Mitochondria-Mediated Azole Drug Resistance and Fungal Pathogenicity: Opportunities for Therapeutic Development

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Role of Fungal Mitochondria in Azole Resistance
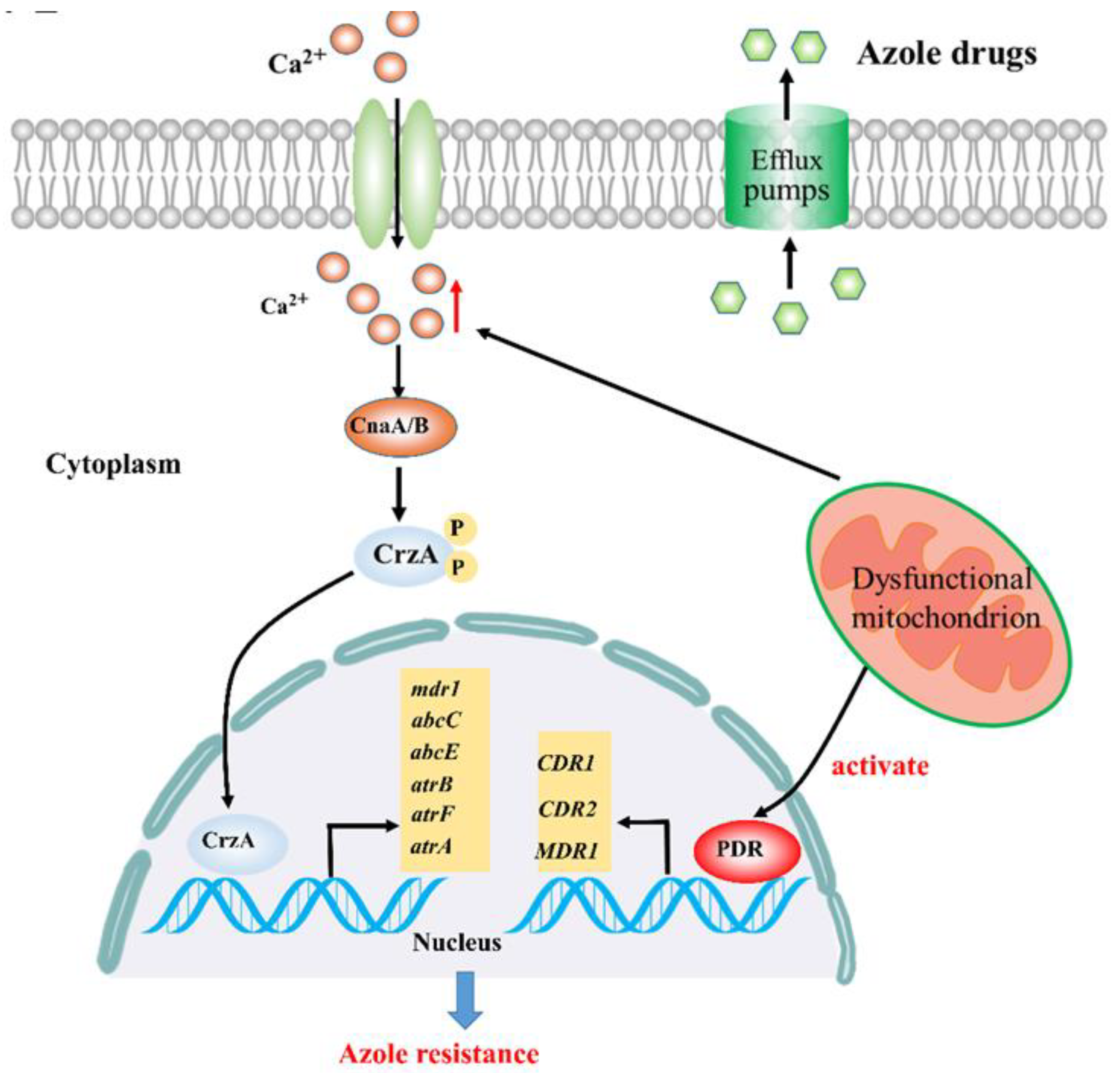
2.1. Calcium Signaling Participates in Mitochondria-Mediated Azole Resistance
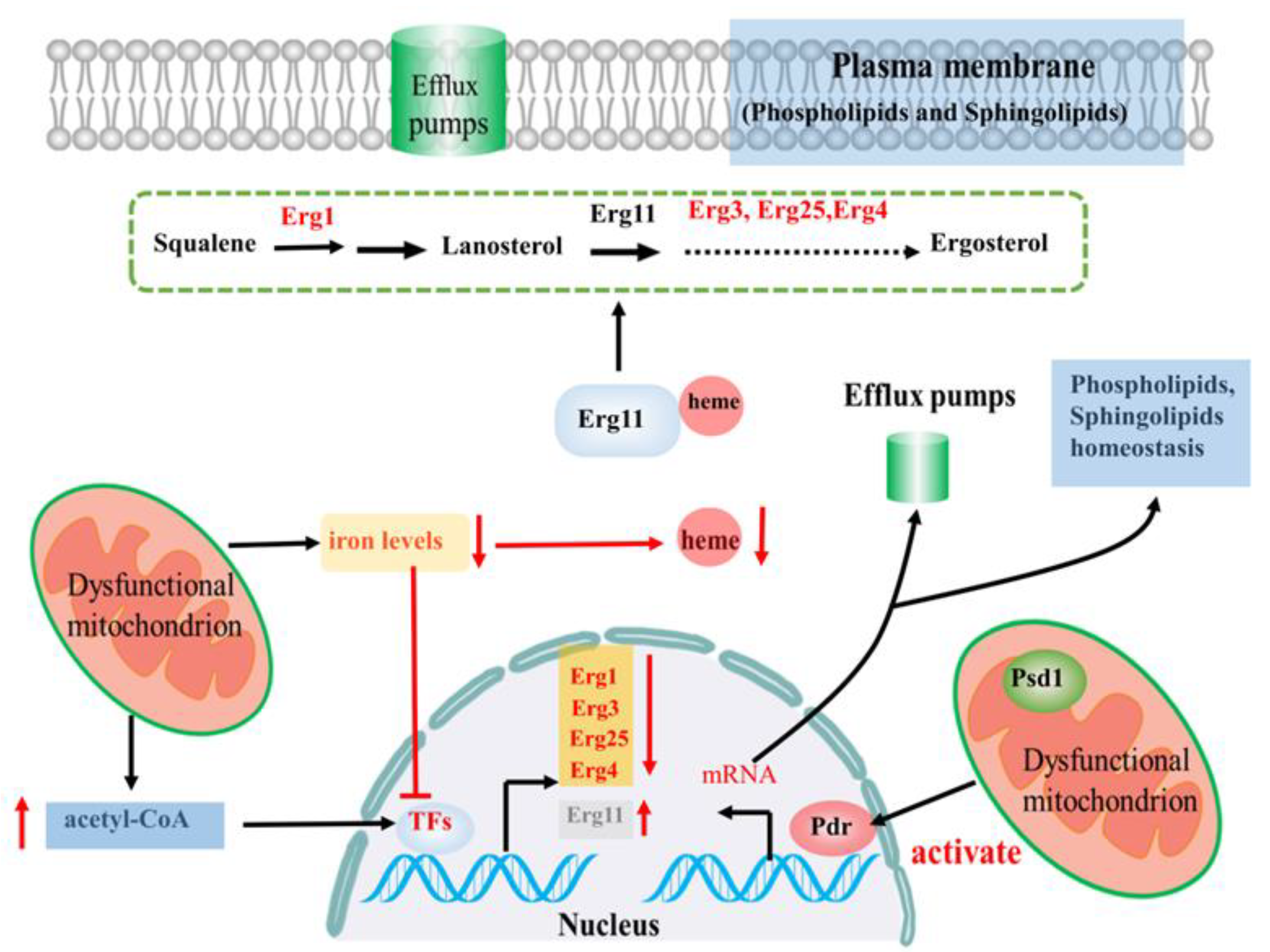
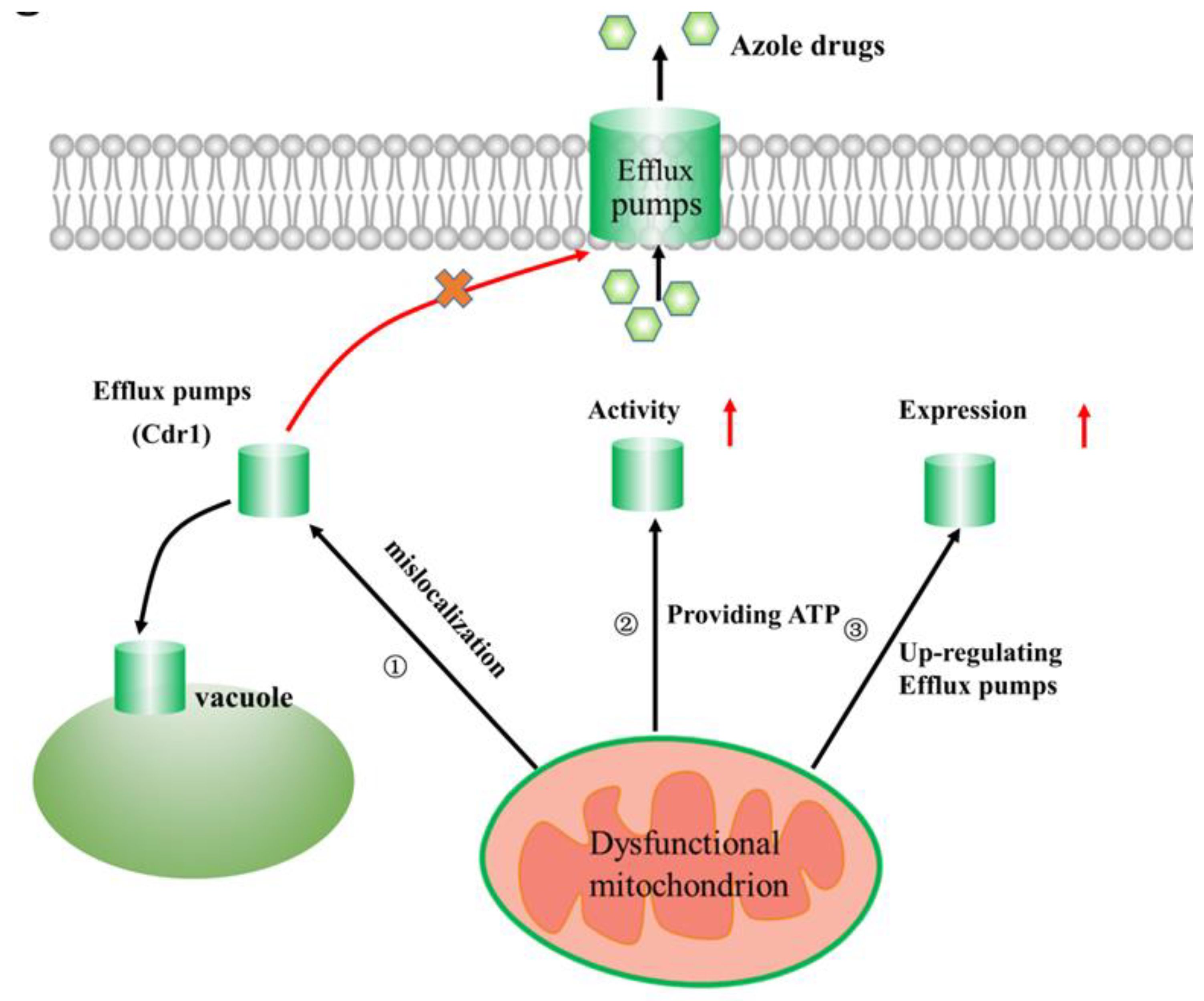
2.2. Linking Efflux Pump Activity and Mitochondrial Function in Azole Drug Resistance
2.3. A Link Between Mitochondrial Dynamics and Azole Resistance
2.4. Linking Fungal Lipid Biosynthesis and Mitochondrial Function in Azole Drug Resistance
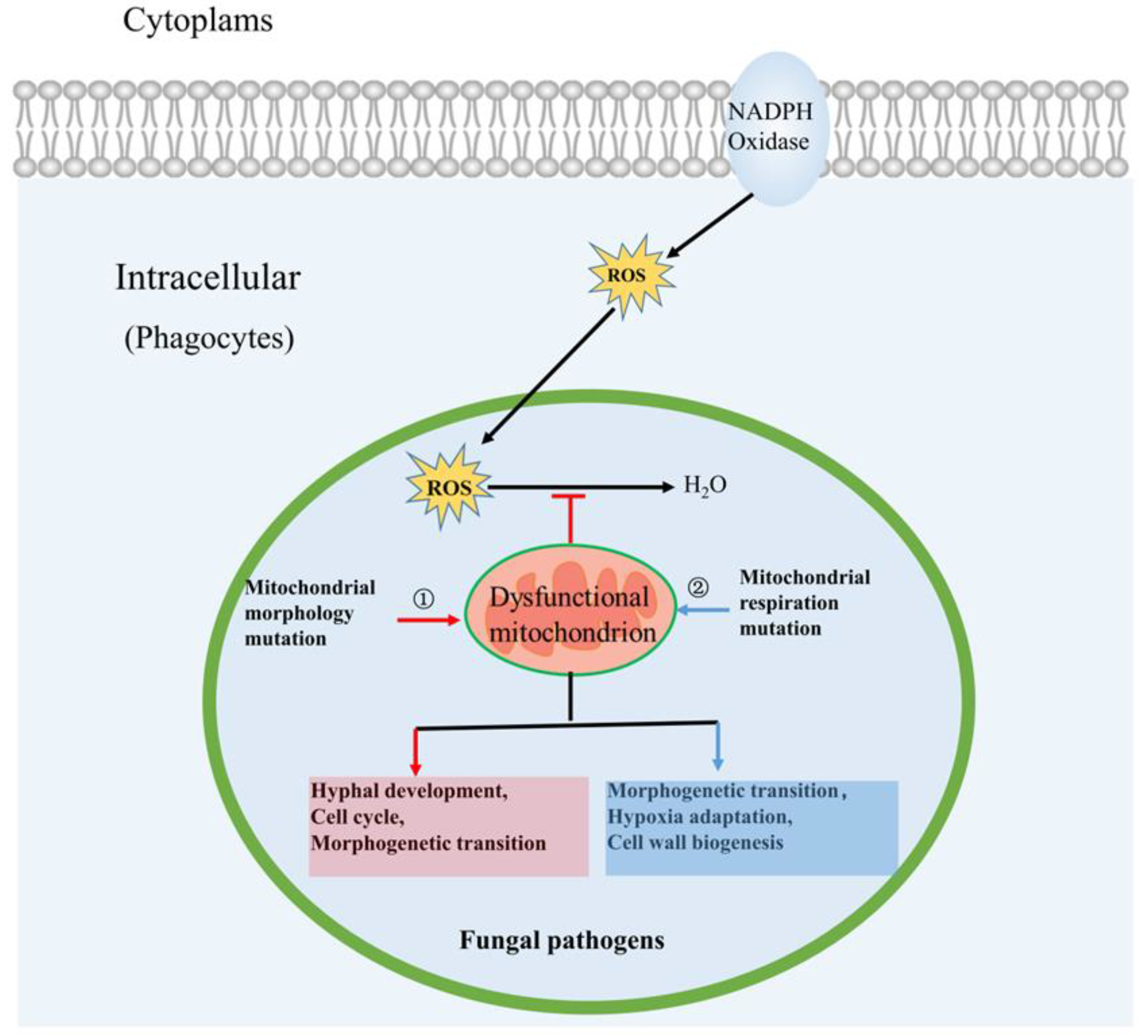
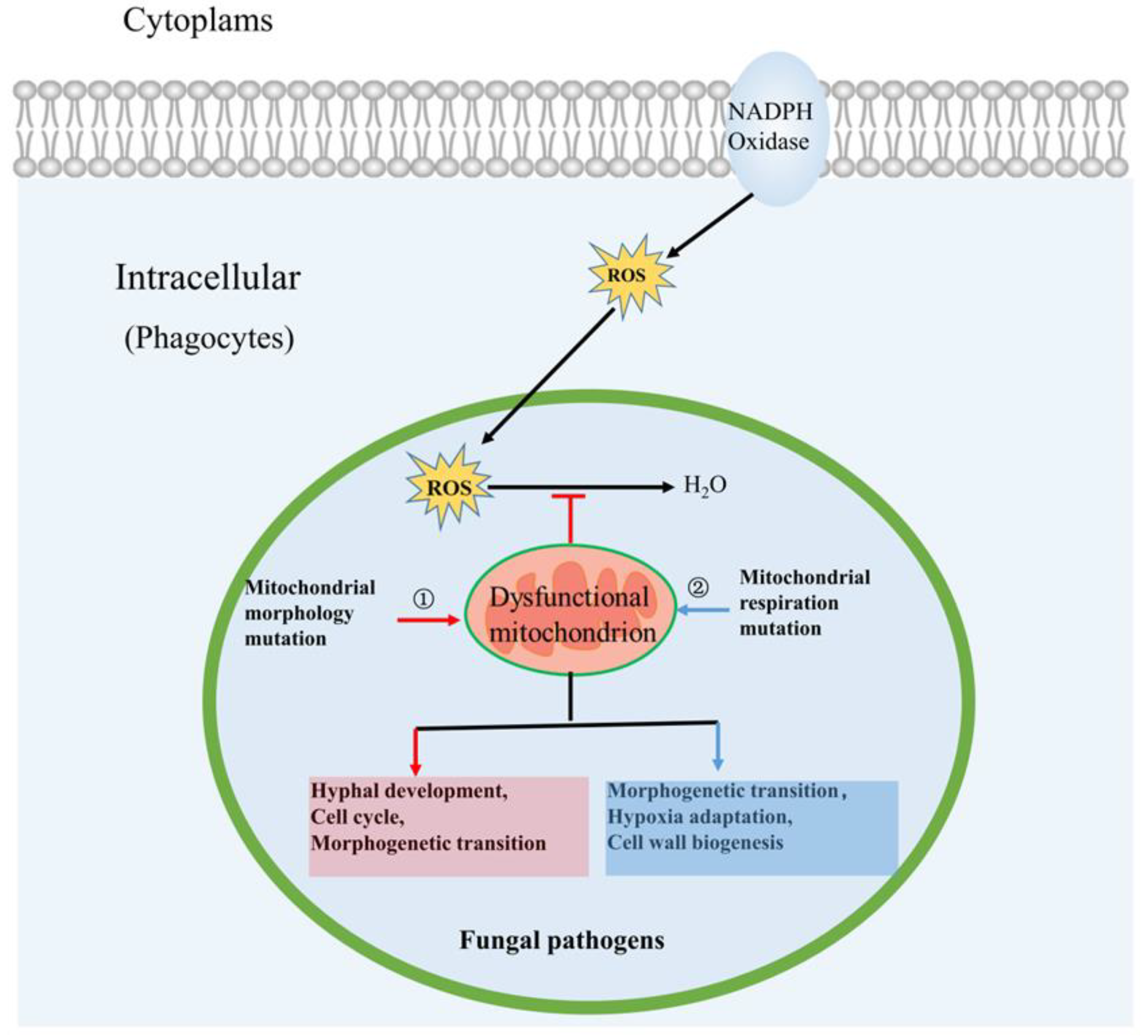
3. Roles of Mitochondria in Fungal Pathogenicity
3.1. Mitochondrial Morphology Influences Fungal Pathogenicity
3.2. Mitochondrial Respiration Influences Fungal Pathogenicity
4. Potential for Mitochondrial Factors as Novel Antifungal Therapeutic Targets
5. Conclusions and Future Prospects
Author Contributions
Funding
Conflicts of Interest
References
- Pianalto, K.M.; Alspaugh, J.A. New Horizons in Antifungal Therapy. J. Fungi 2016, 2, 26. [Google Scholar]
- Houst, J.; Spizek, J.; Havlicek, V. Antifungal Drugs. Metabolites 2020, 10, 106. [Google Scholar]
- Carmona, E.M.; Limper, A.H. Overview of Treatment Approaches for Fungal Infections. Clin. Chest Med. 2017, 38, 393–402. [Google Scholar]
- Song, J.; Zhang, S.; Lu, L. Fungal cytochrome P450 protein Cyp51: What we can learn from its evolution, regulons and Cyp51-based azole resistance. Fungal Biol. Rev. 2018, 32, 131–142. [Google Scholar]
- Geißel, B.; Loiko, V.; Klugherz, I.; Zhu, Z.; Wagener, N.; Kurzai, O.; Hondel, C.A.M.J.J.V.D.; Wagener, J. Azole-induced cell wall carbohydrate patches kill Aspergillus fumigatus. Nat. Commun. 2018, 9, 3098. [Google Scholar]
- Meletiadis, J.; Antachopoulos, C.; Stergiopoulou, T.; Pournaras, S.; Roilides, E.; Walsh, T.J. Differential Fungicidal Activities of Amphotericin B and Voriconazole against Aspergillus Species Determined by Microbroth Methodology. Antimicrob. Agents Chemother. 2007, 51, 3329–3337. [Google Scholar]
- Song, J.; Zhai, P.; Zhang, Y.; Zhang, C.; Sang, H.; Han, G.; Keller, N.P.; Lu, L. TheAspergillus fumigatusDamage Resistance Protein Family Coordinately Regulates Ergosterol Biosynthesis and Azole Susceptibility. mBio 2016, 7, e01919-15. [Google Scholar]
- Shapiro, R.S.; Robbins, N.; Cowen, L.E. Regulatory Circuitry Governing Fungal Development, Drug Resistance, and Disease. Microbiol. Mol. Biol. Rev. 2011, 75, 213–267. [Google Scholar]
- Robbins, N.; Caplan, T.; Cowen, L.E. Molecular Evolution of Antifungal Drug Resistance. Annu. Rev. Microbiol. 2017, 71, 753–775. [Google Scholar]
