community Interaction
Impact of Protozoa on Bacterial Community Diversity
Impact of Protozoa on Bacterial Community Structure
3.2.2. Interaction of Helminths and Bacterial Community
Influence of Helminths on Bacterial Community Diversity
Impact of Helminths on Bacterial Community Structure
3.2.3. Interaction between Fungal and Bacterial Communities
Impact of Fungi on Bacterial Community Diversity
Impact of Fungi on Bacterial Community Structure
4. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Virgin, H.W. The virome in mammalian physiology and disease. Cell 2014, 157, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Boyer, M.; Madoui, M.-A.; Gimenez, G.; Scola, B.L.; Raoult, D. Phylogenetic and Phyletic Studies of Informational Genes in Genomes Highlight Existence of a 4th Domain of Life Including Giant Viruses. PLoS ONE 2010, 5, e15530. [Google Scholar] [CrossRef] [PubMed]
- Raoult, D. TRUC or the Need for a New Microbial Classification. INT 2013, 56, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef] [PubMed]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.-Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking Long-Term Dietary Patterns with Gut Microbial Enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef]
- Xu, Z.; Knight, R. Dietary effects on human gut microbiome diversity. Br. J. Nutr. 2015, 113, S1–S5. [Google Scholar] [CrossRef]
- Falony, G.; Joossens, M.; Vieira-Silva, S.; Wang, J.; Darzi, Y.; Faust, K.; Kurilshikov, A.; Bonder, M.J.; Valles-Colomer, M.; Vandeputte, D.; et al. Population-level analysis of gut microbiome variation. Science 2016, 352, 560–564. [Google Scholar] [CrossRef]
- Vandeputte, D.; Falony, G.; Vieira-Silva, S.; Tito, R.Y.; Joossens, M.; Raes, J. Stool consistency is strongly associated with gut microbiota richness and composition, enterotypes and bacterial growth rates. Gut 2016, 65, 57–62. [Google Scholar] [CrossRef]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef] [PubMed]
- Zhernakova, A.; Kurilshikov, A.; Bonder, M.J.; Tigchelaar, E.F.; Schirmer, M.; Vatanen, T.; Mujagic, Z.; Vila, A.V.; Falony, G.; Vieira-Silva, S.; et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 2016, 352, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Francino, M.P. Antibiotics and the Human Gut Microbiome: Dysbioses and Accumulation of Resistances. Front. Microbiol. 2016, 6, 1543. [Google Scholar] [CrossRef] [PubMed]
- Smits, S.A.; Leach, J.; Sonnenburg, E.D.; Gonzalez, C.G.; Lichtman, J.S.; Reid, G.; Knight, R.; Manjurano, A.; Changalucha, J.; Elias, J.E.; et al. Seasonal cycling in the gut microbiome of the Hadza hunter-gatherers of Tanzania. Science 2017, 357, 802–806. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Liu, Y.; Jiao, N.; Xu, B.; Gu, Z.; Xing, T.; Xiong, J. Bacterial community composition and diversity in Kalakuli, an alpine glacial-fed lake in Muztagh Ata of the westernmost Tibetan Plateau. FEMS Microbiol. Ecol. 2017, 93. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, J.K.; Waters, J.L.; Poole, A.C.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.T.; et al. Human Genetics Shape the Gut Microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef]
- Blekhman, R.; Goodrich, J.K.; Huang, K.; Sun, Q.; Bukowski, R.; Bell, J.T.; Spector, T.D.; Keinan, A.; Ley, R.E.; Gevers, D.; et al. Host genetic variation impacts microbiome composition across human body sites. Genome Biol. 2015, 16, 191. [Google Scholar] [CrossRef]
- Goodrich, J.K.; Davenport, E.R.; Beaumont, M.; Jackson, M.A.; Knight, R.; Ober, C.; Spector, T.D.; Bell, J.T.; Clark, A.G.; Ley, R.E. Genetic Determinants of the Gut Microbiome in UK Twins. Cell Host Microbe 2016, 19, 731–743. [Google Scholar] [CrossRef]
- Frank, D.N.; Pace, N.R. Gastrointestinal microbiology enters the metagenomics era. Curr. Opin. Gastroenterol. 2008, 24, 4–10. [Google Scholar] [CrossRef]
- Li, J.; Jia, H.; Cai, X.; Zhong, H.; Feng, Q.; Sunagawa, S.; Arumugam, M.; Kultima, J.R.; Prifti, E.; Nielsen, T.; et al. An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 2014, 32, 834–841. [Google Scholar] [CrossRef]
- Porter, J.R. Antony van Leeuwenhoekl: Tercentenary of His Discovery of Bacteria. Bacteriol. Rev. 1976, 40, 260. [Google Scholar] [CrossRef] [PubMed]
- Lagier, J.-C.; Edouard, S.; Pagnier, I.; Mediannikov, O.; Drancourt, M.; Raoult, D. Current and Past Strategies for Bacterial Culture in Clinical Microbiology. Clin. Microbiol. Rev. 2015, 28, 208–236. [Google Scholar] [CrossRef] [PubMed]
- Fournier, P.-E.; Drancourt, M.; Colson, P.; Rolain, J.-M.; Scola, B.L.; Raoult, D. Modern clinical microbiology: New challenges and solutions. Nat. Rev. Microbiol. 2013, 11, 574–585. [Google Scholar] [CrossRef] [PubMed]
- Dave, M.; Purohit, T.; Razonable, R.; Loftus, E.V. Opportunistic Infections Due to Inflammatory Bowel Disease Therapy. Inflamm. Bowel Dis. 2014, 20, 196–212. [Google Scholar] [CrossRef] [PubMed]
- Hugon, P.; Dufour, J.-C.; Colson, P.; Fournier, P.-E.; Sallah, K.; Raoult, D. A comprehensive repertoire of prokaryotic species identified in human beings. Lancet Infect. Dis. 2015, 15, 1211–1219. [Google Scholar] [CrossRef]
- Lagier, J.-C.; Drancourt, M.; Charrel, R.; Bittar, F.; La Scola, B.; Ranque, S.; Raoult, D. Many More Microbes in Humans: Enlarging the Microbiome Repertoire. Clin. Infect. Dis. 2017, 65, S20–S29. [Google Scholar] [CrossRef][Green Version]
- O’Brien Andersen, L.; Karim, A.B.; Roager, H.M.; Vigsnæs, L.K.; Krogfelt, K.A.; Licht, T.R.; Stensvold, C.R. Associations between common intestinal parasites and bacteria in humans as revealed by qPCR. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 1427–1431. [Google Scholar] [CrossRef]
- Iebba, V.; Santangelo, F.; Totino, V.; Pantanella, F.; Monsia, A.; Di Cristanziano, V.; Di Cave, D.; Schippa, S.; Berrilli, F.; D’Alfonso, R. Gut microbiota related to Giardia duodenalis, Entamoeba spp. and Blastocystis hominis infections in humans from Côte d’Ivoire. J. Infect. Dev. Ctries. 2016, 10, 1035–1041. [Google Scholar] [CrossRef]
- Audebert, C.; Even, G.; Cian, A.; Loywick, A.; Merlin, S.; Viscogliosi, E.; Chabé, M.; Blastocystis Investigation Group. Colonization with the enteric protozoa Blastocystis is associated with increased diversity of human gut bacterial microbiota. Sci. Rep. 2016, 6, 25255. [Google Scholar] [CrossRef]
- Beatty, J.K.; Akierman, S.V.; Motta, J.-P.; Muise, S.; Workentine, M.L.; Harrison, J.J.; Bhargava, A.; Beck, P.L.; Rioux, K.P.; McKnight, G.W.; et al. Giardia duodenalis induces pathogenic dysbiosis of human intestinal microbiota biofilms. Int. J. Parasitol. 2017, 47, 311–326. [Google Scholar] [CrossRef]
- Gilchrist, C.A.; Petri, S.E.; Schneider, B.N.; Reichman, D.J.; Jiang, N.; Begum, S.; Watanabe, K.; Jansen, C.S.; Elliott, K.P.; Burgess, S.L.; et al. Role of the Gut Microbiota of Children in Diarrhea Due to the Protozoan Parasite Entamoeba histolytica. J. Infect. Dis. 2016, 213, 1579–1585. [Google Scholar] [CrossRef] [PubMed]
- Yooseph, S.; Kirkness, E.F.; Tran, T.M.; Harkins, D.M.; Jones, M.B.; Torralba, M.G.; O’Connell, E.; Nutman, T.B.; Doumbo, S.; Doumbo, O.K.; et al. Stool microbiota composition is associated with the prospective risk of Plasmodium falciparum infection. BMC Genom. 2015, 16, 631. [Google Scholar] [CrossRef] [PubMed]
