Combined Use of Diagnostic Fumarate Addition Metabolites and Genes Provides Evidence for Anaerobic Hydrocarbon Biodegradation in Contaminated Groundwater



Abstract
1. Introduction
2. Materials and Methods
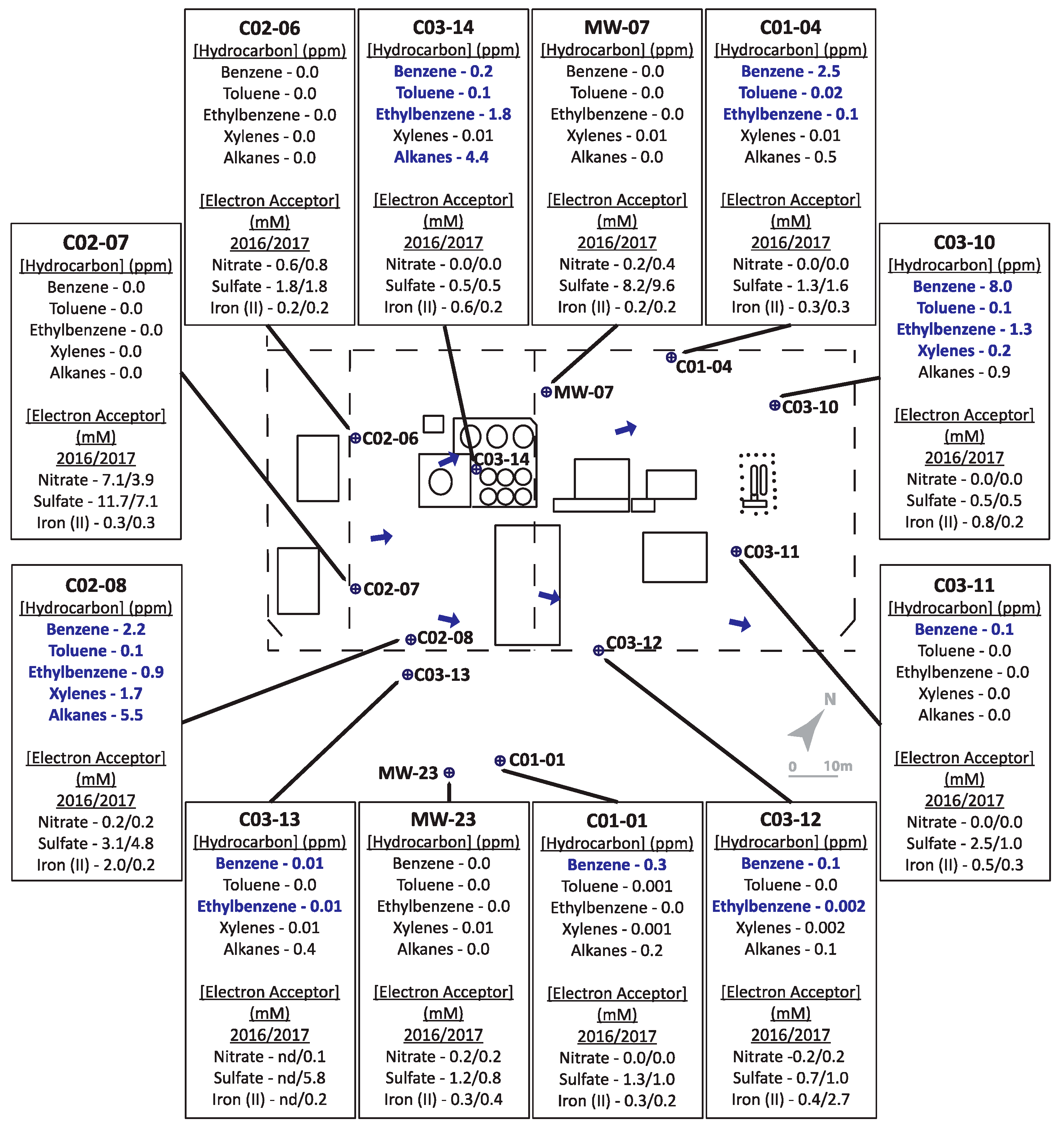
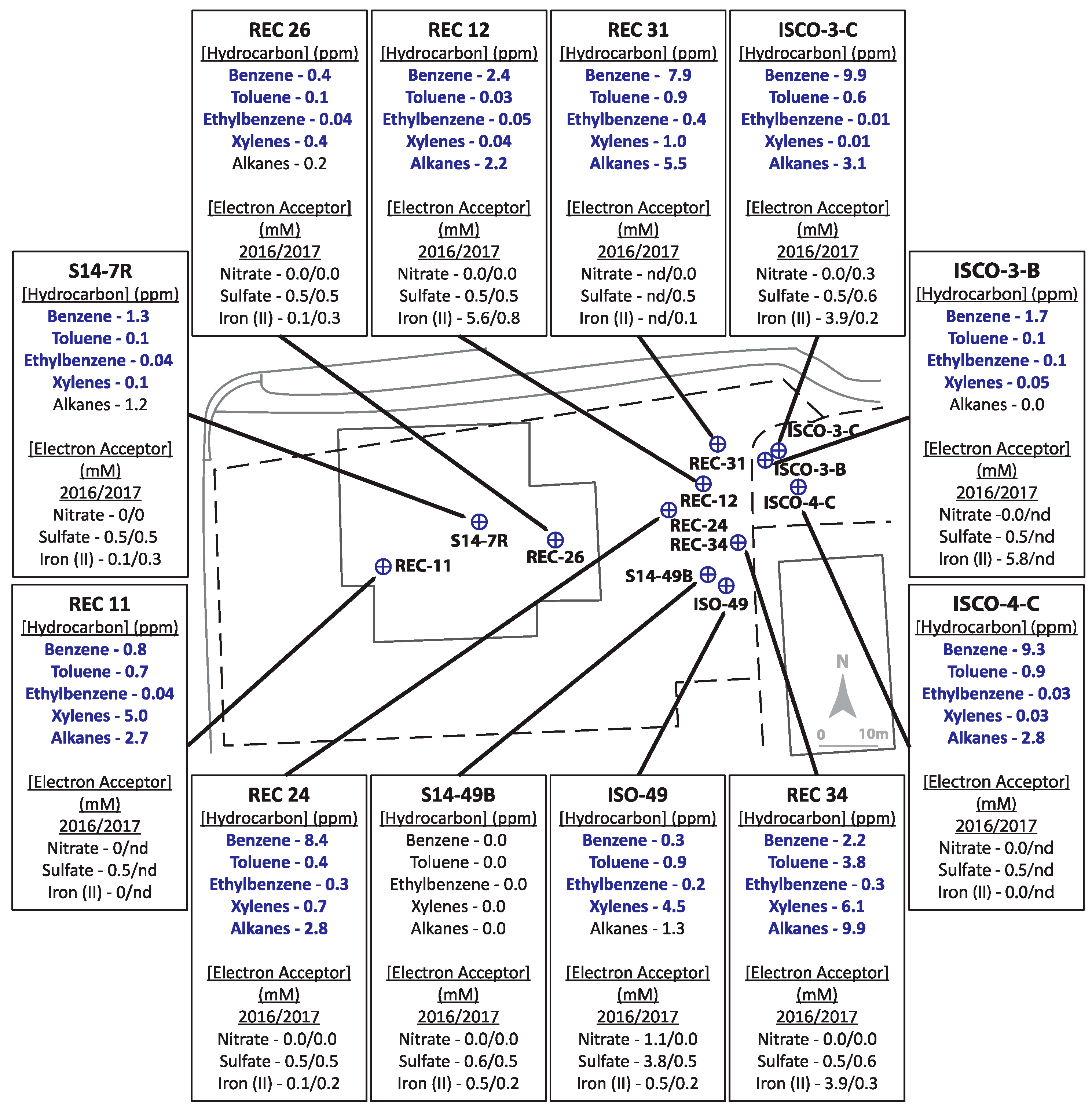
2.1. Site Descriptions
2.2. Sample Collection
2.3. Hydrocarbon Metabolite Analysis
2.4. Biomass Collection and DNA Extraction
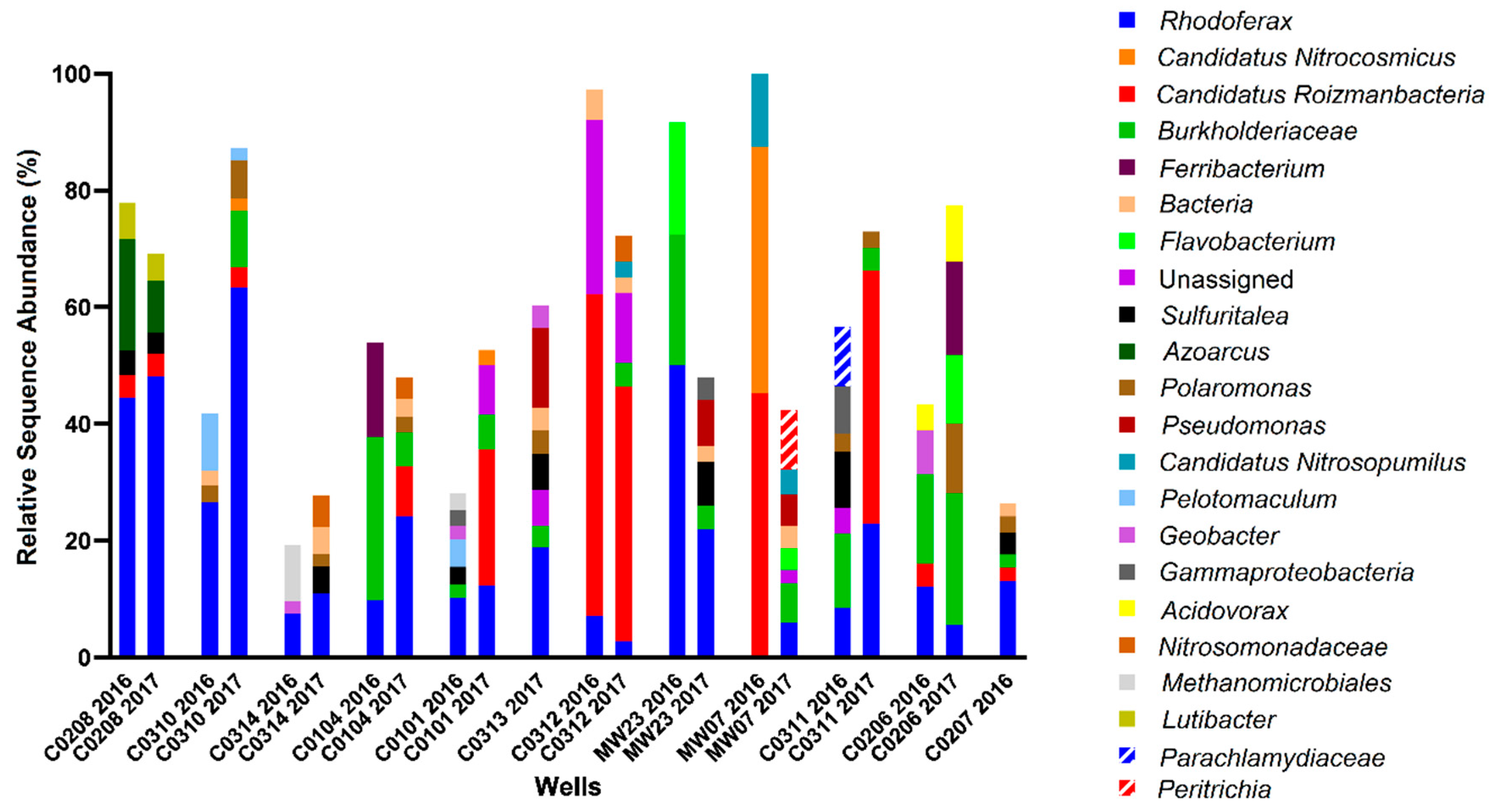
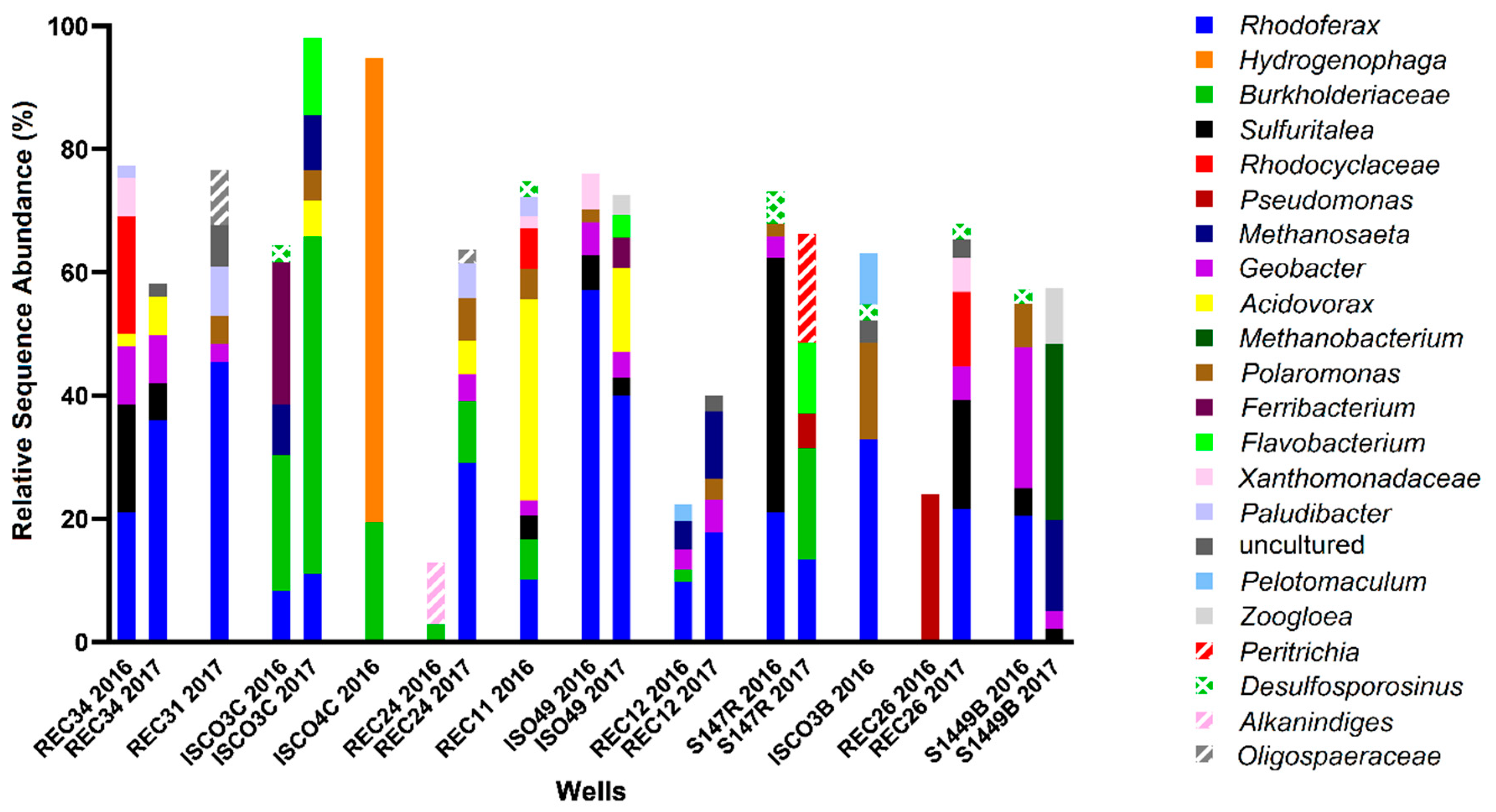
2.5. 16S rRNA Gene Analysis for Microbial Community Profiling
2.6. Qualitative and Quantitative Assays for assA and bssA Genes
2.7. Illumina Sequencing of assA and bssA Amplicons
3. Results
3.1. Hydrocarbons, Putative Electron Acceptors, and Microbial Community Profiles
3.2. Fumarate Addition Metabolites and Biodegradation Genes
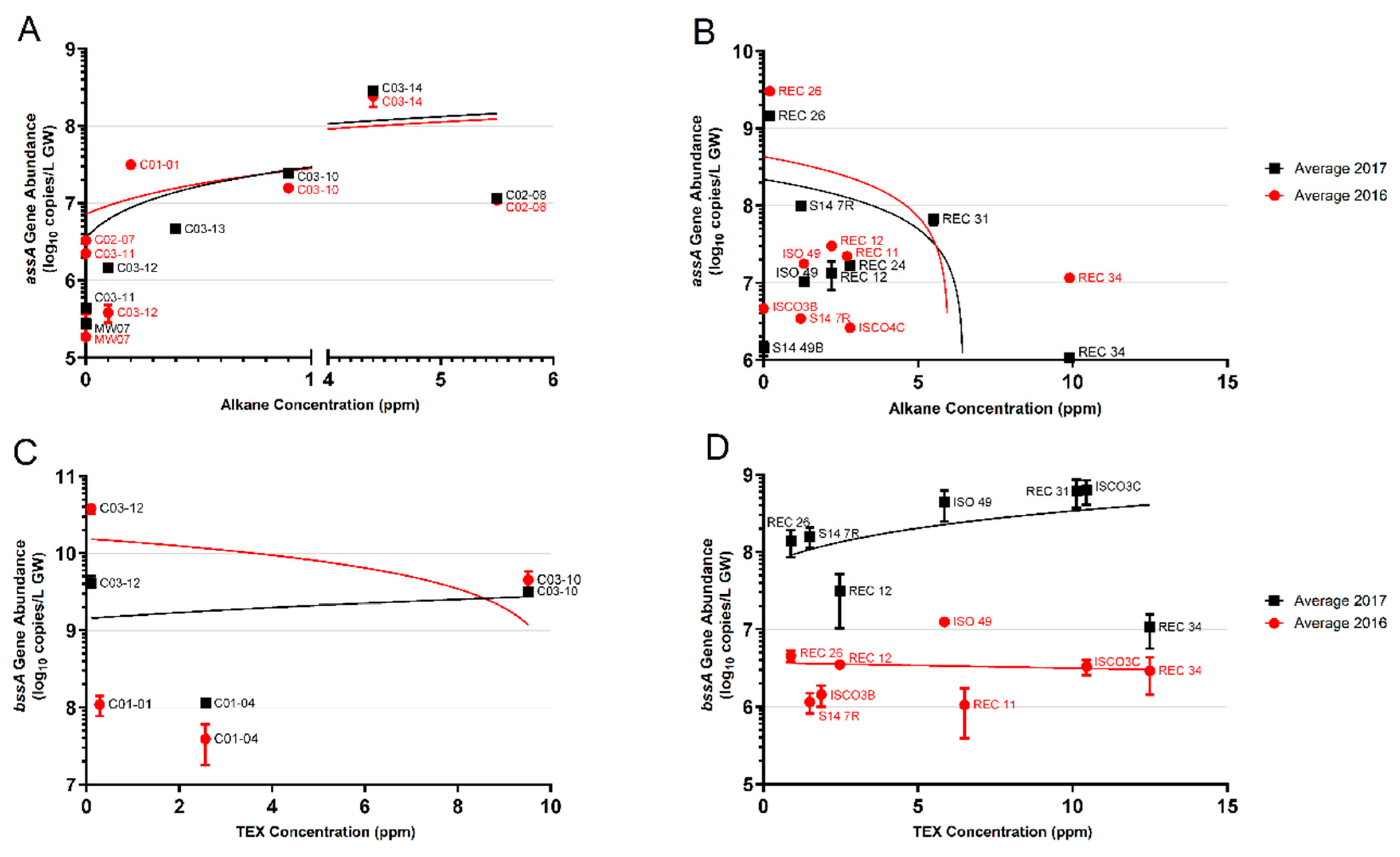
3.2.1. Alkylsuccinates and assA Genes
3.2.2. Benzylsuccinates and bssA Genes
3.3. assA and bssA Amplicon Sequencing Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lynge, E.; Andersen, A.; Nilsson, R.; Barlow, L.; Pukkala, E.; Nordlinder, R.; Boffetta, P.; Grandjean, P.; Heikkilä, P.; Hörte, L.G.; et al. Risk of cancer and exposure to gasoline vapors. Am. J. Epidemiol. 1997, 145, 449–458. [Google Scholar] [PubMed]
- Hilpert, M.; Mora, B.A.; Ni, J.; Rule, A.M.; Nachman, K.E. Hydrocarbon release during fuel storage and transfer at gas stations: Environmental and health effects. Curr. Environ. Health Rep. 2015, 2, 412–422. [Google Scholar]
