Screening of FDA-Approved Drugs Using a 384-Well Plate-Based Biofilm Platform: The Case of Fingolimod


Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Culture Conditions
2.2. Compounds
2.3. Screening of the Compound Library in 384-Well Plates on S. aureus Biofilms
2.4. Resazurin Staining
2.5. Crystal Violet Staining
2.6. Minimum Bactericidal Concentration (MBC) and Biofilm Preventing Concentration (BPC)
2.7. Quantification of the Viable Cells in Biofilms
2.8. Time-Kill Kinetic Assay
2.9. Assessing Resistance Development
2.10. Quorum Sensing Inhibition Assays
2.11. Statistical Analysis
3. Results and Discussion
3.1. Screening FDA-Approved Compounds Identifies 45 Anti-Biofilm Agents against S. aureus
3.2. Hit-to-Lead Identification Process
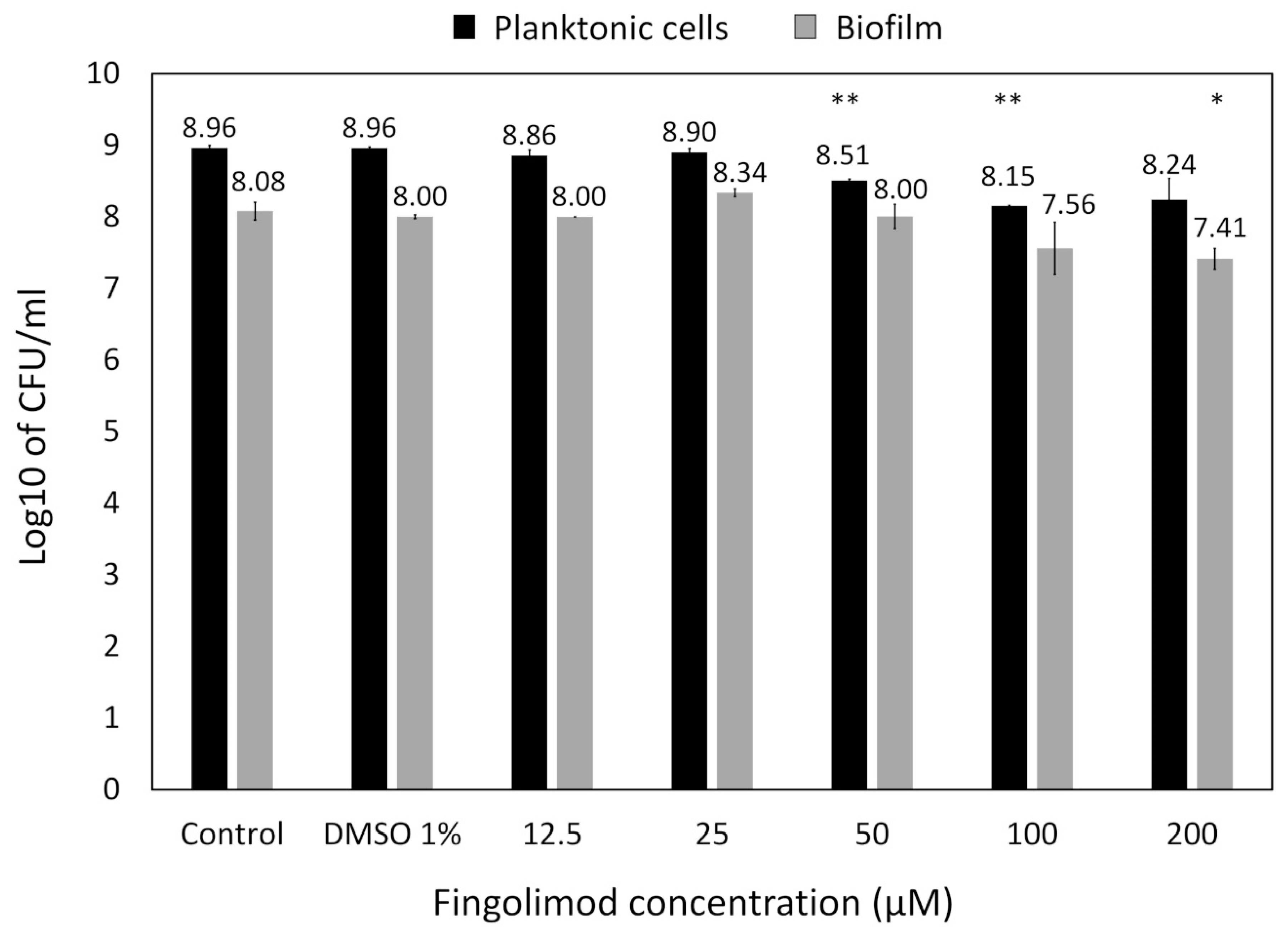
3.3. Concentration–Response Effect of Fingolimod against S. aureus Biofilms
3.4. Reduction in the Count of Viable Cells in Pre-Formed S. aureus Biofilms Exposed to Fingolimod
3.5. Time-Kill Kinetic Effect of Fingolimod against S. aureus
3.6. Resistance Development of S. aureus against Fingolimod
3.7. Activity of Fingolimod on Additional Bacterial Strains
3.8. Quorum Sensing Inhibitory Effect of Fingolimod
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Appendix A


References
- Spellberg, B.; Guidos, R.; Gilbert, D.; Bradley, J.; Boucher, H.W.; Scheld, W.M.; Bartlett, J.G.; Edwards, J., Jr. The epidemic of antibiotic-resistant infections: A call to action for the medical community from the Infectious Diseases Society of America. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2008, 46, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad bugs, no drugs: No ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2009, 48, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ferri, M.; Ranucci, E.; Romagnoli, P.; Giaccone, V. Antimicrobial resistance: A global emerging threat to public health systems. Crit. Rev. Food Sci. Nutr. 2017, 57, 2857–2876. [Google Scholar] [CrossRef] [PubMed]
- Olson, M.E.; Ceri, H.; Morck, D.W.; Buret, A.G.; Read, R.R. Biofilm bacteria: Formation and comparative susceptibility to antibiotics. Can. J. Vet. Res. Rev. Can. Rech. Vet. 2002, 66, 86–92. [Google Scholar]
- Davies, D. Understanding biofilm resistance to antibacterial agents. Nat. Rev. Drug Discov. 2003, 2, 114–122. [Google Scholar] [CrossRef]
- Khan, M.M.; Chattagul, S.; Tran, B.Q.; Freiberg, J.A.; Nita-Lazar, A.; Shirtliff, M.E.; Sermswan, R.W.; Ernst, R.K.; Goodlett, D.R. Temporal proteomic profiling reveals changes that support Burkholderia biofilms. Pathog. Dis. 2019, 77. [Google Scholar] [CrossRef]
- Whiteley, M.; Bangera, M.G.; Bumgarner, R.E.; Parsek, M.R.; Teitzel, G.M.; Lory, S.; Greenberg, E.P. Gene expression in Pseudomonas aeruginosa biofilms. Nature 2001, 413, 860–864. [Google Scholar] [CrossRef]