- Villarino, N.F.; LeCleir, G.R.; Denny, J.E.; Dearth, S.P.; Harding, C.L.; Sloan, S.S.; Gribble, J.L.; Campagna, S.R.; Wilhelm, S.W.; Schmidt, N.W. Composition of the gut microbiota modulates the severity of malaria. Proc. Natl. Acad. Sci. USA 2016, 113, 2235–2240. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-A.; Liang, C.; Lin, C.-L.; Hsiao, C.-T.; Peng, C.-T.; Lin, H.-C.; Chang, J.-G. Impact of Enterobius vermicularis infection and mebendazole treatment on intestinal microbiota and host immune response. PLoS Negl. Trop. Dis. 2017, 11, e0005963. [Google Scholar] [CrossRef]
- Holm, J.B.; Sorobetea, D.; Kiilerich, P.; Ramayo-Caldas, Y.; Estellé, J.; Ma, T.; Madsen, L.; Kristiansen, K.; Svensson-Frej, M. Chronic Trichuris muris Infection Decreases Diversity of the Intestinal Microbiota and Concomitantly Increases the Abundance of Lactobacilli. PLoS ONE 2015, 10, e0125495. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, T.P.; Rathnayaka, Y.; Perera, P.K.; Peachey, L.E.; Nolan, M.J.; Krause, L.; Rajakaruna, R.S.; Cantacessi, C. Infections by human gastrointestinal helminths are associated with changes in faecal microbiota diversity and composition. PLoS ONE 2017, 12, e0184719. [Google Scholar] [CrossRef] [PubMed]
- Lamendella, R.; Wright, J.R.; Hackman, J.; McLimans, C.; Toole, D.R.; Bernard Rubio, W.; Drucker, R.; Wong, H.T.; Sabey, K.; Hegarty, J.P.; et al. Antibiotic Treatments for Clostridium difficile Infection Are Associated with Distinct Bacterial and Fungal Community Structures. mSphere 2018, 3, e00572-17. [Google Scholar] [CrossRef] [PubMed]
- Liguori, G.; Lamas, B.; Richard, M.L.; Brandi, G.; da Costa, G.; Hoffmann, T.W.; Di Simone, M.P.; Calabrese, C.; Poggioli, G.; Langella, P.; et al. Fungal Dysbiosis in Mucosa-associated Microbiota of Crohn’s Disease Patients. J. Crohn’s Colitis 2016, 10, 296–305. [Google Scholar] [CrossRef]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the Human Intestinal Microbial Flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef]
- Costa, M.; Weese, J.S. Methods and basic concepts for microbiota assessment. Vet. J. 2019, 249, 10–15. [Google Scholar] [CrossRef]
- Lagier, J.-C.; Hugon, P.; Khelaifia, S.; Fournier, P.-E.; La Scola, B.; Raoult, D. The Rebirth of Culture in Microbiology through the Example of Culturomics to Study Human Gut Microbiota. Clin. Microbiol. Rev. 2015, 28, 237–264. [Google Scholar] [CrossRef] [PubMed]
- Muyzer, G.; Smalla, K. Application of denaturing gradient gel electrophoresis (DGGE) and temperature gradient gel electrophoresis (TGGE) in microbial ecology. Antonie Van Leeuwenhoek 1998, 73, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Green, S.J.; Leigh, M.B.; Neufeld, J.D. Denaturing Gradient Gel Electrophoresis (DGGE) for Microbial Community Analysis. In Hydrocarbon and Lipid Microbiology Protocols: Microbial Quantitation, Community Profiling and Array Approaches; McGenity, T.J., Timmis, K.N., Nogales, B., Eds.; Springer Protocols Handbooks; Springer: Berlin/Heidelberg, Germany, 2017; pp. 77–99. ISBN 978-3-662-52778-8. [Google Scholar]
- Meroth, C.B.; Walter, J.; Hertel, C.; Brandt, M.J.; Hammes, W.P. Monitoring the bacterial population dynamics in sourdough fermentation processes by using PCR-denaturing gradient gel electrophoresis. Appl. Environ. Microbiol. 2003, 69, 475–482. [Google Scholar] [CrossRef]
- de Souza, F.A.; Kowalchuk, G.A.; Leeflang, P.; van Veen, J.A.; Smit, E. PCR-Denaturing Gradient Gel Electrophoresis Profiling of Inter- and Intraspecies 18S rRNA Gene Sequence Heterogeneity Is an Accurate and Sensitive Method To Assess Species Diversity of Arbuscular Mycorrhizal Fungi of the Genus Gigaspora. Appl. Environ. Microbiol. 2004, 70, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Ariefdjohan, M.W.; Savaiano, D.A.; Nakatsu, C.H. Comparison of DNA extraction kits for PCR-DGGE analysis of human intestinal microbial communities from fecal specimens. Nutr. J. 2010, 9, 23. [Google Scholar] [CrossRef]
- Handschur, M.; Pinar, G.; Gallist, B.; Lubitz, W.; Haslberger, A.G. Culture free DGGE and cloning based monitoring of changes in bacterial communities of salad due to processing. Food Chem. Toxicol. 2005, 43, 1595–1605. [Google Scholar] [CrossRef] [PubMed]
- Riemann, L.; Winding, A. Community Dynamics of Free-living and Particle-associated Bacterial Assemblages during a Freshwater Phytoplankton Bloom. Microb. Ecol. 2001, 42, 274–285. [Google Scholar] [CrossRef]
- Heuer, H.; Hartung, K.; Wieland, G.; Kramer, I.; Smalla, K. Polynucleotide Probes That Target a Hypervariable Region of 16S rRNA Genes to Identify Bacterial Isolates Corresponding to Bands of Community Fingerprints. Appl. Environ. Microbiol. 1999, 65, 1045–1049. [Google Scholar] [CrossRef]
- Theron, J.; Cloete, T.E. Molecular Techniques for Determining Microbial Diversity and Community Structure in Natural Environments. Crit. Rev. Microbiol. 2000, 26, 37–57. [Google Scholar] [CrossRef]
- Wintzingerode, F.V.; Göbel, U.B.; Stackebrandt, E. Determination of microbial diversity in environmental samples: Pitfalls of PCR-based rRNA analysis. FEMS Microbiol. Rev. 1997, 21, 213–229. [Google Scholar] [CrossRef]
- Gelsomino, A.; Keijzer-Wolters, A.C.; Cacco, G.; van Elsas, J.D. Assessment of bacterial community structure in soil by polymerase chain reaction and denaturing gradient gel electrophoresis. J. Microbiol. Methods 1999, 38, 1–15. [Google Scholar] [CrossRef]
- Sanschagrin, S.; Yergeau, E. Next-generation Sequencing of 16S Ribosomal RNA Gene Amplicons. J. Vis. Exp. 2014, 90, e51709. [Google Scholar] [CrossRef] [PubMed]
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Nat. Biotechnol. 2008, 26, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Blazej, R.G.; Kumaresan, P.; Mathies, R.A. Microfabricated bioprocessor for integrated nanoliter-scale Sanger DNA sequencing. Proc. Natl. Acad. Sci. USA 2006, 103, 7240–7245. [Google Scholar] [CrossRef] [PubMed]
- Gresham, D.; Dunham, M.J.; Botstein, D. Comparing whole genomes using DNA microarrays. Nat. Rev. Genet. 2008, 9, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Soni, G.V.; Meller, A. Progress toward Ultrafast DNA Sequencing Using Solid-State Nanopores. Clin. Chem. 2007, 53, 1996–2001. [Google Scholar] [CrossRef] [PubMed]
- Shendure, J. Accurate Multiplex Polony Sequencing of an Evolved Bacterial Genome. Science 2005, 309, 1728–1732. [Google Scholar] [CrossRef]
- Healy, K. Nanopore-based single-molecule DNA analysis. Nanomedicine 2007, 2, 459–481. [Google Scholar] [CrossRef]
- Salipante, S.J.; Kawashima, T.; Rosenthal, C.; Hoogestraat, D.R.; Cummings, L.A.; Sengupta, D.J.; Harkins, T.T.; Cookson, B.T.; Hoffman, N.G. Performance Comparison of Illumina and Ion Torrent Next-Generation Sequencing Platforms for 16S rRNA-Based Bacterial Community Profiling. Appl. Environ. Microbiol. 2014, 80, 7583–7591. [Google Scholar] [CrossRef]
- Kuczynski, J.; Lauber, C.L.; Walters, W.A.; Parfrey, L.W.; Clemente, J.C.; Gevers, D.; Knight, R. Experimental and analytical tools for studying the human microbiome. Nat. Rev. Genet. 2011, 13, 47–58. [Google Scholar] [CrossRef]
