Decoding the Genomic Variability among Members of the Bifidobacterium dentium Species

 ,
, 
 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Bifidobacterial Genome Sequences
2.2. Isolation of B. dentium Strains
2.3. Microbial DNA Extraction
2.4. B. dentium Chromosomal Sequencing and Assemblies
2.5. Comparative Genomic Analysis
2.6. Phylogenetic and Phylogenomic Analyses
2.7. Genomic Analyses
2.8. Carbohydrate Growth Assays
2.9. Statistical Analyses
3. Results and Discussion
3.1. Genomic Overview of the B. dentium Taxon
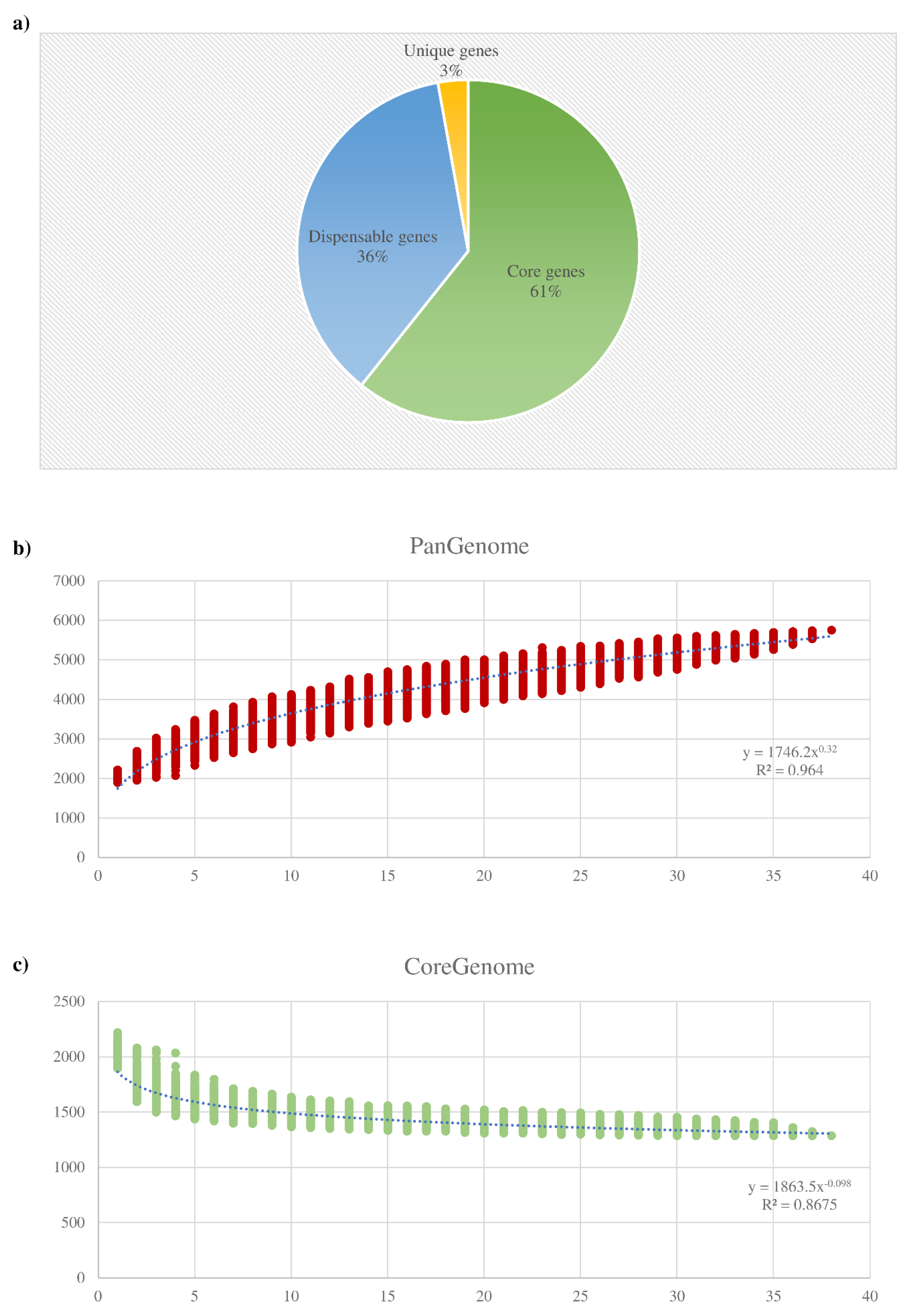
3.2. Pangenome and Core Genome of the B. dentium Species
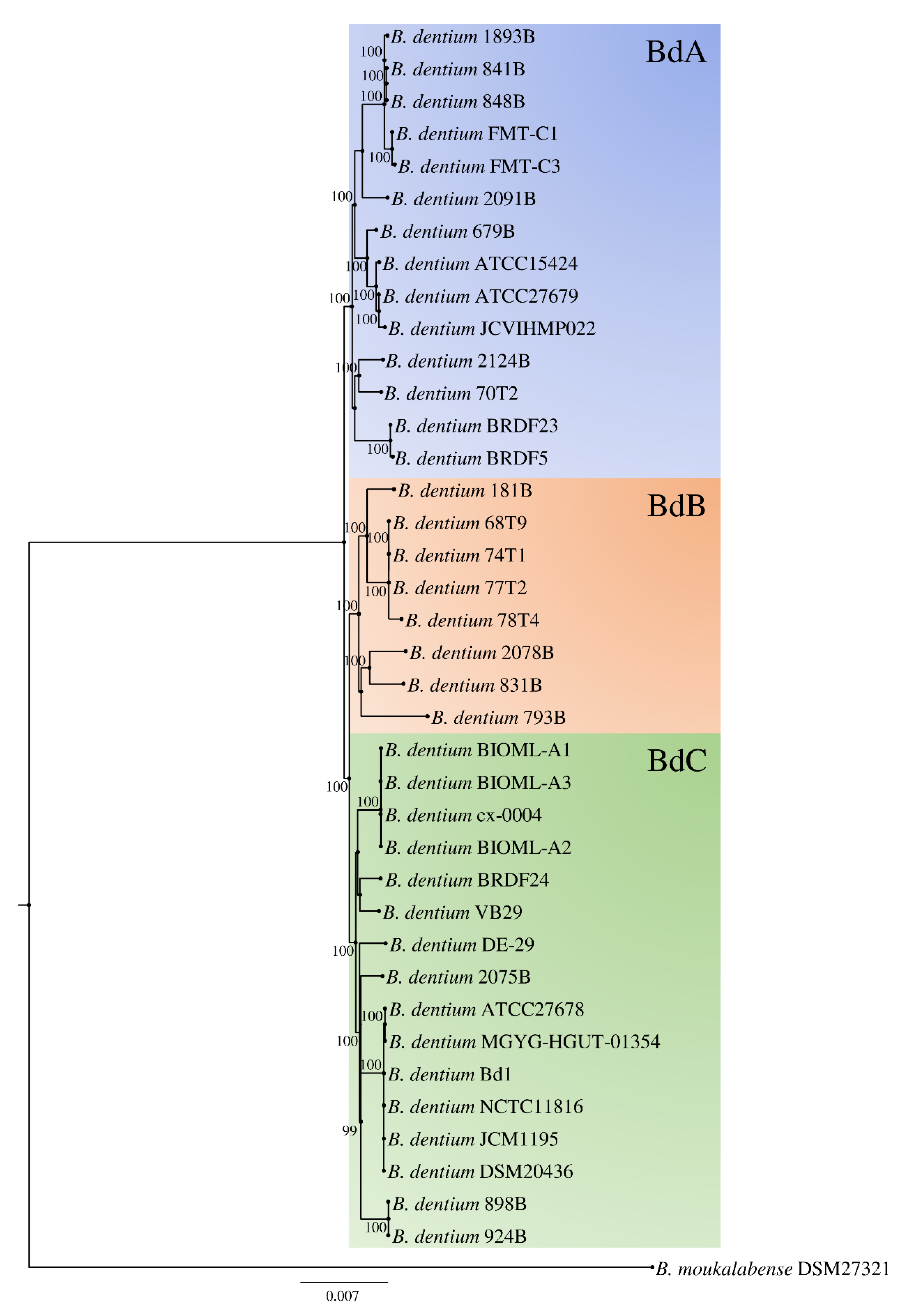
3.3. Phylogenetic Analyses and Evolutionary Development of the B. dentium Taxon
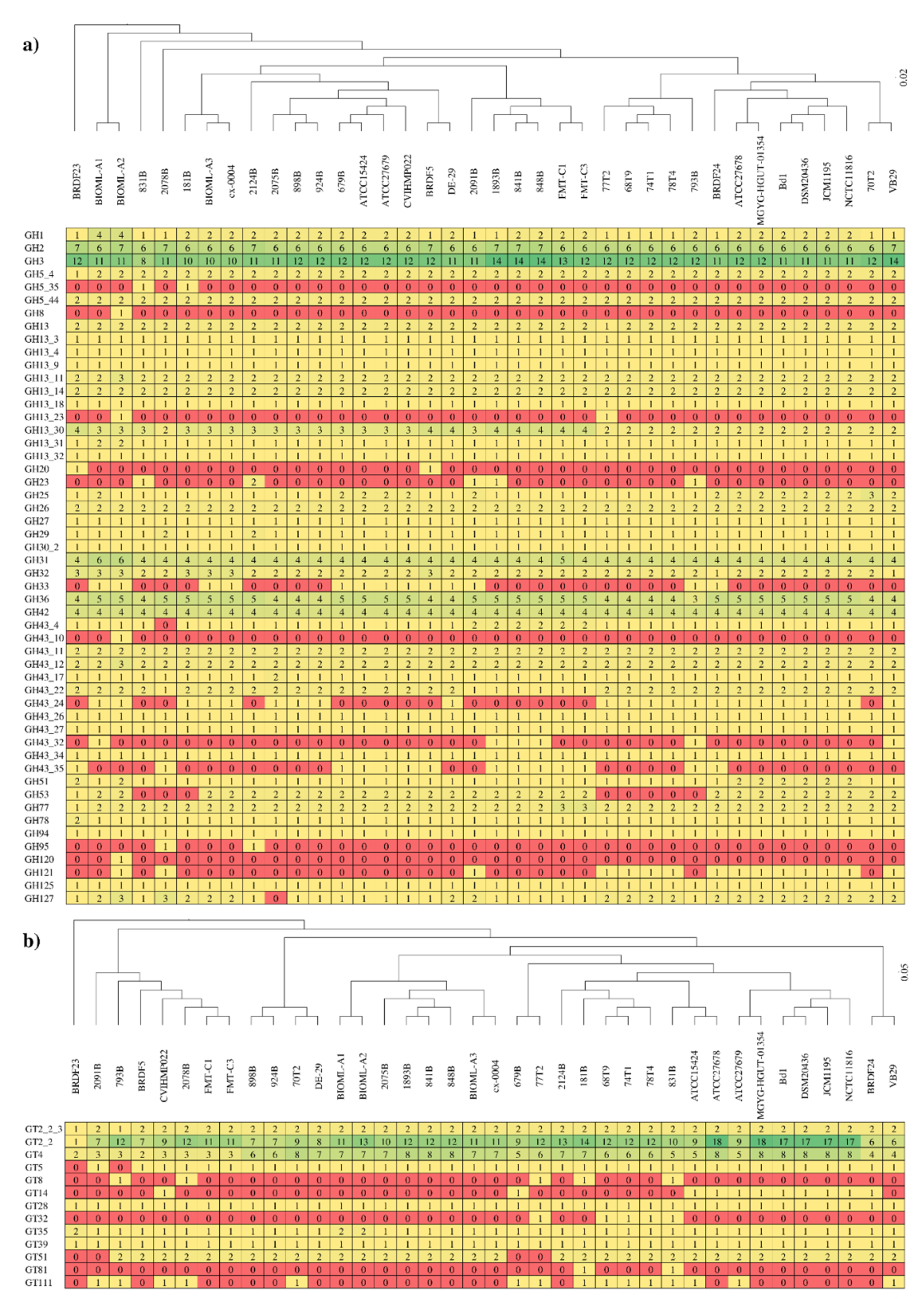
3.4. Carbohydrate-Active Enzymes among Members of the B. dentium Species
3.5. B. dentium Mobilome
3.6. B. dentium Defense Mechanisms
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kostic, A.D.; Howitt, M.R.; Garrett, W.S. Exploring host-microbiota interactions in animal models and humans. Genes Dev. 2013, 27, 701–718. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Donovan, S.M. Introduction to the special focus issue on the impact of diet on gut microbiota composition and function and future opportunities for nutritional modulation of the gut microbiome to improve human health. Gut Microbes 2017, 8, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Milani, C.; Duranti, S.; Bottacini, F.; Casey, E.; Turroni, F.; Mahony, J.; Belzer, C.; Delgado Palacio, S.; Arboleya Montes, S.; Mancabelli, L.; et al. The First Microbial Colonizers of the Human Gut: Composition, Activities, and Health Implications of the Infant Gut Microbiota. Microbiol. Mol. Biol. Rev. Mmbr. 2017, 81. [Google Scholar] [CrossRef] [PubMed]
- Lugli, G.A.; Mangifesta, M.; Duranti, S.; Anzalone, R.; Milani, C.; Mancabelli, L.; Alessandri, G.; Turroni, F.; Ossiprandi, M.C.; van Sinderen, D.; et al. Phylogenetic classification of six novel species belonging to the genus Bifidobacterium comprising Bifidobacterium anseris sp. nov., Bifidobacterium criceti sp. nov., Bifidobacterium imperatoris sp. nov., Bifidobacterium italicum sp. nov., Bifidobacterium margollesii sp. nov. and Bifidobacterium parmae sp. nov. Syst. Appl. Microbiol. 2018, 41, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Lugli, G.A.; Milani, C.; Duranti, S.; Mancabelli, L.; Mangifesta, M.; Turroni, F.; Viappiani, A.; van Sinderen, D.; Ventura, M. Tracking the Taxonomy of the Genus Bifidobacterium Based on a Phylogenomic Approach. Appl. Environ. Microbiol. 2018, 84. [Google Scholar] [CrossRef]
- Duranti, S.; Mangifesta, M.; Lugli, G.A.; Turroni, F.; Anzalone, R.; Milani, C.; Mancabelli, L.; Ossiprandi, M.C.; Ventura, M. Bifidobacterium vansinderenii sp. nov., isolated from faeces of emperor tamarin (Saguinus imperator). Int. J. Syst. Evol. Microbiol. 2017, 67, 3987–3995. [Google Scholar] [CrossRef] [PubMed]
- Modesto, M.; Michelini, S.; Oki, K.; Biavati, B.; Watanabe, K.; Mattarelli, P. Bifidobacterium catulorum sp. nov., a novel taxon from the faeces of the baby common marmoset (Callithrix jacchus). Int. J. Syst. Evol. Microbiol. 2018, 68, 575–581. [Google Scholar] [CrossRef]