- Stewart, P.S. Antimicrobial Tolerance in Biofilms. Microbiol. Spectr. 2015, 3. [Google Scholar] [CrossRef]
- Walters, M.C., 3rd; Roe, F.; Bugnicourt, A.; Franklin, M.J.; Stewart, P.S. Contributions of antibiotic penetration, oxygen limitation, and low metabolic activity to tolerance of Pseudomonas aeruginosa biofilms to ciprofloxacin and tobramycin. Antimicrob. Agents Chemother. 2003, 47, 317–323. [Google Scholar] [CrossRef]
- Costerton, J.W.; Lewandowski, Z.; Caldwell, D.E.; Korber, D.R.; Lappin-Scott, H.M. Microbial biofilms. Annu. Rev. Microbiol. 1995, 49, 711–745. [Google Scholar] [CrossRef]
- James, G.A.; Swogger, E.; Wolcott, R.; Pulcini, E.; Secor, P.; Sestrich, J.; Costerton, J.W.; Stewart, P.S. Biofilms in chronic wounds. Wound Repair Regen 2008, 16, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.L. Nosocomial infections in adult intensive-care units. Lancet 2003, 361, 2068–2077. [Google Scholar] [CrossRef]
- Del Pozo, J.L. Biofilm-related disease. Expert Rev. Anti Infect. Ther. 2018, 16, 51–65. [Google Scholar] [CrossRef] [PubMed]
- McConoughey, S.J.; Howlin, R.; Granger, J.F.; Manring, M.M.; Calhoun, J.H.; Shirtliff, M.; Kathju, S.; Stoodley, P. Biofilms in periprosthetic orthopedic infections. Future Microbiol. 2014, 9, 987–1007. [Google Scholar] [CrossRef]
- Høiby, N.; Bjarnsholt, T.; Moser, C.; Bassi, G.L.; Coenye, T.; Donelli, G.; Hall-Stoodley, L.; Holá, V.; Imbert, C.; Kirketerp-Møller, K.; et al. ESCMID guideline for the diagnosis and treatment of biofilm infections 2014. Clin. Microbiol. Infect. 2015, 21 (Suppl. S1), S1–S25. [Google Scholar] [CrossRef]
- Darouiche, R.O. Treatment of infections associated with surgical implants. N. Engl. J. Med. 2004, 350, 1422–1429. [Google Scholar] [CrossRef]
- Zimmerli, W.; Moser, C. Pathogenesis and treatment concepts of orthopaedic biofilm infections. FEMS Immunol. Med. Microbiol. 2012, 65, 158–168. [Google Scholar] [CrossRef]
- Omar, A.; Wright, J.B.; Schultz, G.; Burrell, R.; Nadworny, P. Microbial Biofilms and Chronic Wounds. Microorganisms 2017, 5, 9. [Google Scholar] [CrossRef]
- Younis, W.; Thangamani, S.; Seleem, M.N. Repurposing Non-Antimicrobial Drugs and Clinical Molecules to Treat Bacterial Infections. Curr. Pharm. Des. 2015, 21, 4106–4111. [Google Scholar] [CrossRef]
- Rangel-Vega, A.; Bernstein, L.R.; Mandujano-Tinoco, E.A.; García-Contreras, S.J.; García-Contreras, R. Drug repurposing as an alternative for the treatment of recalcitrant bacterial infections. Front. Microbiol. 2015, 6, 282. [Google Scholar] [CrossRef]
- Gilbert-Girard, S.; Savijoki, K.; Yli-Kauhaluoma, J.; Fallarero, A. Optimization of a High-Throughput 384-Well Plate-Based Screening Platform with Staphylococcus aureus ATCC 25923 and Pseudomonas aeruginosa ATCC 15442 Biofilms. Int. J. Mol. Sci. 2020, 21, 3034. [Google Scholar] [CrossRef] [PubMed]