- Chowdhary, A.; Meis, J.F. Emergence of azole resistant Aspergillus fumigatus and One Health: Time to implement environmental stewardship. Environ. Microbiol. 2018, 20, 1299–1301. [Google Scholar]
- Revie, N.M.; Iyer, K.R.; Robbins, N.; Cowen, L.E. Antifungal drug resistance: Evolution, mechanisms and impact. Curr. Opin. Microbiol. 2018, 45, 70–76. [Google Scholar]
- Brown, A.J.; Brown, G.D.; Netea, M.G.; Gow, N.A. Metabolism impacts upon Candida immunogenicity and pathogenicity at multiple levels. Trends Microbiol. 2014, 22, 614–622. [Google Scholar]
- Sun, N.; Parrish, R.S.; Calderone, R.A.; Fonzi, W.A. Unique, Diverged, and Conserved Mitochondrial Functions Influencing Candida albicans Respiration. mBio 2019, 10, e00300-19. [Google Scholar]
- Thomas, E.; Roman, E.; Claypool, S.M.; Manzoor, N.; Pla, J.; Panwar, S.L. Mitochondria InfluenceCDR1Efflux Pump Activity, Hog1-Mediated Oxidative Stress Pathway, Iron Homeostasis, and Ergosterol Levels in Candida albicans. Antimicrob. Agents Chemother. 2013, 57, 5580–5599. [Google Scholar]
- Dagley, M.J.; Gentle, I.E.; Beilharz, T.H.; Pettolino, F.A.; Djordjevic, J.T.; Lo, T.L.; Uwamahoro, N.; Rupasinghe, T.; Tull, D.L.; McConville, M.; et al. Cell wall integrity is linked to mitochondria and phospholipid homeostasis in Candida albicans through the activity of the post-transcriptional regulator Ccr4-Pop2. Mol. Microbiol. 2010, 79, 968–989. [Google Scholar]
- Shingu-Vazquez, M.; Traven, A. Mitochondria and Fungal Pathogenesis: Drug Tolerance, Virulence, and Potential for Antifungal Therapy. Eukaryot. Cell 2011, 10, 1376–1383. [Google Scholar]
- Vincent, B.M.; Langlois, J.-B.; Srinivas, R.; Lancaster, A.K.; Scherz-Shouval, R.; Whitesell, L.; Tidor, B.; Buchwald, S.L.; Lindquist, S. A Fungal-Selective Cytochrome bc1 Inhibitor Impairs Virulence and Prevents the Evolution of Drug Resistance. Cell Chem. Biol. 2016, 23, 978–991. [Google Scholar]
- Long, N.; Xu, X.; Qian, H.; Zhang, S.; Lu, L. A Putative Mitochondrial Iron Transporter MrsA in Aspergillus fumigatus Plays Important Roles in Azole-, Oxidative Stress Responses and Virulence. Front. Microbiol. 2016, 7, 716. [Google Scholar]
- Demuyser, L.; Swinnen, E.; Fiori, A.; Herrera-Malaver, B.; Verstrepen, K.J.; Van Dijck, P. Mitochondrial Cochaperone Mge1 Is Involved in Regulating Susceptibility to Fluconazole in Saccharomyces cerevisiae and Candida Species. mBio 2017, 8, e00201-17. [Google Scholar]
- Duvenage, L.; Munro, C.A.; Gourlay, C.W. The potential of respiration inhibition as a new approach to combat human fungal pathogens. Curr. Genet. 2019, 65, 1347–1353. [Google Scholar]