- Head, I.M.; Gray, N.D.; Larter, S.R. Life in the slow lane; biogeochemistry of biodegraded petroleum containing reservoirs and implications for energy recovery and carbon management. Front. Microbiol. 2014, 5, 566. [Google Scholar] [PubMed]
- Lueders, T. The ecology of anaerobic degraders of BTEX hydrocarbons in aquifers. FEMS Microbiol. Ecol. 2017, 93, fiw220. [Google Scholar] [PubMed]
- Gieg, L.M.; Toth, C.R.A. Signature metabolite analysis to determine in situ anaerobic hydrocarbon biodegradation. In Anaerobic Utilization of Hydrocarbons, Oils, and Lipids; Handbook of Hydrocarbon and Lipid Microbiology Book Series; Boll, M., Ed.; Springer: Cham, Switzerland, 2017; Volume 30, 30p. [Google Scholar]
- Rabus, R.; Boll, M.; Heider, J.; Meckenstock, R.U.; Buckel, W.; Einsle, O.; Ermler, U.; Golding, B.T.; Gunsalus, R.P.; Kroneck, P.N.H.; et al. Anaerobic microbial degradation of hydrocarbons: From enzymatic reactions to the environment. J. Mol. Microbiol. Biotechnol. 2016, 26, 5–28. [Google Scholar] [PubMed]
- Zhang, X.; Young, L.Y. Carboxylation as an initial reaction in the anaerobic metabolism of naphthalene and phenanthrene by sulfidogenic consortia. Appl. Environ. Microbiol. 1997, 63, 4759–4764. [Google Scholar]
- Davidova, I.A.; Gieg, L.M.; Duncan, K.E.; Suflita, J.M. Anaerobic phenanthrene mineralization by a carboxylating sulfate-reducing bacterial enrichment. ISME J. 2007, 1, 436–442. [Google Scholar] [PubMed]
- Mouttaki, H.; Johannes, J.; Meckenstock, R.U. Identification of naphthalene carboxylase as a prototype for the anaerobic activation of non-substituted hydrocarbons. Environ. Microbiol. 2012, 14, 2770–2774. [Google Scholar]