- Percival, S.L.; Suleman, L.; Vuotto, C.; Donelli, G. Healthcare-associated infections, medical devices and biofilms: Risk, tolerance and control. J. Med. Microbiol. 2015, 64, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Skogman, M.E.; Kanerva, S.; Manner, S.; Vuorela, P.M.; Fallarero, A. Flavones as Quorum Sensing Inhibitors Identified by a Newly Optimized Screening Platform Using Chromobacterium violaceum as Reporter Bacteria. Molecules 2016, 21, 1211. [Google Scholar] [CrossRef] [PubMed]
- Beus, M.; Savijoki, K.Z.; Patel, J.; Yli-Kauhaluoma, J.; Fallarero, A.; Zorca, B. Chloroquine fumardiamides as novel quorum sensing inhibitors. Bioorg. Med. Chem. Lett. 2020, 30, 127336. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Borgers, M. Mechanism of action of antifungal drugs, with special reference to the imidazole derivatives. Rev. Infect. Dis. 1980, 2, 520–534. [Google Scholar] [CrossRef] [PubMed]
- Vanden Bossche, H.; Willemsens, G.; Marichal, P. Anti-Candida drugs—The biochemical basis for their activity. Crit. Rev. Microbiol. 1987, 15, 57–72. [Google Scholar] [CrossRef]
- Zhang, L.; Peng, X.M.; Damu, G.L.; Geng, R.X.; Zhou, C.H. Comprehensive review in current developments of imidazole-based medicinal chemistry. Med. Res. Rev. 2014, 34, 340–437. [Google Scholar] [CrossRef]
- Zhang, J.; Ba, Y.; Wang, S.; Yang, H.; Hou, X.; Xu, Z. Nitroimidazole-containing compounds and their antibacterial and antitubercular activities. Eur. J. Med. Chem. 2019, 179, 376–388. [Google Scholar] [CrossRef]
- Abdel-Motaal, M.; Almohawes, K.; Tantawy, M.A. Antimicrobial evaluation and docking study of some new substituted benzimidazole-2yl derivatives. Bioorg. Chem. 2020, 101, 103972. [Google Scholar] [CrossRef]
- Bulut, O.; Oktem, H.A.; Yilmaz, M.D. A highly substituted and fluorescent aromatic-fused imidazole derivative that shows enhanced antibacterial activity against methicillin-resistant Staphylococcus aureus (MRSA). J. Hazard. Mater. 2020, 399, 122902. [Google Scholar] [CrossRef] [PubMed]
- Aguinagalde, L.; Díez-Martínez, R.; Yuste, J.; Royo, I.; Gil, C.; Lasa, Í.; Martín-Fontecha, M.; Marín-Ramos, N.I.; Ardanuy, C.; Liñares, J.; et al. Auranofin efficacy against MDR Streptococcus pneumoniae and Staphylococcus aureus infections. J. Antimicrob. Chemother. 2015, 70, 2608–2617. [Google Scholar] [CrossRef] [PubMed]
- May, H.C.; Yu, J.J.; Guentzel, M.N.; Chambers, J.P.; Cap, A.P.; Arulanandam, B.P. Repurposing Auranofin, Ebselen, and PX-12 as Antimicrobial Agents Targeting the Thioredoxin System. Front. Microbiol. 2018, 9, 336. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Inoue, K.; Yamamoto, S.; Ikumoto, T.; Sasaki, S.; Toyama, R.; Chiba, K.; Hoshino, Y.; Okumoto, T. Fungal metabolites. Part 11. A potent immunosuppressive activity found in Isaria sinclairii metabolite. J. Antibiot. 1994, 47, 208–215. [Google Scholar] [CrossRef]