- She, X.; Khamooshi, K.; Gao, Y.; Shen, Y.; Lv, Y.; Calderone, R.; Fonzi, W.; Liu, W.; Li, N. Fungal-specific subunits of the Candida albicans mitochondrial complex I drive diverse cell functions including cell wall synthesis. Cell. Microbiol. 2015, 17, 1350–1364. [Google Scholar]
- Li, S.X.; Song, Y.J.; Zhang, Y.S.; Wu, H.T.; Guo, H.; Zhu, K.J.; Li, D.-M.; Zhang, H. Mitochondrial Complex V proportional to Subunit Is Critical for Candida albicans Pathogenicity through Modulating Multiple Virulence Properties. Front Microbiol. 2017, 8, 285. [Google Scholar]
- Qu, Y.; Jelicic, B.; Pettolino, F.; Perry, A.J.; Lo, T.L.; Hewitt, V.; Bantun, F.; Beilharz, T.H.; Peleg, A.Y.; Lithgow, T.; et al. Mitochondrial Sorting and Assembly Machinery Subunit Sam37 in Candida albicans: Insight into the Roles of Mitochondria in Fitness, Cell Wall Integrity, and Virulence. Eukaryot. Cell 2012, 11, 532–544. [Google Scholar]
- Li, Y.; Zhang, Y.; Zhang, C.; Wang, H.; Wei, X.; Chen, P.; Lu, L. Mitochondrial dysfunctions trigger the calcium signaling-dependent fungal multidrug resistance. Proc. Natl. Acad. Sci. USA 2019, 117, 1711–1721. [Google Scholar]
- Bowyer, P.; Bromley, M.J.; Denning, D.W. Linking calcium signaling and mitochondrial function in fungal drug resistance. Proc. Natl. Acad. Sci. USA 2020, 117, 1254–1256. [Google Scholar]
- Neubauer, M.; Zhu, Z.J.; Penka, M.; Helmschrott, C.; Wagener, N.; Wagener, J. Mitochondrial dynamics in the pathogenic mold Aspergillus fumigatus: Therapeutic and evolutionary implications. Mol. Microbiol. 2015, 98, 930–945. [Google Scholar]
- Li, Y.; Zhang, Y.; Lu, L. Calcium signaling pathway is involved in non-CYP51 azole resistance in Aspergillus fumigatus. Med. Mycol. 2019, 57, S233–S238. [Google Scholar]
- Miyazaki, T.; Yamauchi, S.; Inamine, T.; Nagayoshi, Y.; Saijo, T.; Izumikawa, K.; Seki, M.; Kakeya, H.; Yamamoto, Y.; Yanagihara, K.; et al. Roles of Calcineurin and Crz1 in Antifungal Susceptibility and Virulence of Candida glabrata. Antimicrob. Agents Chemother. 2010, 54, 1639–1643. [Google Scholar]
- Yu, S.-J.; Chang, Y.-L.; Chen, Y.-L. Calcineurin signaling: Lessons from Candida species. FEMS Yeast Res. 2015, 15. [Google Scholar] [CrossRef] [Green Version]
- Edlind, T.; Smith, L.; Henry, K.; Katiyar, S.; Nickels, J. Antifungal activity in Saccharomyces cerevisiae is modulated by calcium signalling. Mol. Microbiol. 2002, 46, 257–268. [Google Scholar]
- Xu, H.; Fang, T.; Omran, R.P.; Whiteway, M.; Jiang, L. RNA sequencing reveals an additional Crz1-binding motif in promoters of its target genes in the human fungal pathogen Candida albicans. Cell Commun. Signal. 2020, 18, 1–14. [Google Scholar]
- Holmes, A.; Cardno, T.S.; Strouse, J.J.; Ivnitski-Steele, I.; Keniya, M.V.; Lackovic, K.; Monk, B.C.; Sklar, L.A.; Cannon, R.D. Targeting efflux pumps to overcome antifungal drug resistance. Future Med. Chem. 2016, 8, 1485–1501. [Google Scholar]