- Luo, F.; Gitiafroz, R.; Devine, C.E.; Gong, Y.; Hug, L.A.; Raskin, L.; Edwards, E.A. Metatranscriptome of an anaerobic benzene-degrading enrichment culture reveals involvement of carboxylation in benzene ring activation. Appl. Environ. Microbiol. 2014, 80, 4095–4107. [Google Scholar]
- Callaghan, A.V. Enzymes involved in the anaerobic oxidation of n-alkanes: From methane to long chain alkanes. Front. Microbiol. 2016, 4, 89. [Google Scholar]
- Biegert, T.; Fuchs, G.; Heider, J. Evidence that anaerobic oxidation of toluene in the denitrifying bacterium Thauera aromatica is initiated by formation of benzylsuccinate from toluene and fumarate. Eur. J. Biochem. 1996, 238, 661–668. [Google Scholar]
- Heider, J.; Szaleniec, M.; Martins, B.M.; Seyhan, D.; Buckel, W.; Golding, B.T. Structure and function of benzylsuccinate synthase and related fumarate-adding glycyl radical enzymes. J. Mol. Microbiol. Biotechnol. 2016, 26, 29–44. [Google Scholar]
- Achong, G.R.; Rodriguez, A.M.; Spormann, A.M. Benzylsuccinate synthase of Azoarcus sp. strain T: Cloning, sequencing, transcriptional organization, and its role in anaerobic toluene and m-xylene mineralization. J. Bacteriol. 2001, 183, 6763–6770. [Google Scholar]
- Kniemeyer, O.; Fischer, T.; Wilkes, H.; Glockner, F.O.; Widdel, F. Anaerobic degradation of ethylbenzene by a new type of marine sulfate-reducing bacterium. Appl. Environ. Microbiol. 2003, 69, 760–768. [Google Scholar]
- Callaghan, A.V.; Wawrik, B.; Ni Chadain, S.M.; Young, L.Y.; Zylstra, G.J. Anaerobic alkane-degrading strain AK-01 contains two alkylsuccinate synthase genes. Biochem. Biophys. Res. Commun. 2008, 3366, 142–148. [Google Scholar]