- Adachi, K.; Kohara, T.; Nakao, N.; Arita, M.; Chiba, K.; Mishina, T.; Sasaki, S.; Fujita, T. Design, synthesis, and structure-activity-relationships of 2-substituted-2-amino-1,3-propanediols—Discovery of a novel immunosuppressant, FTY720. Bioorg. Med. Chem. Lett. 1995, 5, 853–856. [Google Scholar] [CrossRef]
- Brinkmann, V.; Chen, S.; Feng, L.; Pinschewer, D.; Nikolova, Z.; Hof, R. FTY720 alters lymphocyte homing and protects allografts without inducing general immunosuppression. Transplant. Proc. 2001, 33, 530–531. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef]
- Heaver, S.L.; Johnson, E.L.; Ley, R.E. Sphingolipids in host-microbial interactions. Curr. Opin. Microbiol. 2018, 43, 92–99. [Google Scholar] [CrossRef]
- Kester, M.; Kolesnick, R. Sphingolipids as therapeutics. Pharmacol. Res. 2003, 47, 365–371. [Google Scholar] [CrossRef]
- Menaldino, D.S.; Bushnev, A.; Sun, A.; Liotta, D.C.; Symolon, H.; Desai, K.; Dillehay, D.L.; Peng, Q.; Wang, E.; Allegood, J.; et al. Sphingoid bases and de novo ceramide synthesis: Enzymes involved, pharmacology and mechanisms of action. Pharmacol. Res. 2003, 47, 373–381. [Google Scholar] [CrossRef]
- Sahu, S.K.; Hannun, Y.A.; Yao, N. Emergence of membrane sphingolipids as a potential therapeutic target. Biochimie 2019, 158, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.A.; Riethmüller, J.; Seitz, A.P.; Gardner, A.; Boudreau, R.; Kamler, M.; Kleuser, B.; Schuchman, E.; Caldwell, C.C.; Edwards, M.J.; et al. Sphingolipids as targets for inhalation treatment of cystic fibrosis. Adv. Drug Deliv Rev. 2018, 133, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Pewzner-Jung, Y.; Tavakoli Tabazavareh, S.; Grassmé, H.; Becker, K.A.; Japtok, L.; Steinmann, J.; Joseph, T.; Lang, S.; Tuemmler, B.; Schuchman, E.H.; et al. Sphingoid long chain bases prevent lung infection by Pseudomonas aeruginosa. EMBO Mol. Med. 2014, 6, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Tavakoli Tabazavareh, S.; Seitz, A.; Jernigan, P.; Sehl, C.; Keitsch, S.; Lang, S.; Kahl, B.C.; Edwards, M.; Grassmé, H.; Gulbins, E.; et al. Lack of Sphingosine Causes Susceptibility to Pulmonary Staphylococcus Aureus Infections in Cystic Fibrosis. Cell Physiol. Biochem. 2016, 38, 2094–2102. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.E.; Boudreau, R.M.; Couch, C.; Becker, K.A.; Edwards, M.J.; Caldwell, C.C.; Gulbins, E.; Seitz, A. Sphingosine’s role in epithelial host defense: A natural antimicrobial and novel therapeutic. Biochimie 2017, 141, 91–96. [Google Scholar] [CrossRef]
- Bibel, D.J.; Aly, R.; Shah, S.; Shinefield, H.R. Sphingosines: Antimicrobial barriers of the skin. Acta Derm Venereol. 1993, 73, 407–411. [Google Scholar] [CrossRef]