- Paul, R.A.; Rudramurthy, S.M.; Dhaliwal, M.; Singh, P.; Ghosh, A.K.; Kaur, H.; Varma, S.; Agarwal, R.; Chakrabarti, A. Magnitude of Voriconazole Resistance in Clinical and Environmental Isolates of Aspergillus flavus and Investigation into the Role of Multidrug Efflux Pumps. Antimicrob. Agents Chemother. 2018, 62, e01022-18. [Google Scholar]
- Singh, S.; Fatima, Z.; Ahmad, K.; Hameed, S. Fungicidal action of geraniol against Candida albicans is potentiated by abrogated CaCdr1p drug efflux and fluconazole synergism. PLoS ONE 2018, 13, e0203079. [Google Scholar]
- Li, N.; Calderone, R. Exploiting mitochondria as targets for the development of new antifungals. Virulence 2016, 8, 159–168. [Google Scholar]
- Janganan, T.K.; Bavro, V.N.; Zhang, L.; Borges-Walmsley, M.I.; Walmsley, A.R. Tripartite efflux pumps: Energy is required for dissociation, but not assembly or opening of the outer membrane channel of the pump. Mol. Microbiol. 2013, 88, 590–602. [Google Scholar]
- Guo, H.; Xie, S.; Li, S.; Song, Y.; Zhong, X.; Zhang, H. Involvement of mitochondrial aerobic respiratory activity in efflux-mediated resistance of C. albicans to fluconazole. J. Mycol. Médicale 2017, 27, 339–344. [Google Scholar]
- Guo, H.; Xie, S.M.; Li, S.X.; Song, Y.J.; Lv, X.L.; Zhang, H. Synergistic mechanism for tetrandrine on fluconazole against Candida albicans through the mitochondrial aerobic respiratory metabolism pathway. J. Med. Microbiol. 2014, 63, 988–996. [Google Scholar]
- Neubauer, M.; Zhu, Z.; Penka, M.; Helmschrott, C.; Wagener, J. The role of mitochondrial dynamics in the pathogenic mold Aspergillus Fumigatus. Mycoses 2015, 58, 2. [Google Scholar]
- Kaur, R.; Castaño, I.; Cormack, B.P. Functional Genomic Analysis of Fluconazole Susceptibility in the Pathogenic Yeast Candida glabrata: Roles of Calcium Signaling and Mitochondria. Antimicrob. Agents Chemother. 2004, 48, 1600–1613. [Google Scholar]
- Sturm, L.; Geißel, B.; Martin, R.; Wagener, J. Differentially Regulated Transcription Factors and ABC Transporters in a Mitochondrial Dynamics Mutant Can Alter Azole Susceptibility of Aspergillus fumigatus. Front. Microbiol. 2020, 11, 1017. [Google Scholar]
- Truong, T.; Zeng, G.; Lim, T.K.; Cao, T.; Pang, L.M.; Lee, Y.M.; Lin, Q.; Wang, Y.; Seneviratne, C.J. Proteomics Analysis of Candida albicans dnm1 Haploid Mutant Unraveled the Association between Mitochondrial Fission and Antifungal Susceptibility. Proteomics 2019, 20, 1900240. [Google Scholar]
- Horianopoulos, L.C.; Kronstad, J.W. Connecting iron regulation and mitochondrial function in Cryptococcus neoformans. Curr. Opin. Microbiol. 2019, 52, 7–13. [Google Scholar]