- Modesto, M.; Puglisi, E.; Bonetti, A.; Michelini, S.; Spiezio, C.; Sandri, C.; Sgorbati, B.; Morelli, L.; Mattarelli, P. Bifidobacterium primatium sp. nov., Bifidobacterium scaligerum sp. nov., Bifidobacterium felsineum sp. nov. and Bifidobacterium simiarum sp. nov.: Four novel taxa isolated from the faeces of the cotton top tamarin (Saguinus oedipus) and the emperor tamarin (Saguinus imperator). Syst. Appl. Microbiol. 2018, 41, 593–603. [Google Scholar] [CrossRef]
- Modesto, M.; Michelini, S.; Sansosti, M.C.; De Filippo, C.; Cavalieri, D.; Qvirist, L.; Andlid, T.; Spiezio, C.; Sandri, C.; Pascarelli, S.; et al. Bifidobacterium callitrichidarum sp. nov. from the faeces of the emperor tamarin (Saguinus imperator). Int. J. Syst. Evol. Microbiol. 2018, 68, 141–148. [Google Scholar] [CrossRef]
- Michelini, S.; Modesto, M.; Filippini, G.; Spiezio, C.; Sandri, C.; Biavati, B.; Pisi, A.; Mattarelli, P. Corrigendum to “Bifidobacterium aerophilum sp. nov., Bifidobacterium avesanii sp. nov. and Bifidobacterium ramosum sp. nov.: Three novel taxa from the faeces of cotton-top tamarin (Saguinus oedipus L.)” [Syst. Appl. Microbiol. 39 (2016) 229–236]. Syst. Appl. Microbiol. 2018, 41, 528. [Google Scholar] [CrossRef] [PubMed]
- Pechar, R.; Killer, J.; Salmonova, H.; Geigerova, M.; Svejstil, R.; Svec, P.; Sedlacek, I.; Rada, V.; Benada, O. Bifidobacterium apri sp. nov., a thermophilic actinobacterium isolated from the digestive tract of wild pigs (Sus scrofa). Int. J. Syst. Evol. Microbiol. 2017, 67, 2349–2356. [Google Scholar] [CrossRef]
- Alberoni, D.; Gaggia, F.; Baffoni, L.; Modesto, M.M.; Biavati, B.; Di Gioia, D. Bifidobacterium xylocopae sp. nov. and Bifidobacterium aemilianum sp. nov., from the carpenter bee (Xylocopa violacea) digestive tract. Syst. Appl. Microbiol. 2019, 42, 205–216. [Google Scholar] [CrossRef]
- Modesto, M.; Watanabe, K.; Arita, M.; Satti, M.; Oki, K.; Sciavilla, P.; Patavino, C.; Camma, C.; Michelini, S.; Sgorbati, B.; et al. Bifidobacterium jacchi sp. nov., isolated from the faeces of a baby common marmoset (Callithrix jacchus). Int. J. Syst. Evol. Microbiol. 2019, 69, 2477–2485. [Google Scholar] [CrossRef]
- Modesto, M.; Satti, M.; Watanabe, K.; Puglisi, E.; Morelli, L.; Huang, C.H.; Liou, J.S.; Miyashita, M.; Tamura, T.; Saito, S.; et al. Characterization of Bifidobacterium species in feaces of the Egyptian fruit bat: Description of B. vespertilionis sp. nov. and B. rousetti sp. nov. Syst. Appl. Microbiol. 2019, 42, 126017. [Google Scholar] [CrossRef]
- Eckel, V.P.L.; Ziegler, L.M.; Vogel, R.F.; Ehrmann, M. Bifidobacterium tibiigranuli sp. nov. isolated from homemade water kefir. Int. J. Syst. Evol. Microbiol. 2020, 70, 1562–1570. [Google Scholar] [CrossRef] [PubMed]
- Duranti, S.; Lugli, G.A.; Viappiani, A.; Mancabelli, L.; Alessandri, G.; Anzalone, R.; Longhi, G.; Milani, C.; Ossiprandi, M.C.; Turroni, F.; et al. Characterization of the phylogenetic diversity of two novel species belonging to the genus Bifidobacterium: Bifidobacterium cebidarum sp. nov. and Bifidobacterium leontopitheci sp. nov. Int. J. Syst. Evol. Microbiol. 2020, 70, 2288–2297. [Google Scholar] [CrossRef]
- Neuzil-Bunesova, V.; Lugli, G.A.; Modrackova, N.; Makovska, M.; Mrazek, J.; Mekadim, C.; Musilova, S.; Svobodova, I.; Spanek, R.; Ventura, M.; et al. Bifidobacterium canis sp. nov., a novel member of the Bifidobacterium pseudolongum phylogenetic group isolated from faeces of a dog (Canis lupus f. familiaris). Int. J. Syst. Evol. Microbiol. 2020. [Google Scholar] [CrossRef]