- Fischer, C.L.; Walters, K.S.; Drake, D.R.; Blanchette, D.R.; Dawson, D.V.; Brogden, K.A.; Wertz, P.W. Sphingoid bases are taken up by Escherichia coli and Staphylococcus aureus and induce ultrastructural damage. Skin Pharmacol. Physiol. 2013, 26, 36–44. [Google Scholar] [CrossRef]
- Arikawa, J.; Ishibashi, M.; Kawashima, M.; Takagi, Y.; Ichikawa, Y.; Imokawa, G. Decreased levels of sphingosine, a natural antimicrobial agent, may be associated with vulnerability of the stratum corneum from patients with atopic dermatitis to colonization by Staphylococcus aureus. J. Investig. Dermatol. 2002, 119, 433–439. [Google Scholar] [CrossRef]
- Fischer, C.L.; Drake, D.R.; Dawson, D.V.; Blanchette, D.R.; Brogden, K.A.; Wertz, P.W. Antibacterial activity of sphingoid bases and fatty acids against Gram-positive and Gram-negative bacteria. Antimicrob. Agents Chemother. 2012, 56, 1157–1161. [Google Scholar] [CrossRef]
- Cukkemane, N.; Bikker, F.J.; Nazmi, K.; Brand, H.S.; Sotres, J.; Lindh, L.; Arnebrant, T.; Veerman, E.C. Anti-adherence and bactericidal activity of sphingolipids against Streptococcus mutans. Eur. J. Oral Sci. 2015, 123, 221–227. [Google Scholar] [CrossRef]
- Becam, J.; Walter, T.; Burgert, A.; Schlegel, J.; Sauer, M.; Seibel, J.; Schubert-Unkmeir, A. Antibacterial activity of ceramide and ceramide analogs against pathogenic Neisseria. Sci. Rep. 2017, 7, 17627. [Google Scholar] [CrossRef] [PubMed]
- Seitz, A.P.; Schumacher, F.; Baker, J.; Soddemann, M.; Wilker, B.; Caldwell, C.C.; Gobble, R.M.; Kamler, M.; Becker, K.A.; Beck, S.; et al. Sphingosine-coating of plastic surfaces prevents ventilator-associated pneumonia. J. Mol. Med. 2019, 97, 1195–1211. [Google Scholar] [CrossRef] [PubMed]
- Verhaegh, R.; Becker, K.A.; Edwards, M.J.; Gulbins, E. Sphingosine kills bacteria by binding to cardiolipin. J. Biol. Chem. 2020, 295, 7686–7696. [Google Scholar] [CrossRef] [PubMed]
- Huwiler, A.; Zangemeister-Wittke, U. The sphingosine 1-phosphate receptor modulator fingolimod as a therapeutic agent: Recent findings and new perspectives. Pharmacol. Ther. 2018, 185, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Kihara, Y.; Jonnalagadda, D.; Blaho, V.A. Fingolimod: Lessons Learned and New Opportunities for Treating Multiple Sclerosis and Other Disorders. Annu. Rev. Pharm. Toxicol. 2019, 59, 149–170. [Google Scholar] [CrossRef]
- Rumah, K.R.; Vartanian, T.K.; Fischetti, V.A. Oral Multiple Sclerosis Drugs Inhibit the In vitro Growth of Epsilon Toxin Producing Gut Bacterium, Clostridium perfringens. Front. Cell. Infect. Microbiol. 2017, 7, 11. [Google Scholar] [CrossRef]
- Verderosa, A.D.; Totsika, M.; Fairfull-Smith, K.E. Bacterial Biofilm Eradication Agents: A Current Review. Front. Chem. 2019, 7, 824. [Google Scholar] [CrossRef]