- Grundmann, O.; Behrends, A.; Rabus, R.; Amann, J.; Halder, T.; Heider, J.; Widdel, F. Genes encoding the candidate enzyme for anaerobic activation of n-alkanes in the denitrifying bacterium, strain HxN1. Environ. Microbial. 2008, 10, 376–385. [Google Scholar]
- Meckenstock, R.U.; Boll, M.; Mouttaki, H.; Koelschbach, J.S.; Cunha Tarouco, P.; Weyrauch, P.; Dong, X.; Himmelburg, A.M. Anaerobic degradation of benzene and polycyclic aromatic hydrocarbons. J. Mol. Microbiol. Biotechnol. 2016, 26, 92–118. [Google Scholar]
- Winderl, C.; Schaefer, S.; Lueders, T. Detection of anaerobic toluene and hydrocarbon degraders in contaminated aquifers using benzylsuccinate synthase (bssA) genes as a functional marker. Environ. Microbiol. 2007, 9, 1035–1046. [Google Scholar]
- Callaghan, A.V.; Davidova, I.A.; Savage-Ashlock, K.; Parisi, V.A.; Gieg, L.M.; Suflita, J.M.; Kukor, J.J.; Wawrik, B. Diversity of benyzl- and alkylsuccinate synthase genes in hydrocarbon-impacted environments and enrichment cultures. Environ. Sci. Technol. 2010, 44, 7287–7294. [Google Scholar]
- Acosta-Gonzáles, A.; Rosselló-Móra, R.; Marqués, S. Diversity of benzylsuccinate synthase-like (bssA) genes in hydrocarbon-polluted marine sediments suggests substrate-depending clustering. Appl. Environ. Microbiol. 2013, 79, 3667–3676. [Google Scholar]
- Von Netzer, F.; Pilloni, G.; Kleindienst, S.; Krüger, M.; Knittel, K.; Gründger, F.; Lueders, T. Enhanced gene detection assays for fumarate-adding enzymes allow uncovering of anaerobic hydrocarbon degraders in terrestrial and marine systems. Appl. Environ. Microbiol. 2013, 79, 543–552. [Google Scholar]