- Lu, Z.; Jia, X.; Chen, Y.; Han, X.; Chen, F.; Tian, S.; Su, X.; Li, Z.; Zhao, J.; Zhang, X.; et al. Identification and Characterization of Key Charged Residues in the Cofilin Protein Involved in Azole Susceptibility, Apoptosis, and Virulence of Aspergillus fumigatus. Antimicrob. Agents Chemother. 2018, 62, e01659-17. [Google Scholar]
- Gulshan, K.; Schmidt, J.A.; Shahi, P.; Moye-Rowley, W.S. Evidence for the Bifunctional Nature of Mitochondrial Phosphatidylserine Decarboxylase: Role in Pdr3-Dependent Retrograde Regulation of PDR5 Expression. Mol. Cell. Biol. 2008, 28, 5851–5864. [Google Scholar]
- Balzi, E.; Moye-Rowley, W.S. Unveiling the transcriptional control of pleiotropic drug resistance in Saccharomyces cerevisiae: Contributions of André Goffeau and his group. Yeast 2018, 36, 195–200. [Google Scholar]
- Morschhäuser, J. Regulation of multidrug resistance in pathogenic fungi. Fungal Genet. Biol. 2010, 47, 94–106. [Google Scholar]
- Moye-Rowley, W.S. Multiple interfaces control activity of the Candida glabrata Pdr1 transcription factor mediating azole drug resistance. Curr. Genet. 2018, 65, 103–108. [Google Scholar]
- Paul, S.; Schmidt, J.A.; Moye-Rowley, W.S. Regulation of the CgPdr1 Transcription Factor from the Pathogen Candida glabrata. Eukaryot. Cell 2010, 10, 187–197. [Google Scholar]
- Song, J.; Liu, X.; Li, R. Sphingolipids: Regulators of azole drug resistance and fungal pathogenicity. Mol. Microbiol. 2020. [Google Scholar] [CrossRef]
- Suchodolski, J.; Muraszko, J.; Korba, A.; Bernat, P.; Krasowska, A. Lipid composition and cell surface hydrophobicity of Candida albicans influence the efficacy of fluconazole–gentamicin treatment. Yeast 2020, 37, 117–129. [Google Scholar]
- Verma, S.; Shakya, V.P.S.; Idnurm, A. Exploring and exploiting the connection between mitochondria and the virulence of human pathogenic fungi. Virulence 2018, 9, 426–446. [Google Scholar]
- Youle, R.; Van Der Bliek, A.M. Mitochondrial Fission, Fusion, and Stress. Science 2012, 337, 1062–1065. [Google Scholar]
- Chang, A.L.; Doering, T.L. Maintenance of Mitochondrial Morphology inCryptococcus neoformansIs Critical for Stress Resistance and Virulence. mBio 2018, 9, e01375-18. [Google Scholar]
- Sousa, C.A.; Soares, E.V. Mitochondria are the main source and one of the targets of Pb (lead)-induced oxidative stress in the yeast Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 2014, 98, 5153–5160. [Google Scholar]
- Liang, C.; Zhang, B.; Cui, L.; Li, J.; Yu, Q.; Li, M. Mgm1 is required for maintenance of mitochondrial function and virulence in Candida albicans. Fungal Genet. Biol. 2018, 120, 42–52. [Google Scholar]