- Luo, C.; Rodriguez-R, L.M.; Konstantinidis, K.T. A User’s Guide to Quantitative and Comparative Analysis of Metagenomic Datasets. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2013; Volume 531, pp. 525–547. ISBN 978-0-12-407863-5. [Google Scholar]
- Luo, C.; Rodriguez-R, L.M.; Konstantinidis, K.T. MyTaxa: An advanced taxonomic classifier for genomic and metagenomic sequences. Nucleic Acids Res. 2014, 42, e73. [Google Scholar] [CrossRef] [PubMed]
- Sims, D.; Sudbery, I.; Ilott, N.E.; Heger, A.; Ponting, C.P. Sequencing depth and coverage: Key considerations in genomic analyses. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, R.; Rani, A.; Metwally, A.; McGee, H.S.; Perkins, D.L. Analysis of the microbiome: Advantages of whole genome shotgun versus 16S amplicon sequencing. Biochem. Biophys. Res. Commun. 2016, 469, 967–977. [Google Scholar] [CrossRef] [PubMed]
- van Nimwegen, K.J.M.; van Soest, R.A.; Veltman, J.A.; Nelen, M.R.; van der Wilt, G.J.; Vissers, L.E.L.M.; Grutters, J.P.C. Is the $1000 Genome as Near as We Think? A Cost Analysis of Next-Generation Sequencing. Clin. Chem. 2016, 62, 1458–1464. [Google Scholar] [CrossRef] [PubMed]
- Marizzoni, M.; Gurry, T.; Provasi, S.; Greub, G.; Lopizzo, N.; Ribaldi, F.; Festari, C.; Mazzelli, M.; Mombelli, E.; Salvatore, M.; et al. Comparison of Bioinformatics Pipelines and Operating Systems for the Analyses of 16S rRNA Gene Amplicon Sequences in Human Fecal Samples. Front. Microbiol. 2020, 11, 1262. [Google Scholar] [CrossRef]
- Corless, C.E.; Guiver, M.; Borrow, R.; Edwards-Jones, V.; Kaczmarski, E.B.; Fox, A.J. Contamination and Sensitivity Issues with a Real-Time Universal 16S rRNA PCR. J. Clin. Microbiol. 2000, 38, 1747–1752. [Google Scholar] [CrossRef]
- Pereira-Marques, J.; Hout, A.; Ferreira, R.M.; Weber, M.; Pinto-Ribeiro, I.; van Doorn, L.-J.; Knetsch, C.W.; Figueiredo, C. Impact of Host DNA and Sequencing Depth on the Taxonomic Resolution of Whole Metagenome Sequencing for Microbiome Analysis. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef]
- Poretsky, R.; Rodriguez-R, L.M.; Luo, C.; Tsementzi, D.; Konstantinidis, K.T. Strengths and Limitations of 16S rRNA Gene Amplicon Sequencing in Revealing Temporal Microbial Community Dynamics. PLoS ONE 2014, 9, e93827. [Google Scholar] [CrossRef]
- Jones, M.B.; Highlander, S.K.; Anderson, E.L.; Li, W.; Dayrit, M.; Klitgord, N.; Fabani, M.M.; Seguritan, V.; Green, J.; Pride, D.T.; et al. Library preparation methodology can influence genomic and functional predictions in human microbiome research. Proc. Natl. Acad. Sci. USA 2015, 112, 14024–14029. [Google Scholar] [CrossRef]
- Chen, K.; Pachter, L. Bioinformatics for Whole-Genome Shotgun Sequencing of Microbial Communities. PLoS Comput. Biol. 2005, 1, e24. [Google Scholar] [CrossRef]
- Ras, R.; Huynh, K.; Desoky, E.; Badawy, A.; Widmer, G. Perturbation of the intestinal microbiota of mice infected with Cryptosporidium parvum. Int. J. Parasitol. 2015, 45, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, S.; Doğan, İ.; Doğruman-Al, F.; Nalbantoğlu, U.; Üstek, D.; Sarzhanov, F.; Yildirim, S. Association of Enteric Protist Blastocystis spp. and Gut Microbiota with Hepatic Encephalopathy. J. Gastrointest. Liver Dis. 2016, 25, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Morton, E.R.; Lynch, J.; Froment, A.; Lafosse, S.; Heyer, E.; Przeworski, M.; Blekhman, R.; Ségurel, L. Variation in Rural African Gut Microbiota Is Strongly Correlated with Colonization by Entamoeba and Subsistence. PLoS Genet. 2015, 11, e1005658. [Google Scholar] [CrossRef] [PubMed]
- Kelly, P.H.; Bahr, S.M.; Serafim, T.D.; Ajami, N.J.; Petrosino, J.F.; Meneses, C.; Kirby, J.R.; Valenzuela, J.G.; Kamhawi, S.; Wilson, M.E. The Gut Microbiome of the Vector Lutzomyia longipalpis Is Essential for Survival of Leishmania infantum. MBio 2017, 8. [Google Scholar] [CrossRef]
- Heimesaat, M.M.; Dunay, I.R.; Alutis, M.; Fischer, A.; Möhle, L.; Göbel, U.B.; Kühl, A.A.; Bereswill, S. Nucleotide-Oligomerization-Domain-2 Affects Commensal Gut Microbiota Composition and Intracerebral Immunopathology in Acute Toxoplasma gondii Induced Murine Ileitis. PLoS ONE 2014, 9, e105120. [Google Scholar] [CrossRef]
- Brotman, R.M.; Bradford, L.L.; Conrad, M.; Gajer, P.; Ault, K.; Peralta, L.; Forney, L.J.; Carlton, J.M.; Abdo, Z.; Ravel, J. Association between Trichomonas vaginalis and vaginal bacterial community composition among reproductive-age women. Sex. Transm. Dis. 2012, 39, 807–812. [Google Scholar] [CrossRef]
- Šlapeta, J.; Dowd, S.E.; Alanazi, A.D.; Westman, M.E.; Brown, G.K. Differences in the faecal microbiome of non-diarrhoeic clinically healthy dogs and cats associated with Giardia duodenalis infection: Impact of hookworms and coccidia. Int. J. Parasitol. 2015, 45, 585–594. [Google Scholar] [CrossRef]
- Kodio, A.; Coulibaly, D.; Koné, A.K.; Konaté, S.; Doumbo, S.; Guindo, A.; Bittar, F.; Gouriet, F.; Raoult, D.; Thera, M.A.; et al. Blastocystis Colonization Is Associated with Increased Diversity and Altered Gut Bacterial Communities in Healthy Malian Children. Microorganisms 2019, 7, 649. [Google Scholar] [CrossRef]
- Oliveira, B.C.M.; Widmer, G. Probiotic Product Enhances Susceptibility of Mice to Cryptosporidiosis. Appl. Environ. Microbiol. 2018, 84, e01408-18. [Google Scholar] [CrossRef]
- Barash, N.R.; Maloney, J.G.; Singer, S.M.; Dawson, S.C. Giardia Alters Commensal Microbial Diversity throughout the Murine Gut. Infect. Immun. 2017, 85, e00948-16. [Google Scholar] [CrossRef]
- Hatter, J.A.; Kouche, Y.M.; Melchor, S.J.; Ng, K.; Bouley, D.M.; Boothroyd, J.C.; Ewald, S.E. Toxoplasma gondii infection triggers chronic cachexia and sustained commensal dysbiosis in mice. PLoS ONE 2018, 13, e0204895. [Google Scholar] [CrossRef] [PubMed]
- Perez, P.F.; Minnaard, J.; Rouvet, M.; Knabenhans, C.; Brassart, D.; De Antoni, G.L.; Schiffrin, E.J. Inhibition of Giardia intestinalis by Extracellular Factors from Lactobacilli: An In Vitro Study. Appl. Environ. Microbiol. 2001, 67, 5037–5042. [Google Scholar] [CrossRef] [PubMed]
- Alamer, E.; Carpio, V.H.; Ibitokou, S.A.; Kirtley, M.L.; Phoenix, I.R.; Opata, M.M.; Wilson, K.D.; Cong, Y.; Dann, S.M.; Chopra, A.K.; et al. Dissemination of non-typhoidal Salmonella during Plasmodium chabaudi infection affects anti-malarial immunity. Parasitol. Res. 2019, 118, 2277–2285. [Google Scholar] [CrossRef] [PubMed]
- Lappan, R.; Classon, C.; Kumar, S.; Singh, O.P.; de Almeida, R.V.; Chakravarty, J.; Kumari, P.; Kansal, S.; Sundar, S.; Blackwell, J.M. Meta-taxonomic analysis of prokaryotic and eukaryotic gut flora in stool samples from visceral leishmaniasis cases and endemic controls in Bihar State India. PLoS Negl. Trop. Dis. 2019, 13, e0007444. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Li, R.W.; Li, W.; Beshah, E.; Dawson, H.D.; Urban, J.F. Worm Burden-Dependent Disruption of the Porcine Colon Microbiota by Trichuris suis Infection. PLoS ONE 2012, 7, e0035470. [Google Scholar] [CrossRef]
- Li, R.W.; Wu, S.; Li, W.; Navarro, K.; Couch, R.D.; Hill, D.; Urban, J.F. Alterations in the Porcine Colon Microbiota Induced by the Gastrointestinal Nematode Trichuris suis. Infect. Immun. 2012, 80, 2150–2157. [Google Scholar] [CrossRef]