- Michiels, J.E.; Van den Bergh, B.; Fauvart, M.; Michiels, J. Draft genome sequence of Acinetobacter baumannii strain NCTC 13423, a multidrug-resistant clinical isolate. Stand. Genom. Sci. 2016, 11, 57. [Google Scholar] [CrossRef]
- Grosso-Becerra, M.V.; González-Valdez, A.; Granados-Martínez, M.J.; Morales, E.; Servín-González, L.; Méndez, J.L.; Delgado, G.; Morales-Espinosa, R.; Ponce-Soto, G.Y.; Cocotl-Yañez, M.; et al. Pseudomonas aeruginosa ATCC 9027 is a non-virulent strain suitable for mono-rhamnolipids production. Appl. Microbiol. Biotechnol. 2016, 100, 9995–10004. [Google Scholar] [CrossRef]
- Mah, T.F.; O’Toole, G.A. Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol. 2001, 9, 34–39. [Google Scholar] [CrossRef]
- Waters, C.M.; Bassler, B.L. Quorum sensing: Cell-to-cell communication in bacteria. Annu. Rev. Cell Dev. Biol. 2005, 21, 319–346. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.B.; Bassler, B.L. Quorum sensing in bacteria. Annu. Rev. Microbiol. 2001, 55, 165–199. [Google Scholar] [CrossRef] [PubMed]
- McClean, K.H.; Winson, M.K.; Fish, L.; Taylor, A.; Chhabra, S.R.; Camara, M.; Daykin, M.; Lamb, J.H.; Swift, S.; Bycroft, B.W.; et al. Quorum sensing and Chromobacterium violaceum: Exploitation of violacein production and inhibition for the detection of N-acylhomoserine lactones. Microbiology 1997, 143 Pt 12, 3703–3711. [Google Scholar] [CrossRef]
- Hirakawa, H.; Tomita, H. Interference of bacterial cell-to-cell communication: A new concept of antimicrobial chemotherapy breaks antibiotic resistance. Front. Microbiol. 2013, 4, 114. [Google Scholar] [CrossRef]
- Chung, J.; Goo, E.; Yu, S.; Choi, O.; Lee, J.; Kim, J.; Kim, H.; Igarashi, J.; Suga, H.; Moon, J.S.; et al. Small-molecule inhibitor binding to an N-acyl-homoserine lactone synthase. Proc. Natl. Acad. Sci. USA 2011, 108, 12089–12094. [Google Scholar] [CrossRef] [PubMed]
- Defoirdt, T.; Brackman, G.; Coenye, T. Quorum sensing inhibitors: How strong is the evidence? Trends Microbiol. 2013, 21, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Gopu, V.; Meena, C.K.; Shetty, P.H. Quercetin Influences Quorum Sensing in Food Borne Bacteria: In-Vitro and In-Silico Evidence. PLoS ONE 2015, 10, e0134684. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, V.; Zhong, L.; Wang, H.; Yu, G.; Zhang, Y.; Li, A. Virtual Screening and Biomolecular Interactions of CviR-Based Quorum Sensing Inhibitors Against Chromobacterium violaceum. Front. Cell. Infect. Microbiol. 2018, 8, 292. [Google Scholar] [CrossRef]
- Scutera, S.; Zucca, M.; Savoia, D. Novel approaches for the design and discovery of quorum-sensing inhibitors. Expert Opin. Drug Discov. 2014, 9, 353–366. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterial Strains | Provider | Analyses |
|---|---|---|