- Modesto, M.; Satti, M.; Watanabe, K.; Scarafile, D.; Huang, C.H.; Liou, J.S.; Tamura, T.; Saito, S.; Watanabe, M.; Mori, K.; et al. Phylogenetic characterization of two novel species of the genus Bifidobacterium: Bifidobacterium saimiriisciurei sp. nov. and Bifidobacterium platyrrhinorum sp. nov. Syst. Appl. Microbiol. 2020, 43, 126111. [Google Scholar] [CrossRef]
- Modesto, M.; Biavati, B.; Mattarelli, P. Occurrence of the family bifidobacteriaceae in human dental caries and plaque. Caries Res. 2006, 40, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Mantzourani, M.; Fenlon, M.; Beighton, D. Association between Bifidobacteriaceae and the clinical severity of root caries lesions. Oral Microbiol. Immunol. 2009, 24, 32–37. [Google Scholar] [CrossRef]
- Mantzourani, M.; Gilbert, S.C.; Fenlon, M.; Beighton, D. Non-oral bifidobacteria and the aciduric microbiota of the denture plaque biofilm. Mol. Oral Microbiol. 2010, 25, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Ventura, M.; Turroni, F.; Zomer, A.; Foroni, E.; Giubellini, V.; Bottacini, F.; Canchaya, C.; Claesson, M.J.; He, F.; Mantzourani, M.; et al. The Bifidobacterium dentium Bd1 genome sequence reflects its genetic adaptation to the human oral cavity. PLoS Genet. 2009, 5, e1000785. [Google Scholar] [CrossRef]
- Henne, K.; Rheinberg, A.; Melzer-Krick, B.; Conrads, G. Aciduric microbial taxa including Scardovia wiggsiae and Bifidobacterium spp. in caries and caries free subjects. Anaerobe 2015, 35, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Neves, B.G.; Stipp, R.N.; Bezerra, D.D.S.; Guedes, S.F.F.; Rodrigues, L.K.A. Quantitative analysis of biofilm bacteria according to different stages of early childhood caries. Arch. Oral Biol. 2018, 96, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Milani, C.; Mangifesta, M.; Mancabelli, L.; Lugli, G.A.; James, K.; Duranti, S.; Turroni, F.; Ferrario, C.; Ossiprandi, M.C.; van Sinderen, D.; et al. Unveiling bifidobacterial biogeography across the mammalian branch of the tree of life. Isme J. 2017, 11, 2834–2847. [Google Scholar] [CrossRef]
- Duranti, S.; Milani, C.; Lugli, G.A.; Turroni, F.; Mancabelli, L.; Sanchez, B.; Ferrario, C.; Viappiani, A.; Mangifesta, M.; Mancino, W.; et al. Insights from genomes of representatives of the human gut commensal Bifidobacterium bifidum. Environ. Microbiol. 2015, 17, 2515–2531. [Google Scholar] [CrossRef]
- Duranti, S.; Milani, C.; Lugli, G.A.; Mancabelli, L.; Turroni, F.; Ferrario, C.; Mangifesta, M.; Viappiani, A.; Sanchez, B.; Margolles, A.; et al. Evaluation of genetic diversity among strains of the human gut commensal Bifidobacterium adolescentis. Sci. Rep. 2016, 6, 23971. [Google Scholar] [CrossRef]
- O’Callaghan, A.; Bottacini, F.; O’Connell Motherway, M.; van Sinderen, D. Pangenome analysis of Bifidobacterium longum and site-directed mutagenesis through by-pass of restriction-modification systems. Bmc Genom. 2015, 16, 832. [Google Scholar] [CrossRef]
- Lugli, G.A.; Duranti, S.; Albert, K.; Mancabelli, L.; Napoli, S.; Viappiani, A.; Anzalone, R.; Longhi, G.; Milani, C.; Turroni, F.; et al. Unveiling Genomic Diversity among Members of the Species Bifidobacterium pseudolongum, a Widely Distributed Gut Commensal of the Animal Kingdom. Appl. Environ. Microbiol. 2019, 85. [Google Scholar] [CrossRef]