- Cooper, P.; Walker, A.W.; Reyes, J.; Chico, M.; Salter, S.J.; Vaca, M.; Parkhill, J. Patent Human Infections with the Whipworm, Trichuris trichiura, Are Not Associated with Alterations in the Faecal Microbiota. PLoS ONE 2013, 8, e76573. [Google Scholar] [CrossRef]
- Lee, S.C.; Tang, M.S.; Lim, Y.A.L.; Choy, S.H.; Kurtz, Z.D.; Cox, L.M.; Gundra, U.M.; Cho, I.; Bonneau, R.; Blaser, M.J.; et al. Helminth Colonization Is Associated with Increased Diversity of the Gut Microbiota. PLoS Negl. Trop. Dis. 2014, 8, e2880. [Google Scholar] [CrossRef]
- Rosa, B.A.; Supali, T.; Gankpala, L.; Djuardi, Y.; Sartono, E.; Zhou, Y.; Fischer, K.; Martin, J.; Tyagi, R.; Bolay, F.K.; et al. Differential human gut microbiome assemblages during soil-transmitted helminth infections in Indonesia and Liberia. Microbiome 2018, 6, 33. [Google Scholar] [CrossRef]
- Houlden, A.; Hayes, K.S.; Bancroft, A.J.; Worthington, J.J.; Wang, P.; Grencis, R.K.; Roberts, I.S. Chronic Trichuris muris Infection in C57BL/6 Mice Causes Significant Changes in Host Microbiota and Metabolome: Effects Reversed by Pathogen Clearance. PLoS ONE 2015, 10, e0125945. [Google Scholar] [CrossRef]
- Williams, A.R.; Krych, L.; Ahmad, H.F.; Nejsum, P.; Skovgaard, K.; Nielsen, D.S.; Thamsborg, S.M. A polyphenol-enriched diet and Ascaris suum infection modulate mucosal immune responses and gut microbiota composition in pigs. PLoS ONE 2017, 12, e0186546. [Google Scholar] [CrossRef] [PubMed]
- Wegener Parfrey, L.; Jirků, M.; Šíma, R.; Jalovecká, M.; Sak, B.; Grigore, K.; Jirků Pomajbíková, K. A benign helminth alters the host immune system and the gut microbiota in a rat model system. PLoS ONE 2017, 12, e0182205. [Google Scholar] [CrossRef] [PubMed]
- McKenney, E.A.; Williamson, L.; Yoder, A.D.; Rawls, J.F.; Bilbo, S.D.; Parker, W. Alteration of the rat cecal microbiome during colonization with the helminth Hymenolepis diminuta. Gut Microbes 2015, 6, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Giacomin, P.; Zakrzewski, M.; Croese, J.; Su, X.; Sotillo, J.; McCann, L.; Navarro, S.; Mitreva, M.; Krause, L.; Loukas, A.; et al. Experimental hookworm infection and escalating gluten challenges are associated with increased microbial richness in celiac subjects. Sci. Rep. 2015, 5, 13797. [Google Scholar] [CrossRef]
- Vicente, C.S.L.; Ozawa, S.; Hasegawa, K. Composition of the Cockroach Gut Microbiome in the Presence of Parasitic Nematodes. Microbes Environ. 2016, 31, 314–320. [Google Scholar] [CrossRef]
- El-Ashram, S.; Suo, X. Exploring the microbial community (microflora) associated with ovine Haemonchus contortus (macroflora) field strains. Sci. Rep. 2017, 7, 70. [Google Scholar] [CrossRef]
- Kay, G.L.; Millard, A.; Sergeant, M.J.; Midzi, N.; Gwisai, R.; Mduluza, T.; Ivens, A.; Nausch, N.; Mutapi, F.; Pallen, M. Differences in the Faecal Microbiome in Schistosoma haematobium Infected Children vs. Uninfected Children. PLoS Negl. Trop. Dis. 2015, 9, e0003861. [Google Scholar] [CrossRef]
- Peachey, L.E.; Molena, R.A.; Jenkins, T.P.; Di Cesare, A.; Traversa, D.; Hodgkinson, J.E.; Cantacessi, C. The relationships between faecal egg counts and gut microbial composition in UK Thoroughbreds infected by cyathostomins. Int. J. Parasitol. 2018, 48, 403–412. [Google Scholar] [CrossRef]
- Rausch, S.; Held, J.; Fischer, A.; Heimesaat, M.M.; Kühl, A.A.; Bereswill, S.; Hartmann, S. Small Intestinal Nematode Infection of Mice Is Associated with Increased Enterobacterial Loads alongside the Intestinal Tract. PLoS ONE 2013, 8, e74026. [Google Scholar] [CrossRef]
- Kreisinger, J.; Bastien, G.; Hauffe, H.C.; Marchesi, J.; Perkins, S.E. Interactions between multiple helminths and the gut microbiota in wild rodents. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20140295. [Google Scholar] [CrossRef]
- Cattadori, I.M.; Sebastian, A.; Hao, H.; Katani, R.; Albert, I.; Eilertson, K.E.; Kapur, V.; Pathak, A.; Mitchell, S. Impact of Helminth Infections and Nutritional Constraints on the Small Intestine Microbiota. PLoS ONE 2016, 11, e0159770. [Google Scholar] [CrossRef] [PubMed]
- Duarte, A.M.; Jenkins, T.P.; Latrofa, M.S.; Giannelli, A.; Papadopoulos, E.; de Carvalho, L.M.; Nolan, M.J.; Otranto, D.; Cantacessi, C. Helminth infections and gut microbiota—A feline perspective. Parasites Vectors 2016, 9, 625. [Google Scholar] [CrossRef] [PubMed]
- Schneeberger, P.H.H.; Coulibaly, J.T.; Panic, G.; Daubenberger, C.; Gueuning, M.; Frey, J.E.; Keiser, J. Investigations on the interplays between Schistosoma mansoni, praziquantel and the gut microbiome. Parasit Vectors 2018, 11, 168. [Google Scholar] [CrossRef] [PubMed]
- Broadhurst, M.J.; Ardeshir, A.; Kanwar, B.; Mirpuri, J.; Gundra, U.M.; Leung, J.M.; Wiens, K.E.; Vujkovic-Cvijin, I.; Kim, C.C.; Yarovinsky, F.; et al. Therapeutic Helminth Infection of Macaques with Idiopathic Chronic Diarrhea Alters the Inflammatory Signature and Mucosal Microbiota of the Colon. PLoS Pathog. 2012, 8, e1003000. [Google Scholar] [CrossRef]
- Walk, S.T.; Blum, A.M.; Ewing, S.A.-S.; Weinstock, J.V.; Young, V.B. Alteration of the murine gut microbiota during infection with the parasitic helminth, Heligmosomoides polygyrus. Inflamm. Bowel Dis. 2010, 16, 1841–1849. [Google Scholar] [CrossRef]
- Shimokawa, C.; Obi, S.; Shibata, M.; Olia, A.; Imai, T.; Suzue, K.; Hisaeda, H. Suppression of Obesity by an Intestinal Helminth through Interactions with Intestinal Microbiota. Infect. Immun. 2019, 87, e00042-19. [Google Scholar] [CrossRef]
- Jenkins, T.P.; Peachey, L.E.; Ajami, N.J.; MacDonald, A.S.; Hsieh, M.H.; Brindley, P.J.; Cantacessi, C.; Rinaldi, G. Schistosoma mansoni infection is associated with quantitative and qualitative modifications of the mammalian intestinal microbiota. Sci. Rep. 2018, 8, 12072. [Google Scholar] [CrossRef]
- Williamson, L.L.; McKenney, E.A.; Holzknecht, Z.E.; Belliveau, C.; Rawls, J.F.; Poulton, S.; Parker, W.; Bilbo, S.D. Got worms? Perinatal exposure to helminths prevents persistent immune sensitization and cognitive dysfunction induced by early-life infection. Brain Behav. Immun. 2016, 51, 14–28. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, F.; Urban, J.F.; Paerewijck, O.; Geldhof, P.; Li, R.W. Ascaris suum infection was associated with a worm-independent reduction in microbial diversity and altered metabolic potential in the porcine gut microbiome. Int. J. Parasitol. 2019, 49, 247–256. [Google Scholar] [CrossRef]
- Li, S.; Sha, Z.; Wang, X.; Bu, Z.; Wang, L.; Guan, X.; Lang, X.; Wang, X. Yeast Surface Display of Escherichia coli Enterotoxin and Its Effects of Intestinal Microflora and Mucosal Immunity. Curr. Microbiol. 2017, 74, 854–862. [Google Scholar] [CrossRef]
- Pinheiro, I.; Robinson, L.; Verhelst, A.; Marzorati, M.; Winkens, B.; den Abbeele, P.V.; Possemiers, S. A yeast fermentate improves gastrointestinal discomfort and constipation by modulation of the gut microbiome: Results from a randomized double-blind placebo-controlled pilot trial. BMC Complement. Altern. Med. 2017, 17, 441. [Google Scholar] [CrossRef] [PubMed]