- Leipheimer, J.; Bloom, A.L.M.; Campomizzi, C.S.; Salei, Y.; Panepinto, J.C. Translational Regulation Promotes Oxidative Stress Resistance in the Human Fungal Pathogen Cryptococcus neoformans. mBio 2019, 10, e02143-19. [Google Scholar]
- McClelland, E.E.; Ramagopal, U.A.; Rivera, J.; Cox, J.; Nakouzi, A.; Prabu, M.M.; Almo, S.C.; Casadevall, A. A Small Protein Associated with Fungal Energy Metabolism Affects the Virulence of Cryptococcus neoformans in Mammals. PLOS Pathog. 2016, 12, e1005849. [Google Scholar]
- Paulussen, C.; Hallsworth, J.E.; Álvarez-Pérez, S.; Nierman, W.C.; Hamill, P.G.; Blain, D.; Rediers, H.; Lievens, B. Ecology of aspergillosis: Insights into the pathogenic potency ofAspergillus fumigatusand some otherAspergillusspecies. Microb. Biotechnol. 2016, 10, 296–322. [Google Scholar]
- Grahl, N.; Dinamarco, T.M.; Willger, S.D.; Goldman, G.H.; Cramer, R.A. Aspergillus fumigatusmitochondrial electron transport chain mediates oxidative stress homeostasis, hypoxia responses and fungal pathogenesis. Mol. Microbiol. 2012, 84, 383–399. [Google Scholar]
- She, X.; Zhang, P.; Gao, Y.; Zhang, L.; Wang, Q.; Chen, H.; Calderone, R.; Liu, W.; Li, N. A mitochondrial proteomics view of complex I deficiency in Candida albicans. Mitochondrion 2018, 38, 48–57. [Google Scholar]
- Schrettl, M.; Beckmann, N.; Varga, J.; Eheinekamp, T.; Jacobsen, I.D.; Jöchl, C.; Moussa, T.A.; Wang, S.; Gsaller, F.; Blatzer, M.; et al. HapX-Mediated Adaption to Iron Starvation Is Crucial for Virulence of Aspergillus fumigatus. PLoS Pathog. 2010, 6, e1001124. [Google Scholar]
- Kroll, K.; Pähtz, V.; Hillmann, F.; Vaknin, Y.; Schmidt-Heck, W.; Roth, M.; Jacobsen, I.D.; Osherov, N.; Brakhage, A.A.; Kniemeyer, O. Identification of Hypoxia-Inducible Target Genes of Aspergillus fumigatus by Transcriptome Analysis Reveals Cellular Respiration as an Important Contributor to Hypoxic Survival. Eukaryot. Cell 2014, 13, 1241–1253. [Google Scholar]
- Grahl, N.; Demers, E.G.; Lindsay, A.K.; Harty, C.E.; Willger, S.D.; Piispanen, A.E.; Hogan, D.A. Mitochondrial Activity and Cyr1 Are Key Regulators of Ras1 Activation of C. albicans Virulence Pathways. PLoS Pathog. 2015, 11, e1005133. [Google Scholar]
- Silao, F.G.S.; Ward, M.; Ryman, K.; Wallstrom, A.; Brindefalk, B.; Udekwu, K.; Ljungdahl, P.O. Mitochondrial proline catabolism activates Ras1/cAMP/PKA-induced filamentation in Candida albicans. PLoS Genet. 2019, 15, e1007976. [Google Scholar]
- Basso, V.; D’Enfert, C.; Znaidi, S.; Bachellier-Bassi, S. From Genes to Networks: The Regulatory Circuitry Controlling Candida albicans Morphogenesis. Fungal Physiol. Immunopathog. 2018, 422, 61–99. [Google Scholar]