- Ventura, M.; Zink, R.; Fitzgerald, G.F.; van Sinderen, D. Gene structure and transcriptional organization of the dnaK operon of Bifidobacterium breve UCC 2003 and application of the operon in bifidobacterial tracing. Appl. Environ. Microbiol. 2005, 71, 487–500. [Google Scholar] [CrossRef]
- Strandwitz, P.; Kim, K.H.; Terekhova, D.; Liu, J.K.; Sharma, A.; Levering, J.; McDonald, D.; Dietrich, D.; Ramadhar, T.R.; Lekbua, A.; et al. GABA-modulating bacteria of the human gut microbiota. Nat. Microbiol. 2019, 4, 396–403. [Google Scholar] [CrossRef]
- Duranti, S.; Ruiz, L.; Lugli, G.A.; Tames, H.; Milani, C.; Mancabelli, L.; Mancino, W.; Longhi, G.; Carnevali, L.; Sgoifo, A.; et al. Bifidobacterium adolescentis as a key member of the human gut microbiota in the production of GABA. Sci. Rep. 2020, 10, 14112. [Google Scholar] [CrossRef]
- Pokusaeva, K.; Johnson, C.; Luk, B.; Uribe, G.; Fu, Y.; Oezguen, N.; Matsunami, R.K.; Lugo, M.; Major, A.; Mori-Akiyama, Y.; et al. GABA-producing Bifidobacterium dentium modulates visceral sensitivity in the intestine. Neurogastroenterol. Motil. Off. J. Eur. Gastrointest. Motil. Soc. 2017, 29. [Google Scholar] [CrossRef]
- Engevik, M.A.; Luk, B.; Chang-Graham, A.L.; Hall, A.; Herrmann, B.; Ruan, W.; Endres, B.T.; Shi, Z.; Garey, K.W.; Hyser, J.M.; et al. Bifidobacterium dentium Fortifies the Intestinal Mucus Layer via Autophagy and Calcium Signaling Pathways. mBio 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Turroni, F.; Marchesi, J.R.; Foroni, E.; Gueimonde, M.; Shanahan, F.; Margolles, A.; van Sinderen, D.; Ventura, M. Microbiomic analysis of the bifidobacterial population in the human distal gut. Isme J. 2009, 3, 745–751. [Google Scholar] [CrossRef]
- Milani, C.; Lugli, G.A.; Turroni, F.; Mancabelli, L.; Duranti, S.; Viappiani, A.; Mangifesta, M.; Segata, N.; van Sinderen, D.; Ventura, M. Evaluation of bifidobacterial community composition in the human gut by means of a targeted amplicon sequencing (ITS) protocol. Fems Microbiol. Ecol. 2014, 90, 493–503. [Google Scholar] [CrossRef]
- Lugli, G.A.; Milani, C.; Mancabelli, L.; van Sinderen, D.; Ventura, M. MEGAnnotator: A user-friendly pipeline for microbial genomes assembly and annotation. Fems Microbiol. Lett. 2016, 363. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. A J. Comput. Mol. Cell Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Zhao, Y.; Tang, H.; Ye, Y. RAPSearch2: A fast and memory-efficient protein similarity search tool for next-generation sequencing data. Bioinformatics 2012, 28, 125–126. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rodland, E.A.; Staerfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Brooks, L.; Kaze, M.; Sistrom, M. A Curated, Comprehensive Database of Plasmid Sequences. Microbiol. Resour. Announc. 2019, 8. [Google Scholar] [CrossRef]