- Borgo, F.; Verduci, E.; Riva, A.; Lassandro, C.; Riva, E.; Morace, G.; Borghi, E. Relative Abundance in Bacterial and Fungal Gut Microbes in Obese Children: A Case Control Study. Child. Obes. 2017, 13, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Zeber-Lubecka, N.; Kulecka, M.; Ambrozkiewicz, F.; Paziewska, A.; Lechowicz, M.; Konopka, E.; Majewska, U.; Borszewska-Kornacka, M.; Mikula, M.; Cukrowska, B.; et al. Effect of Saccharomyces boulardii and Mode of Delivery on the Early Development of the Gut Microbial Community in Preterm Infants. PLoS ONE 2016, 11, e0150306. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Leducq, V.; Aschard, H.; Pham, H.-P.; Jegou, S.; Landman, C.; Cohen, D.; Liguori, G.; Bourrier, A.; Nion-Larmurier, I.; et al. Fungal microbiota dysbiosis in IBD. Gut 2017, 66, 1039–1048. [Google Scholar] [CrossRef]
- Frykman, P.K.; Nordenskjöld, A.; Kawaguchi, A.; Hui, T.T.; Granström, A.L.; Cheng, Z.; Tang, J.; Underhill, D.M.; Iliev, I.; Funari, V.A.; et al. Characterization of Bacterial and Fungal Microbiome in Children with Hirschsprung Disease with and without a History of Enterocolitis: A Multicenter Study. PLoS ONE 2015, 10, e0124172. [Google Scholar] [CrossRef]
- Chang, C.-J.; Lin, C.-S.; Lu, C.-C.; Martel, J.; Ko, Y.-F.; Ojcius, D.M.; Tseng, S.-F.; Wu, T.-R.; Chen, Y.-Y.M.; Young, J.D.; et al. Ganoderma lucidum reduces obesity in mice by modulating the composition of the gut microbiota. Nat. Commun. 2015, 6, 7489. [Google Scholar] [CrossRef]
- Shelburne, S.A.; Ajami, N.J.; Chibucos, M.C.; Beird, H.C.; Tarrand, J.; Galloway-Peña, J.; Albert, N.; Chemaly, R.F.; Ghantoji, S.S.; Marsh, L.; et al. Implementation of a Pan-Genomic Approach to Investigate Holobiont-Infecting Microbe Interaction: A Case Report of a Leukemic Patient with Invasive Mucormycosis. PLoS ONE 2015, 10, e0139851. [Google Scholar] [CrossRef]
- Hoarau, G.; Mukherjee, P.K.; Gower-Rousseau, C.; Hager, C.; Chandra, J.; Retuerto, M.A.; Neut, C.; Vermeire, S.; Clemente, J.; Colombel, J.F.; et al. Bacteriome and Mycobiome Interactions Underscore Microbial Dysbiosis in Familial Crohn’s Disease. MBio 2016, 7, e01250-16. [Google Scholar] [CrossRef]
- Strati, F.; Cavalieri, D.; Albanese, D.; De Felice, C.; Donati, C.; Hayek, J.; Jousson, O.; Leoncini, S.; Pindo, M.; Renzi, D.; et al. Altered gut microbiota in Rett syndrome. Microbiome 2016, 4, 41. [Google Scholar] [CrossRef]
- Tan, S.-Q.; Zhang, K.-Q.; Chen, H.-X.; Ge, Y.; Ji, R.; Shi, W.-P. The mechanism for microsporidian parasite suppression of the hindgut bacteria of the migratory locust Locusta migratoria manilensis. Sci. Rep. 2015, 5, 17365. [Google Scholar] [CrossRef]
- Shao, M.-W.; Lu, Y.-H.; Miao, S.; Zhang, Y.; Chen, T.-T.; Zhang, Y.-L. Diversity, Bacterial Symbionts and Antibacterial Potential of Gut-Associated Fungi Isolated from the Pantala flavescens Larvae in China. PLoS ONE 2015, 10, e0134542. [Google Scholar] [CrossRef] [PubMed]
- van Leeuwen, P.T.; van der Peet, J.M.; Bikker, F.J.; Hoogenkamp, M.A.; Oliveira Paiva, A.M.; Kostidis, S.; Mayboroda, O.A.; Smits, W.K.; Krom, B.P. Interspecies Interactions between Clostridium difficile and Candida albicans. mSphere 2016, 1, e00187-16. [Google Scholar] [CrossRef] [PubMed]
- Sovran, B.; Planchais, J.; Jegou, S.; Straube, M.; Lamas, B.; Natividad, J.M.; Agus, A.; Dupraz, L.; Glodt, J.; Da Costa, G.; et al. Enterobacteriaceae are essential for the modulation of colitis severity by fungi. Microbiome 2018, 6, 152. [Google Scholar] [CrossRef] [PubMed]
- Valentine, M.; Benadé, E.; Mouton, M.; Khan, W.; Botha, A. Binary interactions between the yeast Candida albicans and two gut-associated Bacteroides species. Microb. Pathog. 2019, 135, 103619. [Google Scholar] [CrossRef]
- Markey, L.; Shaban, L.; Green, E.R.; Lemon, K.P.; Mecsas, J.; Kumamoto, C.A. Pre-colonization with the commensal fungus Candida albicans reduces murine susceptibility to Clostridium difficile infection. Gut Microbes 2018, 9, 497–509. [Google Scholar] [CrossRef]
- Panpetch, W.; Somboonna, N.; Palasuk, M.; Hiengrach, P.; Finkelman, M.; Tumwasorn, S.; Leelahavanichkul, A. Oral Candida administration in a Clostridium difficile mouse model worsens disease severity but is attenuated by Bifidobacterium. PLoS ONE 2019, 14, e0210798. [Google Scholar] [CrossRef]
- Robino, P.; Ferrocino, I.; Rossi, G.; Dogliero, A.; Alessandria, V.; Grosso, L.; Galosi, L.; Tramuta, C.; Cocolin, L.; Nebbia, P. Changes in gut bacterial communities in canaries infected by Macrorhabdus ornithogaster. Avian Pathol. 2019, 48, 111–120. [Google Scholar] [CrossRef]
- Solano-Aguilar, G.I.; Jang, S.; Lakshman, S.; Gupta, R.; Beshah, E.; Sikaroodi, M.; Vinyard, B.; Molokin, A.; Gillevet, P.M.; Urban, J.F. The Effect of Dietary Mushroom Agaricus bisporus on Intestinal Microbiota Composition and Host Immunological Function. Nutrients 2018, 10, 1721. [Google Scholar] [CrossRef]
- Mueller, K.D.; Zhang, H.; Serrano, C.R.; Billmyre, R.B.; Huh, E.Y.; Wiemann, P.; Keller, N.P.; Wang, Y.; Heitman, J.; Lee, S.C. Gastrointestinal microbiota alteration induced by Mucor circinelloides in a murine model. J. Microbiol. 2019, 57, 509–520. [Google Scholar] [CrossRef]
- Rouzé, R.; Moné, A.; Delbac, F.; Belzunces, L.; Blot, N. The Honeybee Gut Microbiota Is Altered after Chronic Exposure to Different Families of Insecticides and Infection by Nosema ceranae. Microbes Environ. 2019, 34, 226–233. [Google Scholar] [CrossRef]
- Briand, F.; Sulpice, T.; Giammarinaro, P.; Roux, X. Saccharomyces boulardii CNCM I-745 changes lipidemic profile and gut microbiota in a hamster hypercholesterolemic model. Benef. Microbes 2019, 10, 555–567. [Google Scholar] [CrossRef] [PubMed]
- García, G.; Dogi, C.; de Moreno de LeBlanc, A.; Greco, C.; Cavaglieri, L. Gut-borne Saccharomyces cerevisiae, a promising candidate for the formulation of feed additives, modulates immune system and gut microbiota. Benef. Microbes 2016, 7, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Ducray, H.A.G.; Globa, L.; Pustovyy, O.; Morrison, E.; Vodyanoy, V.; Sorokulova, I. Yeast fermentate prebiotic improves intestinal barrier integrity during heat stress by modulation of the gut microbiota in rats. J. Appl. Microbiol. 2019, 127, 1192–1206. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Haku, A.; Bi, B.; Takahashi, H.; Kamada, N.; Yaguchi, T.; Saijo, S.; Yoneyama, M.; Goto, Y. Fecal microbiota transplantation prevents Candida albicans from colonizing the gastrointestinal tract. Microbiol. Immunol. 2019, 63, 155–163. [Google Scholar] [CrossRef]
- Charlet, R.; Pruvost, Y.; Tumba, G.; Istel, F.; Poulain, D.; Kuchler, K.; Sendid, B.; Jawhara, S. Remodeling of the Candida glabrata cell wall in the gastrointestinal tract affects the gut microbiota and the immune response. Sci. Rep. 2018, 8, 3316. [Google Scholar] [CrossRef]
- Wang, J.; Liu, S.; Wang, H.; Xiao, S.; Li, C.; Li, Y.; Liu, B. Xanthophyllomyces dendrorhous-Derived Astaxanthin Regulates Lipid Metabolism and Gut Microbiota in Obese Mice Induced by A High-Fat Diet. Mar. Drugs 2019, 17, 337. [Google Scholar] [CrossRef]