- Gittel, A.; Donhauser, J.; Roy, H.; Girguis, P.R.; Jorgensen, B.B.; Kjeldsen, K.U. Ubiquitous presence and novel diversity of anaerobic alkane degraders in cold marine sediments. Front. Microbiol. 2015, 6, 1414. [Google Scholar] [PubMed]
- Stagars, M.H.; Ruff, S.E.; Amann, R.; Knittel, K. High diversity of anaerobic alkane-degrading microbial communities in marine seep sediments based on (1-methylalkyl)succinate synthase genes. Front. Microbiol. 2016, 6, 1511. [Google Scholar]
- Bombach, P.; Richnow, H.H.; Kästner, M.; Fischer, A. Current approaches for the assessment of in situ biodegradation. Appl. Microbiol. Biotechnol. 2010, 86, 839–852. [Google Scholar] [PubMed]
- Gieg, L.M.; Kolhatkar, R.V.; McInerney, M.J.; Tanner, R.S.; Harris, S.H., Jr.; Sublette, K.; Sulfita, J.M. Intrinsic bioremedation of petroleum hydrocarbons in a gas condensate-contaminated groundwater. Environ. Sci. Technol. 1999, 33, 2550–2560. [Google Scholar]
- Beller, H.R.; Ding, W.H.; Reinhard, M. By-products of anaerobic alkylbenzene metabolism useful as indicators of in-situ bioremediation. Environ. Sci. Technol. 1995, 29, 2864–2870. [Google Scholar]
- Jobelius, C.; Ruth, B.; Griebler, C.; Meckenstock, R.U.; Hollender, J.; Reineke, A.; Frimmel, F.H.; Zwiener, C. Metabolites indicate hot spots of biodegradation and biogeochemical monitoring well gradients in a high-resolution monitoring well. Environ. Sci. Technol. 2011, 45, 474–481. [Google Scholar]