- Arredondo-Alonso, S.; Rogers, M.R.C.; Braat, J.C.; Verschuuren, T.D.; Top, J.; Corander, J.; Willems, R.J.L.; Schurch, A.C. mlplasmids: A user-friendly tool to predict plasmid- and chromosome-derived sequences for single species. Microb. Genom. 2018, 4. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, J.; Yang, J.; Sun, S.; Xiao, J.; Yu, J. PGAP: Pan-genomes analysis pipeline. Bioinformatics 2012, 28, 416–418. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Vlietstra, W.J.; Zielman, R.; van Dongen, R.M.; Schultes, E.A.; Wiesman, F.; Vos, R.; van Mulligen, E.M.; Kors, J.A. Automated extraction of potential migraine biomarkers using a semantic graph. J. Biomed. Inf. 2017, 71, 178–189. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Chenna, R.; Sugawara, H.; Koike, T.; Lopez, R.; Gibson, T.J.; Higgins, D.G.; Thompson, J.D. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003, 31, 3497–3500. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez, R.L.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, T.J.; Eddy, S.R. nhmmer: DNA homology search with profile HMMs. Bioinformatics 2013, 29, 2487–2489. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef]
- Roberts, R.J.; Vincze, T.; Posfai, J.; Macelis, D. REBASE—A database for DNA restriction and modification: Enzymes, genes and genomes. Nucleic Acids Res 2015, 43, D298–D299. [Google Scholar] [CrossRef]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Neron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef]
- Waack, S.; Keller, O.; Asper, R.; Brodag, T.; Damm, C.; Fricke, W.F.; Surovcik, K.; Meinicke, P.; Merkl, R. Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinform. 2006, 7, 142. [Google Scholar] [CrossRef]
- Milani, C.; Lugli, G.A.; Duranti, S.; Turroni, F.; Bottacini, F.; Mangifesta, M.; Sanchez, B.; Viappiani, A.; Mancabelli, L.; Taminiau, B.; et al. Genomic encyclopedia of type strains of the genus Bifidobacterium. Appl. Environ. Microbiol. 2014, 80, 6290–6302. [Google Scholar] [CrossRef]
- Lugli, G.A.; Milani, C.; Turroni, F.; Tremblay, D.; Ferrario, C.; Mancabelli, L.; Duranti, S.; Ward, D.V.; Ossiprandi, M.C.; Moineau, S.; et al. Prophages of the genus Bifidobacterium as modulating agents of the infant gut microbiota. Environ. Microbiol. 2016, 18, 2196–2213. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef]
- Lakin, S.M.; Dean, C.; Noyes, N.R.; Dettenwanger, A.; Ross, A.S.; Doster, E.; Rovira, P.; Abdo, Z.; Jones, K.L.; Ruiz, J.; et al. MEGARes: An antimicrobial resistance database for high throughput sequencing. Nucleic Acids Res. 2017, 45, D574–D580. [Google Scholar] [CrossRef] [PubMed]
- Van Heel, A.J.; de Jong, A.; Montalban-Lopez, M.; Kok, J.; Kuipers, O.P. BAGEL3: Automated identification of genes encoding bacteriocins and (non-)bactericidal posttranslationally modified peptides. Nucleic Acids Res. 2013, 41, W448–W453. [Google Scholar] [CrossRef]
- Xie, Y.; Wei, Y.; Shen, Y.; Li, X.; Zhou, H.; Tai, C.; Deng, Z.; Ou, H.Y. TADB 2.0: An updated database of bacterial type II toxin-antitoxin loci. Nucleic Acids Res. 2018, 46, D749–D753. [Google Scholar] [CrossRef]
- Saeed, A.I.; Sharov, V.; White, J.; Li, J.; Liang, W.; Bhagabati, N.; Braisted, J.; Klapa, M.; Currier, T.; Thiagarajan, M.; et al. TM4: A free, open-source system for microarray data management and analysis. BioTechniques 2003, 34, 374–378. [Google Scholar] [CrossRef]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S.; et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial "pan-genome". Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef] [PubMed]
- Albert, K.; Rani, A.; Sela, D.A. Comparative Pangenomics of the Mammalian Gut Commensal Bifidobacterium longum. Microorganisms 2019, 8, 7. [Google Scholar] [CrossRef]