- Tripathi, A.; Liverani, E.; Tsygankov, A.; Puri, S. Iron alters the cell wall composition and intracellular lactate to affect Candida albicans susceptibility to antifungals and host immune response. J. Biol. Chem. 2020, 295. [Google Scholar] [CrossRef]
- Netea, M.G.; Joosten, L.A.B.; Van Der Meer, J.W.M.; Kullberg, B.-J.; Van De Veerdonk, F.L. Immune defence against Candida fungal infections. Nat. Rev. Immunol. 2015, 15, 630–642. [Google Scholar]
- Pradhan, A.; Avelar, G.M.; Bain, J.M.; Childers, D.S.; Larcombe, D.E.; Netea, M.G.; Shekhova, E.; Munro, C.A.; Brown, G.D.; Erwig, L.P.; et al. Hypoxia Promotes Immune Evasion by Triggering beta-Glucan Masking on the Candida albicans Cell Surface via Mitochondrial and cAMP-Protein Kinase A Signaling. mbio 2018, 9, e01318-18. [Google Scholar]
- Koch, B.; Traven, A. Mitochondrial Control of Fungal Cell Walls: Models and Relevance in Fungal Pathogens. Future HIV-1 Ther. 2019, 425, 1–20. [Google Scholar]
- Li, D.; She, X.; Calderone, R. Functional diversity of complex I subunits in Candida albicans mitochondria. Curr. Genet. 2015, 62, 87–95. [Google Scholar]
- She, X.D.; Zhang, L.L.; Chen, H.; Calderone, R.; Li, D.M. Cell surface changes in the Candida albicans mitochondrial mutant goa1 Delta are associated with reduced recognition by innate immune cells. Cell Microbiol. 2013, 15, 1572–1584. [Google Scholar]
- Satish, S.; Jiménez-Ortigosa, C.; Zhao, Y.; Lee, M.H.; Dolgov, E.; Krüger, T.; Park, S.; Denning, D.W.; Kniemeyer, O.; Brakhage, A.A.; et al. Stress-Induced Changes in the Lipid Microenvironment of β-(1,3)-D-Glucan Synthase Cause Clinically Important Echinocandin Resistance in Aspergillus fumigatus. mBio 2019, 10, e00779-19. [Google Scholar]
- Mottie, S.; Riemer, J.; Wideman, J.G.; McBride, H.M. A new mitofusin topology places the redox-regulated C terminus in the mitochondrial intermembrane space. J. Cell Biol. 2018, 217, 507–515. [Google Scholar]
- Bambach, A.; Fernandes, M.P.; Ghosh, A.; Kruppa, M.; Alex, D.; Li, D.; Fonzi, W.A.; Chauhan, N.; Sun, N.; Agrellos, O.A.; et al. Goa1p of Candida albicans Localizes to the Mitochondria during Stress and Is Required for Mitochondrial Function and Virulence. Eukaryot. Cell 2009, 8, 1706–1720. [Google Scholar]
- Li, D.M.; Chen, H.; Florentino, A.; Alex, D.; Sikorski, P.; Fonzi, W.A.; Calderone, R. Enzymatic Dysfunction of Mitochondrial Complex I of the Candida albicans goa1 Mutant Is Associated with Increased Reactive Oxidants and Cell Death. Eukaryot. Cell 2011, 10, 672–682. [Google Scholar]
- Lin, Z.; Wu, J.; Jamieson, P.A.; Zhang, C.Q. Alternative Oxidase Is Involved in the Pathogenicity, Development, and Oxygen Stress Response of Botrytis cinerea. Phytopathology 2019, 109, 1679–1688. [Google Scholar]