- Winderl, C.; Anneser, B.; Griebler, C.; Meckenstock, R.U.; Lueders, T. Depth-resolved quantification of anaerobic toluene degraders and aquifer microbial community patterns in distinct redox zones of a tar oil contaminant plume. Appl. Environ. Microbiol. 2008, 74, 792–801. [Google Scholar]
- Beller, H.R.; Kane, S.R.; Legler, T.C.; McKelvie, J.R.; Sherwood Lollar, B.; Pearson, F.; Balser, L.; Mackay, D.M. Comparative assessments of benzene, toluene, and xylene natural attenuation by quantitative polymerase chain reaction analysis of a catabolic gene, signature metabolites, and compound-specific isotope analysis. Environ. Sci. Technol. 2008, 42, 6065–6072. [Google Scholar]
- Johnson, J.M.; Wawrik, B.; Isom, C.; Boling, B.; Callaghan, A.V. Interrogation of Chesapeake Bay sediment microbial communities for intrinsic alkane-utilizing potential under anaerobic conditions. FEMS Microbiol. Ecol. 2015, 91, 1–14. [Google Scholar] [PubMed]
- Oka, A.R.; Phelps, C.D.; Zhu, X.; Saber, D.L.; Young, L.Y. Dual biomarkers of anaerobic hydrocarbon degradation in historically contaminated groundwater. Environ. Sci. Technol. 2011, 45, 3407–3414. [Google Scholar]
- Okpala, G.N.; Chen, C.; Fida, T.; Voordouw, G. Effect of thermophilic nitrate reduction on sulfide production in high temperature oil reservoir samples. Front. Microbiol. 2017, 8, 1573. [Google Scholar]
- Lovley, D.R.; Phillips, E.J.P. Rapid assay for microbially reducible ferric iron in aquatic sediments. Appl. Environ. Microbiol. 1987, 53, 1536–1540. [Google Scholar] [PubMed]
- Toth, C.R.A.; Gieg, L.M. Time-course dependent methanogenic crude oil biodegradation: Dynamics of fumarate addition metabolites, biodegradative genes, and microbial community composition. Front. Microbiol. 2018, 8, 2610. [Google Scholar] [PubMed]