- Bottacini, F.; O’Connell Motherway, M.; Kuczynski, J.; O’Connell, K.J.; Serafini, F.; Duranti, S.; Milani, C.; Turroni, F.; Lugli, G.A.; Zomer, A.; et al. Comparative genomics of the Bifidobacterium breve taxon. BMC Genom. 2014, 15, 170. [Google Scholar] [CrossRef]
- Lugli, G.A.; Milani, C.; Turroni, F.; Duranti, S.; Mancabelli, L.; Mangifesta, M.; Ferrario, C.; Modesto, M.; Mattarelli, P.; Jiri, K.; et al. Comparative genomic and phylogenomic analyses of the Bifidobacteriaceae family. BMC Genom. 2017, 18, 568. [Google Scholar] [CrossRef]
- Lugli, G.A.; Milani, C.; Turroni, F.; Duranti, S.; Ferrario, C.; Viappiani, A.; Mancabelli, L.; Mangifesta, M.; Taminiau, B.; Delcenserie, V.; et al. Investigation of the evolutionary development of the genus Bifidobacterium by comparative genomics. Appl. Environ. Microbiol. 2014, 80, 6383–6394. [Google Scholar] [CrossRef]
- Richter, M.; Rossello-Mora, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef]
- Milani, C.; Lugli, G.A.; Duranti, S.; Turroni, F.; Mancabelli, L.; Ferrario, C.; Mangifesta, M.; Hevia, A.; Viappiani, A.; Scholz, M.; et al. Bifidobacteria exhibit social behavior through carbohydrate resource sharing in the gut. Sci. Rep. 2015, 5, 15782. [Google Scholar] [CrossRef]
- Botstein, D. A theory of modular evolution for bacteriophages. Ann. N.Y. Acad. Sci. 1980, 354, 484–490. [Google Scholar] [CrossRef]
- Bernheim, A.; Sorek, R. The pan-immune system of bacteria: Antiviral defence as a community resource. Nat. Rev. Microbiol. 2020, 18, 113–119. [Google Scholar] [CrossRef]
- Briner, A.E.; Lugli, G.A.; Milani, C.; Duranti, S.; Turroni, F.; Gueimonde, M.; Margolles, A.; van Sinderen, D.; Ventura, M.; Barrangou, R. Occurrence and Diversity of CRISPR-Cas Systems in the Genus Bifidobacterium. PLoS ONE 2015, 10, e0133661. [Google Scholar] [CrossRef]
- Ershova, A.S.; Rusinov, I.S.; Spirin, S.A.; Karyagina, A.S.; Alexeevski, A.V. Role of Restriction-Modification Systems in Prokaryotic Evolution and Ecology. Biochemistry (Moscow) 2015, 80, 1373–1386. [Google Scholar] [CrossRef]
- Duranti, S.; Lugli, G.A.; Mancabelli, L.; Turroni, F.; Milani, C.; Mangifesta, M.; Ferrario, C.; Anzalone, R.; Viappiani, A.; van Sinderen, D.; et al. Prevalence of Antibiotic Resistance Genes among Human Gut-Derived Bifidobacteria. Appl. Environ. Microbiol. 2017, 83. [Google Scholar] [CrossRef]
- Mancino, W.; Lugli, G.A.; Sinderen, D.V.; Ventura, M.; Turroni, F. Mobilome and Resistome Reconstruction from Genomes Belonging to Members of the Bifidobacterium Genus. Microorganisms 2019, 7, 638. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Strain | Host | Isolation Source | Cluster | Number of Bases | COVERAGE | Contigs | GC Content | ORFs | rRNA loci | tRNA | TUGs | GHs Number | GH Index (%) | HGT (%) | Transposase | Prophages | RM Systems | CRISPR System | Accession Number |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Newly sequenced genomes | |||||||||||||||||||
| 68T9 | Macaca silenus | Stool sample | BdB | 2467165 | 74 | 13 | 58.43 | 1997 | 4 | 55 | 23 | 77 | 3.9 | 7 | 0 | 2 | 1 | TypeIU | PRJNA666310 |
| 70T2 | Pan troglodytes | Stool sample | BdA | 2487785 | 96 | 35 | 58.31 | 2046 | 4 | 57 | 114 | 78 | 3.8 | 7.6 | 2 | 3 | 0 | TypeIU | PRJNA666310 |