- Duvenage, L.; Walker, L.A.; Bojarczuk, A.; Johnston, S.A.; Maccallum, D.M.; Munro, C.A.; Gourlay, C.W. Inhibition of Classical and Alternative Modes of Respiration in Candida albicans Leads to Cell Wall Remodeling and Increased Macrophage Recognition. mBio 2019, 10, e02535-18. [Google Scholar]
- Ebiloma, G.U.; Balogun, E.O.; Cueto-Díaz, E.J.; De Koning, H.P.; Dardonville, C. Alternative oxidase inhibitors: Mitochondrion-targeting as a strategy for new drugs against pathogenic parasites and fungi. Med. Res. Rev. 2019, 39, 1553–1602. [Google Scholar]
- Yan, L.; Li, M.; Cao, Y.; Gao, P.; Cao, Y.; Wang, Y.; Jiang, Y. The alternative oxidase of Candida albicans causes reduced fluconazole susceptibility. J. Antimicrob. Chemother. 2009, 64, 764–773. [Google Scholar]
- Ott, R.; Chibale, K.; Anderson, S.; Chipeleme, A.; Chaudhuri, M.; Guerrah, A.; Colowick, N.; Hill, G.C. Novel inhibitors of the trypanosome alternative oxidase inhibit Trypanosoma brucei brucei growth and respiration. Acta Trop. 2006, 100, 172–184. [Google Scholar]
- Barsottini, M.R.; A Pires, B.; Vieira, M.L.; Pereira, J.G.; Costa, P.C.; Sanitá, J.; Coradini, A.; Mello, F.; Marschalk, C.; Silva, E.M.; et al. Synthesis and testing of novel alternative oxidase (AOX) inhibitors with antifungal activity against Moniliophthora perniciosa (Stahel), the causal agent of witches’ broom disease of cocoa, and other phytopathogens. Pest Manag. Sci. 2018, 75, 1295–1303. [Google Scholar]
- Kroll, K.; Shekhova, E.; Mattern, D.J.; Thywissen, A.; Jacobsen, I.D.; Strassburger, M.; Eheinekamp, T.; Shelest, E.; Brakhage, A.A.; Kniemeyer, O. The hypoxia-induced dehydrogenase HorA is required for coenzyme Q10 biosynthesis, azole sensitivity and virulence ofAspergillus fumigatus. Mol. Microbiol. 2016, 101, 92–108. [Google Scholar]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, J.; Zhou, J.; Zhang, L.; Li, R. Mitochondria-Mediated Azole Drug Resistance and Fungal Pathogenicity: Opportunities for Therapeutic Development. Microorganisms 2020, 8, 1574. https://doi.org/10.3390/microorganisms8101574
Song J, Zhou J, Zhang L, Li R. Mitochondria-Mediated Azole Drug Resistance and Fungal Pathogenicity: Opportunities for Therapeutic Development. Microorganisms. 2020; 8(10):1574. https://doi.org/10.3390/microorganisms8101574
Chicago/Turabian StyleSong, Jinxing, Jingwen Zhou, Lei Zhang, and Rongpeng Li. 2020. "Mitochondria-Mediated Azole Drug Resistance and Fungal Pathogenicity: Opportunities for Therapeutic Development" Microorganisms 8, no. 10: 1574. https://doi.org/10.3390/microorganisms8101574
APA StyleSong, J., Zhou, J., Zhang, L., & Li, R. (2020). Mitochondria-Mediated Azole Drug Resistance and Fungal Pathogenicity: Opportunities for Therapeutic Development. Microorganisms, 8(10), 1574. https://doi.org/10.3390/microorganisms8101574