- Gieg, L.M.; Suflita, J.M. Detection of anaerobic metabolites of saturated and aromatic hydrocarbons in petroleum-contaminated aquifers. Environ. Sci. Technol. 2002, 36, 3755–3762. [Google Scholar] [CrossRef]
- An, D.; Caffrey, S.M.; Soh, J.; Agrawal, A.; Brown, D.C.; Budwill, K.; Dong, X.; Dunfield, P.F.; Foght, J.; Gieg, L.M.; et al. Metagenomics of hydrocarbon resource environments indicates aerobic taxa and genes to be unexpectedly common. Environ. Sci. Technol. 2013, 47, 10708–10717. [Google Scholar]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd merger. Bioinformatics 2014, 30, 614–620. [Google Scholar]
- Dong, X.; Kleiner, M.; Sharp, C.E.; Thorson, E.; Li, C.; Liu, D.; Strous, M. Fast and simple analysis of MiSeq amplicon sequencing data with MetaAmp. Front. Microbiol. 2017, 8, 1461. [Google Scholar] [PubMed]
- Aitken, C.M.; Jones, D.M.; Maguire, M.J.; Gray, N.D.; Sherry, A.; Bowler, B.F.J.; Ditchfield, A.K.; Larter, S.R.; Head, I.M. Evidence that crude oil alkane activation proceeds by different mechanisms under sulfate-reducing and methanogenic conditions. Geochim. Cosmochim. Acta 2013, 109, 162–174. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTALW: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [PubMed]
- Hordijk, W.; Gascuel, O. Improving the efficiency of SPR moves in phylogenetic tree search methods based on maximum likelihood. Bioinformatics 2005, 21, 4338–4347. [Google Scholar] [PubMed]
- Alberta Environment and Parks (AEP). Alberta Tier 1 Soil and Groundwater Remediation Guidelines; Land Policy Branch, Policy and Planning Division: Edmonton, AB, Canada, 2019.