| 74T1 | Eulemur macaco | Stool sample | BdB | 2469902 | 40 | 15 | 58.43 | 2005 | 4 | 55 | 26 | 77 | 3.8 | 7.3 | 0 | 2 | 1 | TypeIU | PRJNA666310 |
| 77T2 | Lemur catta | Stool sample | BdB | 2472493 | 70 | 37 | 58.43 | 2012 | 4 | 56 | 33 | 77 | 3.8 | 7.3 | 0 | 2 | 1 | TypeIU | PRJNA666310 |
| 78T4 | Colobus guereza | Stool sample | BdB | 2468941 | 44 | 17 | 58.41 | 2007 | 4 | 55 | 31 | 77 | 3.8 | 7.4 | 0 | 2 | 1 | TypeIU | PRJNA666310 |
| 181B | Homo sapiens | Stool sample | BdB | 2523858 | 99 | 10 | 58.45 | 2066 | 4 | 56 | 65 | 78 | 3.8 | 9.7 | 0 | 1 | 3 | TypeIC | PRJNA666310 |
| 679B | Homo sapiens | Stool sample | BdA | 2697950 | 42 | 15 | 58.46 | 2255 | 3 | 56 | 85 | 81 | 3.6 | 9.3 | 2 | 3 | 3 | - | PRJNA666310 |
| 793B | Homo sapiens | Colonoscopy | BdB | 2605010 | 73 | 19 | 58.7 | 2178 | 4 | 53 | 219 | 79 | 3.6 | 9.6 | 1 | 2 | 2 | TypeIC | PRJNA666310 |
| 831B | Homo sapiens | Colonoscopy | BdB | 2479317 | 40 | 14 | 58.38 | 2007 | 4 | 56 | 77 | 72 | 3.6 | 7.4 | 0 | 2 | 2 | TypeIU | PRJNA666310 |
| 841B | Homo sapiens | Stool sample | BdA | 2580247 | 53 | 25 | 58.38 | 2090 | 4 | 56 | 22 | 84 | 4.0 | 7.6 | 1 | 1 | 2 | TypeIIC/TypeIU | PRJNA666310 |
| 848B | Homo sapiens | Colonoscopy | BdA | 2582570 | 122 | 21 | 58.35 | 2091 | 4 | 56 | 25 | 84 | 4.0 | 7.8 | 1 | 1 | 2 | TypeIIC/TypeIU | PRJNA666310 |
| 898B | Homo sapiens | Colonoscopy | BdC | 2459079 | 40 | 24 | 58.49 | 1971 | 4 | 57 | 27 | 78 | 4.0 | 5.6 | 0 | 0 | 3 | TypeIU | PRJNA666310 |
| 924B | Homo sapiens | Colonoscopy | BdC | 2456361 | 97 | 15 | 58.5 | 1966 | 4 | 55 | 23 | 78 | 4.0 | 5.4 | 0 | 0 | 3 | TypeIU | PRJNA666310 |
| 2075B | Pongo pygmaeus | Stool sample | BdC | 2531372 | 255 | 14 | 58.64 | 2078 | 3 | 57 | 100 | 77 | 3.7 | 9.9 | 0 | 2 | 1 | TypeIIC/TypeIIA | PRJNA666310 |
| 2078B | Pongo pygmaeus | Stool sample | BdB | 2612253 | 211 | 19 | 58.57 | 2140 | 4 | 58 | 74 | 79 | 3.7 | 11.3 | 2 | 1 | 1 | TypeIE | PRJNA666310 |
| 2091B | Pongo pygmaeus | Stool sample | BdA | 2537119 | 218 | 34 | 58.68 | 2084 | 4 | 56 | 86 | 81 | 3.9 | 10.9 | 1 | 3 | 3 | - | PRJNA666310 |
| 2124B | Ursus arctos | Stool sample | BdA | 2627726 | 120 | 14 | 58.59 | 2120 | 4 | 55 | 68 | 82 | 3.9 | 8.4 | 2 | 0 | 2 | TypeIC | PRJNA666310 |
| VB29 | Macaca silenus | Stool sample | BdC | 2431695 | 76 | 14 | 58.46 | 1936 | 4 | 55 | 74 | 82 | 4.2 | 7.5 | 1 | 1 | 3 | TypeIC | PRJNA666310 |
| Public genomes | |||||||||||||||||||
| 1893B | Homo sapiens | Stool sample | BdA | 2571068 | - | 24 | 58.22 | 2070 | 4 | 56 | 47 | 84 | 4.1 | 8.4 | 2 | 0 | 3 | TypeIIC/TypeIU | NAQE.1 |
| ATCC15424 | Homo sapiens | Pleural fluid | BdA | 2624481 | - | 19 | 58.48 | 2174 | 4 | 56 | 77 | 81 | 3.7 | 8.4 | 1 | 2 | 3 | TypeIU | SQQK.1 |
| ATCC27678 | Homo sapiens | Stool sample | BdC | 2642081 | - | 2 | 58.52 | 2137 | 4 | 56 | 24 | 82 | 3.8 | 8 | 0 | 2 | 2 | TypeIC/TypeIIC | ABIX.2 |
| ATCC27679 | Homo sapiens | Vaginal tract | BdA | 2644565 | - | 8 | 58.45 | 2200 | n.p. | 58 | 34 | 81 | 3.7 | 9.5 | 1 | 2 | 2 | TypeIU | AEEQ.1 |
| Bd1 | Homo sapiens | Dental caries | BdC | 2636367 | - | 1 | 58.54 | 2142 | 4 | 56 | 28 | 81 | 3.8 | 8.2 | 0 | 2 | 2 | TypeIC/TypeIIC | CP001750.1 |
| BIOML-A1 | Homo sapiens | Stool sample | BdC | 2752113 | - | 88 | 58.22 | 2288 | 3 | 60 | 98 | 88 | 3.8 | 10 | 1 | 1 | 3 | TypeIC | WDPC.1 |
| BIOML-A2 | Homo sapiens | Stool sample | BdC | 2868671 | - | 100 | 58.09 | 2371 | 4 | 61 | 130 | 96 | 4.0 | 9.9 | 1 | 1 | 3 | TypeIC | WDPD.1 |