- Swidsinski, A.; Loening-Baucke, V.; Schulz, S.; Manowsky, J.; Verstraelen, H.; Swidsinski, S. Functional anatomy of the colonic bioreactor: Impact of antibiotics and Saccharomyces boulardii on bacterial composition in human fecal cylinders. Syst. Appl. Microbiol. 2016, 39, 67–75. [Google Scholar] [CrossRef]
- Lemoinne, S.; Kemgang, A.; Belkacem, K.B.; Straube, M.; Jegou, S.; Corpechot, C.; Network, S.-A.I.; Chazouillères, O.; Housset, C.; Sokol, H. Fungi participate in the dysbiosis of gut microbiota in patients with primary sclerosing cholangitis. Gut 2020, 69, 92–102. [Google Scholar] [CrossRef]
- Coker, O.O.; Nakatsu, G.; Dai, R.Z.; Wu, W.K.K.; Wong, S.H.; Ng, S.C.; Chan, F.K.L.; Sung, J.J.Y.; Yu, J. Enteric fungal microbiota dysbiosis and ecological alterations in colorectal cancer. Gut 2019, 68, 654–662. [Google Scholar] [CrossRef]
- Sam, Q.H.; Chang, M.W.; Chai, L.Y.A. The Fungal Mycobiome and Its Interaction with Gut Bacteria in the Host. Int. J. Mol. Sci. 2017, 18, 330. [Google Scholar] [CrossRef]
- Hoffmann, C.; Dollive, S.; Grunberg, S.; Chen, J.; Li, H.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Archaea and Fungi of the Human Gut Microbiome: Correlations with Diet and Bacterial Residents. PLoS ONE 2013, 8, e66019. [Google Scholar] [CrossRef] [PubMed]
- Mwangi, W.; de Figueiredo, P.; Criscitiello, M.F. One Health: Addressing Global Challenges at the Nexus of Human, Animal, and Environmental Health. PLoS Pathog. 2016, 12, e1005731. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.; Kim, B.I.; Lim, J.-S.; Tan, C.S.; Chun, B.C. One Health Perspectives on Emerging Public Health Threats. J. Prev. Med. Public Health 2017, 50, 411–414. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Protozoa | Host | Type of Sample | Site of Sample | Bacterial Microbiota Method | Bacterial Microbiota Change | Diversity Profile | Reference | |
|---|---|---|---|---|---|---|---|---|
| Blastocystis spp. | Cirrhotic patients | Faeces | High-throughput sequencing of 16S rRNA amplicons (Illumina) | Family: Enterobacteriaceae↑, Ruminococaceae↓ Genus: Lactobacillus↑, clostridial cluster XIV↓ | ↓alpha diversity | [74] | ||
| Healthy human | High-throughput sequencing of 16S rRNA amplicons (Ion Torrent) | Class: Clostridia↑, Mollicutes↑ Order: Clostridiales↑, Lactobacillales↓ Family: Enterobacteriaceae↓, Enterococcaceae↓, Streptococcaceae↓, Lactobacillaceae↓, Ruminococcaceae↑, Prevotellaceae↑ Genus: Acetanaerobacterium↑, Acetivibrio↑, Coprococcus↑, Hespellia↑, Oscillibacter↑, Papillibacter↑, Sporobacter↑, Ruminococcus↑, Prevotella↑, Roseburia↓, Faecalibacterium↓ | ↑alpha diversity | [29] | ||||
| High-throughput sequencing of 16S rRNA amplicons (Illumina) | Phylum: Firmicutes↑, Elusimicrobia↑, Lentisphaerae↑, Euryarchaeota↑, Actinobacteria↓, Proteobacteria↓, unassigned bacteria↓, Deinococcus–Thermus↓ Class: Clostridia↑, IHU_PC_PC_Bacteria↑, Elusimicrobia↑, Lentisphaeria↑, Metanobacteria↑, Deltaproteobacteria↑, Planctomycetacia↓, Rubrobacteria↓, Deinococci↓, Gammaproteobacteria↓, Actinobacteria↓, unassigned bacteria↓, Bacilli↓ Order: Clostridiales↑, IHU_PO_Bacteria↑, Victivallales↑, Methanobacteriales↑, Elusimicrobiales↑, Aeromonadales↑, Acidaminococcales↑, Desulfovibrionales↑, Planctomycetales↓, Rhodobacterales↓, Sphingomonadales↓, Rubrobacterales↓, Veillonellales↓, Pasteurellales↓, Micrococcales↓, Pseudonocardiales↓, Enterobacteriales↓, Myxococcales↓, Bifidobacteriales↓, unassigned bacteria↓, Lactobacillales↓ Family: Clostridiaceae↑, Ruminococcaceae↑, Lachnospiraceae↑, Streptococcaceae↓, Bifidobacteriaceae↓, Enterobacteriaceae↓, Leuconostocaceae↓ Genus: Ruminococcus↑, Clostridium↑, Streptococcus↓, Bifidobacterium↓, Shigella↓ Species: Clostridium saudii↑, Methanobrevibacter smithii↑, Streptococcus sp.↓, Bifidobacterium sp.↓, Shigella sp.↓ | ↑alpha diversity | [80] | |||||
| Blastocystis spp. with or not Dientamoeba fragilis | Healthy human | Faeces | qPCR | Genus: Bacteroides↓, Clostridial cluster XIVa↓, Prevotella↑ | Not evaluated | [27] | ||
| Cryptosporidium parvum | CD-1 mice | Faeces | High-throughput sequencing of 16S rRNA amplicons (Illumina) | Phylum: Unclassified Bacteroidetes↑ Family: Porphyromonadaceae↑, Prevotellaceae↑ | Not Change | [73] | ||
| Phylum: Proteobacteria↑, Firmicutes↓ | ↓alpha diversity | [81] | ||||||
| Entamoeba histolytica | Children | Faeces | Quantitative polymerase chain reaction (qPCR) | Species: Prevotella copri↑ | Not evaluated | [31] | ||
| Entamoeba (E. dispar, E. histolytica, or both) | Pygmy hunter-gatherers Bantu individuals | High-throughput Sequencing of 16S rRNA amplicons (Illumina) | Phylum: Actinobacteria↓, Bacteroidetes↓, Cyanobacteria↑, Elusimicrobia↑, Euryarchaeota↑, Firmicutes↑, Fusobacteria↓, Lentisphaerae↓, Spirochaetes↑, Tenericutes↑, Verrucomicrobia↑ Order: Clostridiales↑, Elusimicrobiales↑, Treponema↑ Family: Christensenellaceae↑, Elusimicrobiaceae↑, Spirochaetaceae↑ Specie: Prevotella copri↓ | ↑alpha diversity | [75] | |||
| Giardia duodenalis, Ancylostoma caninum, Cystoisospora, Giardia cati | Dog Cat | Faeces | High-throughput sequencing of 16S rRNA amplicons (Ion Torrent) | Dog: Giardia Phylum: Firmicutes↑, Bacteroidetes↓, Proteobacteria↑ Family: Erysipelotrichaceae↑, Bacteroidaceae↓, Lachnospiraceae↓, Pseudomonadaceae↑ Genus: Catenibacterium↑, Pseudomonas↑, Howardella↑, Bacteroides↓, Pseudobutyrivibrio↓, | No change | [79] | ||
| Cat: Cystoisospora Phylum: Actinobacteria↑, Firmicutes↑ Deinococcus-Thermus↑, Proteobacteria↑ Family: Bifidobacteriaceae↑, Coriobacteriaceae↑, Veillonellaceae↑, Bacillaceae↑, Thermaceae↑, Xanthomonadaceae↑, Comamonadaceae↑, Beijerinckiaceae↑, Xanthomonadaceae↑ Genus: Bifidobacterium↑, Olsenella↑, Megamonas↑, Geobacillus↑, Meiothermus↑, Bacillus↑, Thermomonas↑ Schlegelella↑, Chelatococcus↑, Silanimonas↑ | ||||||||
| Cat: Giardia Phylum: Firmicutes↑ Family: Lachnospiraceae↑, Ruminococcaceae↓ Genus: Roseburia↑, Subdoligranulum↓ | ||||||||
| Giardia lamblia | C57BL/6J mice | Mucosal and Luminal proximal Small intestine, Mucosal and luminal Distal small intestine, Cecal contents and colonic contents | Foregut, hindgut | High-throughput sequencing of 16S rRNA amplicons (Illumina) | Phylum: Melainabacteria↓ Order: Clostridiales↓, Family: Rhodocylaceae↑, Moraxellaceae↑ | ↓alpha diversity | [82] | |
| Giardia duodenalis | Healthy human | Mucosal biopsies | Colon | High-throughput sequencing of 16S rRNA amplicons (Illumina) | Phylum: Firmicutes↑ Order: Clostridiales↑ Genus: Phascolarctobacterium↓ | ↑alpha diversity | [30] | |
| Giardia spp., Entamoeba spp./ Blastocystis hominis | Human with or without symptoms | Faeces | qPCR | Genus: Bifidobacterium↑, Species: Escherichia coli↑, Faecalibacterium prausnitzii-/Escherichia coli ratio↑ | Not evaluated | [28] | ||
| Leishmania infantum | Lutzomyia longipalpis | midguts | 16Sv4 rRNA gene sequencing | Family: Pseudomonadaceae↑, Acetobacteraceae↓ | ↓alpha diversity | [76] | ||
| Plasmodium yoelii | BALB/c mice Resistant, C57BL/6 mice susceptible | Cecum, colon | Distal small intestine | High-throughput sequencing of 16S rRNA amplicons (Illumina) | Phylum: Firmicutes↑, Bacteroidetes↓ Family: Clostridiaceae↑, Erysipelotrichaceae↑, Lactobacillaceae↑, Peptostreptococcaceae↑ Bacteroidaceae↓, Prevotellaceae↓, Sutterellaceae↓ Genus: Lactobacillus↑, Bifidobacterium↑ | ↓alpha diversity | [33] | |