| Staphylococcus aureus ATCC 25923 | Faculty of Pharmacy of the University of Helsinki, Helsinki, Finland | Primary screening and concentration–response follow-up, Minimum Bactericidal Concentration (MBC) and Biofilm Preventing Concentration (BPC) determination, time-kill kinetic, resistance development |
| Staphylococcus epidermidis ATCC 35984 (RP62A) | Concentration–response | |
| Escherichia coli K12 | HAMBI collection, Faculty of Agriculture and Forestry, University of Helsinki, Helsinki, Finland | Concentration–response, post-exposure CFU count |
| Acinetobacter baumannii NCTC 13423 | National Collection of Type Culture (NCTC), Salisbury, United Kingdom | Concentration–response, MBC and BPC determination |
| Acinetobacter baumannii NCTC 12156 | Concentration–response | |
| Pseudomonas aeruginosa ATCC 9027 | American Type Culture Collection (ATCC), Wesel, Germany | |
| Pseudomonas aeruginosa ATCC 700829 | ||
| Pseudomonas aeruginosa ATCC 15442 | ||
| Pseudomonas aeruginosa PAO1 | Laboratory of Microbiology, Parasitology and Hygiene (LMPH), University of Antwerp, Antwerp, Belgium | |
| Chromobacterium violaceum ATCC 31532 | ATCC, Wesel, Germany | Quorum sensing inhibition |
| Chromobacterium violaceum NCTC 13278 (Tn5-mutant CV026) | NCTC, Salisbury, United Kingdom |
| % of Replicates with >99% Cells Killed | ||
|---|---|---|
| (Fingolimod) | Planktonic Cells | Adherent Cells |
| 10 µM | 0 | 0 |
| 12.5 µM | 11 | 11 |
| 15 µM | 100 (MBC) | 100 (BPC) |
| 25 µM | 100 | 89 1 |
| 50 µM | 100 | 89 |
| % of Replicates with >99% Cells Killed | ||
|---|---|---|
| (Fingolimod) | Planktonic Cells | Adherent Cells |
| 12.5 µM | 0 | 0 |
| 25 µM | 44 | 11 |
| 50 µM | 56 | 11 |
| 100 µM | 78 | 11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gilbert-Girard, S.; Savijoki, K.; Yli-Kauhaluoma, J.; Fallarero, A. Screening of FDA-Approved Drugs Using a 384-Well Plate-Based Biofilm Platform: The Case of Fingolimod. Microorganisms 2020, 8, 1834. https://doi.org/10.3390/microorganisms8111834
Gilbert-Girard S, Savijoki K, Yli-Kauhaluoma J, Fallarero A. Screening of FDA-Approved Drugs Using a 384-Well Plate-Based Biofilm Platform: The Case of Fingolimod. Microorganisms. 2020; 8(11):1834. https://doi.org/10.3390/microorganisms8111834
Chicago/Turabian StyleGilbert-Girard, Shella, Kirsi Savijoki, Jari Yli-Kauhaluoma, and Adyary Fallarero. 2020. "Screening of FDA-Approved Drugs Using a 384-Well Plate-Based Biofilm Platform: The Case of Fingolimod" Microorganisms 8, no. 11: 1834. https://doi.org/10.3390/microorganisms8111834
APA StyleGilbert-Girard, S., Savijoki, K., Yli-Kauhaluoma, J., & Fallarero, A. (2020). Screening of FDA-Approved Drugs Using a 384-Well Plate-Based Biofilm Platform: The Case of Fingolimod. Microorganisms, 8(11), 1834. https://doi.org/10.3390/microorganisms8111834