| BIOML-A3 | Homo sapiens | Stool sample | BdC | 2590214 | - | 13 | 58.46 | 2130 | 4 | 53 | 19 | 80 | 3.8 | 10.5 | 0 | 1 | 2 | TypeIC | WDPE.1 |
| BRDF5 | Bradypus | Stool sample | BdA | 2629155 | - | 16 | 58.59 | 2147 | n.p. | 53 | 26 | 82 | 3.8 | 9.4 | 2 | 0 | 1 | TypeIU | VYSF.1 |
| BRDF23 | Bradypus | Stool sample | BdA | 2557123 | - | 14 | 58.5 | 2068 | 4 | 54 | 20 | 81 | 3.9 | 8.3 | 1 | 0 | 1 | TypeIU | VYSE.1 |
| BRDF24 | Bradypus | Stool sample | BdC | 2511344 | - | 22 | 58.64 | 2049 | 3 | 55 | 82 | 80 | 3.9 | 8.1 | 0 | 1 | 1 | TypeIC | VYSD.1 |
| JCVIHMP022 | Homo sapiens | Urogenital tract | BdA | 2633876 | - | 31 | 58.45 | 2257 | n.p. | 56 | 142 | 81 | 3.6 | 9.1 | 1 | 2 | 2 | TypeIU | AEHJ.1 |
| cx-0004 | Homo sapiens | Stool sample | BdC | 2589878 | - | 19 | 58.46 | 2129 | 4 | 53 | 20 | 80 | 3.8 | 10.3 | 0 | 1 | 2 | TypeIC | RCXJ.1 |
| DE-29 | Homo sapiens | Stool sample | BdC | 2551991 | - | 25 | 58.53 | 2088 | 4 | 55 | 107 | 80 | 3.8 | 8.2 | 4 | 0 | 2 | TypeIC | BCYE.1 |
| DSM20436 | Homo sapiens | Dental caries | BdC | 2668067 | - | 2 | 58.55 | 2177 | 4 | 56 | 42 | 81 | 3.7 | 7.7 | 0 | 2 | 2 | TypeIC/TypeIIC | FNSE.1 |
| FMT-C1 | Homo sapiens | Stool sample | BdA | 2538385 | - | 29 | 58.49 | 2146 | 4 | 56 | 70 | 83 | 3.9 | 7.6 | 1 | 1 | 2 | TypeIIC/TypeIU | JAAWWM0.1 |
| FMT-C3 | Homo sapiens | Stool sample | BdA | 2538722 | - | 32 | 58.49 | 2156 | 4 | 54 | 79 | 81 | 3.8 | 7.5 | 1 | 1 | 2 | TypeIIC/TypeIU | JAAWWN0.1 |
| JCM1195 | Homo sapiens | Dental caries | BdC | 2635669 | - | 1 | 58.54 | 2141 | 4 | 56 | 24 | 81 | 3.8 | 8.2 | 0 | 2 | 2 | TypeIC | AP012326.1 |
| MGYG-HGUT-01354 | Homo sapiens | Stool sample | BdC | 2642081 | - | 2 | 58.52 | 2137 | 4 | 56 | 24 | 82 | 3.8 | 8 | 0 | 2 | 2 | TypeIC/TypeIIC | CABKPB0.1 |
| NCTC11816 | Homo sapiens | Dental caries | BdC | 2635828 | - | 1 | 58.54 | 2140 | 4 | 56 | 24 | 81 | 3.8 | 8.2 | 0 | 2 | 2 | TypeIC/TypeIIC | LR134349.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lugli, G.A.; Tarracchini, C.; Alessandri, G.; Milani, C.; Mancabelli, L.; Turroni, F.; Neuzil-Bunesova, V.; Ruiz, L.; Margolles, A.; Ventura, M. Decoding the Genomic Variability among Members of the Bifidobacterium dentium Species. Microorganisms 2020, 8, 1720. https://doi.org/10.3390/microorganisms8111720
Lugli GA, Tarracchini C, Alessandri G, Milani C, Mancabelli L, Turroni F, Neuzil-Bunesova V, Ruiz L, Margolles A, Ventura M. Decoding the Genomic Variability among Members of the Bifidobacterium dentium Species. Microorganisms. 2020; 8(11):1720. https://doi.org/10.3390/microorganisms8111720
Chicago/Turabian StyleLugli, Gabriele Andrea, Chiara Tarracchini, Giulia Alessandri, Christian Milani, Leonardo Mancabelli, Francesca Turroni, Vera Neuzil-Bunesova, Lorena Ruiz, Abelardo Margolles, and Marco Ventura. 2020. "Decoding the Genomic Variability among Members of the Bifidobacterium dentium Species" Microorganisms 8, no. 11: 1720. https://doi.org/10.3390/microorganisms8111720
APA StyleLugli, G. A., Tarracchini, C., Alessandri, G., Milani, C., Mancabelli, L., Turroni, F., Neuzil-Bunesova, V., Ruiz, L., Margolles, A., & Ventura, M. (2020). Decoding the Genomic Variability among Members of the Bifidobacterium dentium Species. Microorganisms, 8(11), 1720. https://doi.org/10.3390/microorganisms8111720