| Trichomonas vaginalis | North American women | Vaginal swabs | High-throughput Sequencing of 16S rRNA amplicons (454 pyrosequencinq) | Genus: Lactobacillus↓, Mycoplasma↑, Parvimonas↑, Sneathia↑ | Not evaluated | [78] | ||
| Toxoplasma gondii | NOD2−/− mice | Feces | Real Time-PCR | Order: Enterobacteria↑ Genus: Lactobacillus↑, Bifidobacteria↓, Enterococci↑, Bacteroides/Prevotella spp.↑, eubacterial↑ | Not evaluated | [77] | ||
| C57BL/6 mice | High-throughput sequencing of 16S rRNA amplicons (Illumina) | Phylum: Firmicutes↑ Genus: Clostridia↑ | [83] | |||||
| Helminths | Host | Type of Sample | Site of Sample | Bacterial Microbiota Method | Bacterial Microbiota Change | Diversity Profile | Reference |
|---|---|---|---|---|---|---|---|
| Ascaris, Trichuris, Hookworm | Human volunteers | Fresh stool | High-throughput sequencing of 16S rRNA amplicons (Illumina) | Class: Verrucomicrobiae↑ Order: Verrucomicrobiales↑ Family: Bacteroidaceae↑, Prevotellaceae↓ Verrucomicrobiaceae↑, Enterobacteriaceae↑, Leuconostocaceae↓, Bacteroidaceae↓ Genus: Lactococcus↑, Akkermansia↑, Enterobacteriaceae↑, Bacteroides↓ Species: Prevotella copri↑ | No change | [36] | |
| Ascaris lumbricoides (“Ascaris”), Necator Americanus (“Necator”), Trichuris trichiura (“Trichuris”) | Subject cohort | Feces | High-throughput sequencing of 16S rRNA amplicons (Illumina) | Phylum: Firmicutes↑, Bacteroidetes↓, Actinobacteria↑ Family: Lachnospiraceae↑, Erysipelotrichaceae↑ Genus: Succinivibrio↑, Solobacterium↑, Desulfovibrio↑, Allobaculum↑, Rhodococcus↑, Lachnospiraceae incertae sedis↑, Olsenella↑, Flavonifractor↑, Enterococcus↑. | ↑alpha diversity | [91] | |
| Ascaris suum | Pigs | Digesta | Proximal Colon | High-throughput Sequencing of 16S rRNA amplicons (Illumina) | Genus: ↑Prevotella, ↑Facklamia, ↑, Turicibacter, ↓Ruminicoccus, ↓Lactobacillus, ↑Treponema, ↑Campylobacter | ↑alpha Diversity with Basal diet No change with Grape pomace diet | [93] |
| Cyathostomins spp. (Eggs hight versus low) | Equines | Feces | High-throughput Sequencing of 16S rRNA amplicons (Illumina) | Class: Methanomicrobia↓, Dehobacterium↓ | No difference | [100] | |
| Enterobius vermicularis | School children | Feces | High-throughput Sequencing of 16S rRNA amplicons (Illumina) | Phylum: ↓Fusobacteria, ↑Actinobacteria Genus: ↑Bifidobacterium, ↑Alistipes, ↑Faecalibacterium, ↓Fusobacterium, ↓Veilonella, ↓Megasphaera, ↓Acidaminococcus Species: ↑Faecalibacterium prausnitzii, ↑Ruminococcus flavefaciens, ↑Alistipes purtredinis, ↑Bifidobacterium longum, ↑uncultured Oscillospira sp., ↓Acidaminococcus intestine, ↓Megasphaera elsdenii, ↓Veillonella dispar, ↓Fusobacterium varium | ↑alpha diversity | [34] | |
| Heligmosomoides Polygyrus bakeri | IL-4Rα−/− mice; C57BL/6 mice | Lumen | Caecum; ileum; colon | Culture; Cloned 16S rRNA amplicon; qPCR; Denaturing gradient gel electrophoresis | γ-Proteobacteria/Enterobacteriaceae ratio↑, Bacteroides/Prevotella ratio↑ | Not evaluated | [101] |
| Heligmosomoides Polygyrus, Syphacia spp., Hymenolepis spp. | Wild mouse (Apodemus flavicollis) | Lumen, mucosa | Stomach, ileum, caecum, colon | High-throughput Sequencing of 16S rRNA amplicons (454) | Hymenolepis spp. Family: Ruminococcaceae↓, Acetobacteraceae↓, Sphingomonadaceae↓, S24-7 family (Bacteroidetes)↑ | No change | [102] |
| Heligmosomoides Polygyrus Family: Lachnospiraceae↑, S24-7 OTUs (Bacteroidetes)↑ Genus: Lactobacillus↑ | |||||||
| Syphacia spp. Family: Lachnospiraceae↓, Lactobacillaceae↓, S24-7 family (Bacteroidetes)↓ Genus: Lactobacillus↓ | |||||||
| Leidynema appendiculatum; Leidynema appendiculatum; Hammerschmidtiella diesingi; Thelastoma bulhoesi | Periplaneta fuliginosa Periplaneta americana | Faeces | Foregut; Midgut; Hindgut | High-throughput Sequencing of 16S rRNA amplicons (Illumina) | Phylum: ↓Firmicutes, ↑Proteobacteria, ↓Bacteroidetes, ↑Actinobacteria Genus: ↑Bacillales, ↑Brevibacterium, ↓Gordonia, ↑Xylanimicrobium, ↓Bacteroides Order: ↑Lactobacillales, ↓Enterobacteriales Family: ↓Lachnospiraceae, ↓Ruminococcaceae, ↑Porphyromonadaceae, ↑Desulfovibrionaceae, ↓Weeksellaceae, ↓Bacteroidaceae Periplaneta americana Phylum: ↑Bacteroidetes, ↑Firmicutes, ↑Proteobacteria Family: ↑Porphyromonadaceae, ↓Bacteroidaceae, ↑Ruminococcaceae, ↓Lachnospiraceae, ↑Desulfovibrionaceae | ↑alpha diversity | [97] |
| Necator americanus | Patients with coeliac disease | Faeces | High-throughput Sequencing of 16S rRNA Amplicons (454) | Phylum: Firmicutes↓, Bacteroides, ↑Tenericutes, RF39↓ Class: Bacteroidia↑, Erysipelotrichi↓, Clostridia↓ Genre: Ruminococcus↓, Lachnospira↓ | ↑alpha diversity | [96] | |
| Trichuris muris | C57BL/6 mice | Faeces, lumen | Caecum | High-throughput Sequencing of 16S rRNA amplicons (Illumina) | Alistipes, Odoribacter, and Parasutterella↑, Allobaculum↓, Barnesiella↓ | ↓alpha diversity | [35] |
| Faeces | Denaturing gradient gel electrophoresis; High-throughput sequencing of 16S rRNA amplicons (454) | Phylum: Bacteroidetes↑; Genus: Prevotella↑, Parabacteroides↑ | [92] | ||||
| Trichostrongylus retortaeformis | Rabbits (Oryctolagus cuniculus) | Mucosa | Duodenal | High-throughput Sequencing of 16S rRNA amplicons (Illumina) | Phylum: ↑Proteobacteria, ↑Spirochaetes, ↓Firmicutes Family: Leptospiraceae↑, ↑Ruminococcaceae, ↑Phyromonadaceae, ↑Desulfobacteraceae, ↑Bacteroidaceae Genus: ↑Leptomena, ↑Desulfocella, ↓Bacteroides ↓Ruminococcus | ↓alpha diversity | [103] |
| Toxocara cati | Cat (Felis catus) | Faeces | High-throughput Sequencing of 16S rRNA amplicons (Illumina) | Phylum: ↑Actinobacteria Class: ↑Coreobacteriia, ↓Gammaproteobacteria Order: ↑Lactobacillales, ↑Coribacteriales Family: ↑Enterococcaceae, ↑Coreobacteriaceae Genus: ↑Collinsella, ↑Enterococcus, ↑Dorea, ↑Lactobacillus, ↑Ruminococcus, ↓Bulleidia, ↓Jeotgalicoccus | No change | [104] | |
| Trichuris suis | Pigs (Sus scrofa domestica) | Faeces | Proximal colon | Whole metagenome shotgun Sequencing (Illumina) | Phylum: ↓Fibrobacteres, ↓Spirochaetes, ↓Tenericutes, ↓ Gemmatimonadetes Genus: ↑Fibrobacter, ↑Campylobacter, ↓Treponema, ↓Dorea, ↓Ruminococcus | Not evaluated | [87] |
| Pigs (Sus scrofa domestica) | Lumen | Proximal colon | Whole metagenome shotgun 454 sequencing High-throughput sequencing of 16S rRNA amplicons (454) | Phylum: ↓Proteobacteria, ↓Deferribacteres, ↑Euryarchaeota Genus: ↑Prevotella, ↓Succinivibrio, ↓Mucispirillum, ↓Oscillibacter, ↑Paraprevotella, ↑Desulfovibrio, ↑Heliobacter | No change | [88] | |
| Trichuris trichiura, Ascaris lumbricoides | School children | Faeces | High-throughput Sequencing of 16S rRNA Amplicons (454) | Phylum: ↓Firmicutes Class: ↓Clostridia, ↑streptococci Genus: ↓Clostridium sensu stricto, ↓uncharacterised clostridial cluster IX, ↑Streptococcus, ↓Roseburia | ↓alpha diversity | [89] | |
| Trichuris spp., Ascaris spp., hookworm | Indigenous community | Faeces | High-throughput Sequencing of 16S rRNA Amplicons (Illumina) | Phylum: ↑Mollicutes, ↑Bacteroidales, ↑Alphaproteobacteria Family: ↑Paraprevotellaceae, ↑Lachnospiraceae, ↑Prevotellaceae Genus: ↓Bifidobacterium | ↑alpha diversity | [90] | |
| Schistosoma haematobium, Schistosoma mansoni | Children (six months to 13 years old) | Urine, Stool | High-throughput sequencing of 16S rRNA amplicons (Illumina) | Phylum: Bacteroidetes↑, Firmicutes↑, Proteobacteria↑ Genus: Prevotella↑ | ↑alpha diversity | [99] | |
| Schistosoma mansoni | Children from Côte d’Ivoire | Faeces | High-throughput sequencing of 16S rRNA amplicons (Illumina) | Phylum: Proteobacteria↑ Family: Cerasicoccaceae↑, Anaeroplasmataceae↑, Campylobacteraceae↑, Peptococcaceae↑ Genus: Klebsiella↑, Enterobacter arachidis↑, Fructobacillus↓ | ↓alpha diversity | [105] | |
| Trichuris trichiura | Rhesus monkeys | Colon mucosa | High-throughput sequencing of 16S rRNA amplicons (Illumina) | Phylum: ↓Cyanobacteria, ↑Firmicutes, ↑Bacteroidetes, ↑Tenericutes, ↓unclassified bacteria taxon ZB2 Genus: ↓Streptophyta | No change | [106] |
| Fungi | Host | Type of Sample | Site of Sample | Bacterial Microbiota Method | Bacterial Microbiota Change | Diversity Profile | Reference |
|---|---|---|---|---|---|---|---|
| Candida albicans | Culture | Clostridium difficile↑ | Not evaluated | [124] | |||
| Culture; qPCR | Family: Enterobacteriaceae↑ | Not evaluated | [125] | ||||
| Culture | Bacteroides fragilis↑, Bacteroides vulgatus↑ | Not evaluated | [126] | ||||
| Clostridium difficile in C57BL/6 mice | Distal Cecum, contents | High-throughput sequencing of 16S rRNA amplicons (Illimuna) | Phylum: Verrucomicrobia↑, Proteobacteria↑, Actinobacteria↑, Firmicutes↑, Bacteroidetes↑ Family: Comamonadaceae↑, Erysipelotrichacea↑, S24-7↑ Genus: Akkermansia sp.↑, Sutterella sp.↑, Bifidobacterium sp.↑, Adlercreutzia sp.↑ | ↓alpha diversity | [127] | ||
| Mouse model of Clostridium difficile | Faeces | Colon | Culture | Clostridium difficile↑ | Not evaluated | [128] | |
| Debaryomyces hansenii↑ Candida spp.↓, and Saccharomyces spp.↓ | Obese children | Faeces | Denaturing gradient gel electrophoresis (DGGE) qPCR | Species: Akkermansia muciniphila↓, Faecalibacterium prausnitzii↓ | ↑alpha diversity | [114] | |
| Ganoderma lucidum mycelium | High-Fat Diet (HFD)-fed Mice Chow mice | Faeces | Caecal | Pyrosequencing of bacterial 16S rRNA | Phylum: Firmicutes-to-Bacteroidetes ratios↓, Proteobacteria↓ Species: Parabacteroides goldsteinii↑, Bacteroides↑, Anaerotruncus colihominis↑, Roseburia hominis↑, Clostridium↑, Clostridium methylpentosum↑, Clostridium XIVa and XVIII↑, Eubacterium coprostanoligenes↑ | ↑alpha diversity | [118] |
| Macrorhabdus ornithogaster | Canaries (Serinus canaria domestica) | Faeces | PCR-DGGE, High-throughput sequencing of 16S rRNA amplicons (Illimuna) | Phylum: Acidobacteria↑, Actinobacteria↑, Cyanobacteria↑, Planctomycetes↑ Family: Lachnospiraceae↓, Enterobacteriaceae↓ Genus: Lactobacillus↑, Streptococcus↑, Clostridium↓, Lactococcus↓, Pseudomonas↓, Acinetobacter↓, Weissella↓, Propionibacterium↓ Species: Candidatus Arthromitus↑ | ↓alpha diversity | [129] | |
| Mushroom (Agaricus bisporus) | Pigs | Faeces, Proximal colon contents | High-throughput sequencing of 16S rRNA amplicons (Ion Torrent) | Family: Lachnospiraceae↑, Ruminococcaceae↑ Order: Clostridiales↑ | No change | [130] | |
| Mucor circinelloides | BALB/C mice | Faeces | High-throughput sequencing of 16S rRNA amplicons (Illimuna) | Genus: Bacteroides↑ Species: Akkermansia muciniphila↑ | ↑alpha diversity | [131] | |
| Mucor velutinosus | Old man with Onychomycosis and acute myelogenous leukemia | Oral Stool | High-throughput sequencing of 16S rRNA amplicons (Illimuna) | staphylococci↑ | ↓alpha diversity | [119] | |
| Malassezia↓, Saccharomyces sp.↓ | Children with Hirschsprung disease | Faeces | High-throughput sequencing of 16S rRNA amplicons (Illimuna; Ion Torrent) | Phylum: Firmicutes↓, Verrucomicrobia↓, Bacteroidetes↓, Proteobacteria↓ | ↓alpha diversity | [117] | |
| Nosema ceranae | Adult workers Honeybees (Apis mellifera) | Hindguts | qPCR | Lactobacillus spp.↓ and Bifidobacterium spp.↓, Snodgrassella alvi↑, Gilliamella apicola↑ | Not evaluated | [132] | |
| Paranosema locustae | Locusta migratoria manilensis | Faeces | Hindgut | High-throughput Pyrosequencing of 16S rRNA amplicons (454) | Genus: Citrobacter↑, Lactococcus↑, Raoultella↑ Species: Corynebacterium sp. WA7↓, Raoultella terrigena↓ | ↓alpha diversity | [122] |
| Sordariomycetes Eurotiomycetes Dothideomycetes Leotiomycetes | Pantala flavescens | Fresh mycelia | Larvae | Culture; High-throughput sequencing of 16S rRNA | Phylum: Proteobacteria↑, Firmicutes↑ Genus: Sphingomonas, Methylobacterium, Burkholderia, Pantoea, Enterobacter↑, Leclercia, and Serratia, Oceanobacillus Species: Leclercia sp., Oceanobacillus oncorhynchi, Methylobacterium extorquens | Not evaluated | [123] |
| Saccharomyces boulardii | Premature infants | Faeces | High-throughput sequencing of 16S rRNA amplicons (Ion Torrent) | Phylum: Proteobacteria↓, Bacteroidetes↓, Actinobacteria↓ Genus: Escherichia↑, Enterococcus↓, Veilonella↑, Clostridium↑, Bifidobacteriu↑ | ↑alpha diversity | [115] | |
| Hamster hypercholesterolemic model | High-throughput Sequencing of 16S rRNA amplicons (Illumina) | Phylum: Firmicutes↓, Tenericutes↓, TM7↓, Proteobacteria↑, Lentispharerae↑, unknown phyla↑ Genus: Allobaculum↑, CF231↑ | No difference | [133] | |||
| Saccharomyces cerevisiae | Male BALB/c mice | Caecum | Culture | Family: Enterobacteriaceae↓ | Not evaluated | [134] | |
| Rats | Faeces | Terminal restriction Fragment length Polymorphism (T-RFLP) analysis | Genus: Bacteroides↑, Fusobacterium↑, Bifidobacterium↑, Lactobacilli↑, Enterococcus↑ | ↑alpha diversity | [112] | ||
| Colon | Cuture High-throughput Sequencing of 16S rRNA amplicons (Illumina) | Genus: Bifidobacterium↓, Allobaculum↓, Acetanaerobacterium↑, Bacteroides↑, Eubacterium↑, Johnsonella↑, Lactococcus↑, Oscillospira↑, Roseburia↑, Vallitalea↑ Species: Staphylococcus spp.↑, haemolytic bacteria↑ | Not evaluated | [135] | |||
| EpiCor fermentate (dried yeast fermentate made using Saccharomy cescerevisiae) | Healthy volunteers (symptoms of gastrointestinal discomfort and reduced bowel movements) | Faeces | High-throughput Sequencing of 16S rRNA amplicons (Illumina and 454) | Phylum: Firmicutes↓, Bacteroidetes↑ Family: Bacteroidaceae↑, Porphyromonadaceae↑, Prevotellaceae↑ Genus: Propionibacterium↑, Paraprevotella↑, Oscillibacter↑, Barnesiella↑, Prevotella↑, Akkermansia↑, Odoribacte↑, Anaerostipes↑, Blautia↓, Roseburia↓ Specis: Akkermansia muciniphila↑ | Community evenness↑ | [113] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kodio, A.; Menu, E.; Ranque, S. Eukaryotic and Prokaryotic Microbiota Interactions. Microorganisms 2020, 8, 2018. https://doi.org/10.3390/microorganisms8122018
Kodio A, Menu E, Ranque S. Eukaryotic and Prokaryotic Microbiota Interactions. Microorganisms. 2020; 8(12):2018. https://doi.org/10.3390/microorganisms8122018
Chicago/Turabian StyleKodio, Aly, Estelle Menu, and Stéphane Ranque. 2020. "Eukaryotic and Prokaryotic Microbiota Interactions" Microorganisms 8, no. 12: 2018. https://doi.org/10.3390/microorganisms8122018
APA StyleKodio, A., Menu, E., & Ranque, S. (2020). Eukaryotic and Prokaryotic Microbiota Interactions. Microorganisms, 8(12), 2018. https://doi.org/10.3390/microorganisms8122018