- Cravo-Laureau, C.; Grossi, V.; Raphel, D.; Matheron, R.; Hirschler-Réa, A. Anaerobic n-alkane metabolism by a Desulfatibacillum aliphaticivorans strain CV2803 T. Appl. Environ. Microbiol. 2005, 71, 3458–3467. [Google Scholar]
- Cravo-Laureau, C.; Matheron, R.; Cayol, J.L.; Joulian, C.; Hirschler-Réa, A. Desulfatibacillum aliphaticivorans gen. nov., sp. nov., an n-alkane- and n-alkene-degrading, sulfate-reducing bacterium. Int. J. Syst. Evol. Microbiol. 2004, 54, 77–83. [Google Scholar] [PubMed]
- Jeon, C.O.; Park, W.; Ghiorse, W.C.; Madsen, E.L. Polaromonas naphthalenivorans sp. nov., a naphthalene-degrading bacterium from naphthalene-contaminated sediment. Int. J. Syst. Evol. Microbiol. 2004, 54, 93–97. [Google Scholar]
- Sun, W.; Xie, S.; Luo, C.; Cupples, A.M. Direct link between toluene degradation in contaminated-site microcosms and a Polaromonas strain. Appl. Environ. Microbiol. 2010, 76, 956–959. [Google Scholar]
- Ehrenreich, P.; Behrends, A.; Harder, J.; Widdel, F. Anaerobic oxidation of alkanes by newly isolated denitrifying bacteria. Arch. Microbiol. 2000, 173, 58–64. [Google Scholar] [PubMed]
- Iffis, B.; St-Arnaud, M.; Hijri, M. Bacteria associated with arbuscular mycorrhizal fungi within roots of plants growing in a soil highly contaminated with aliphatic and aromatic petroleum hydrocarbons. FEMS Microbiol. Lett. 2014, 358, 44–54. [Google Scholar]
- Coschigano, P.W. Transcriptional analysis of the tutE tutFDGH gene cluster from Thauera aromatica strain T1. Appl. Environ. Microbiol. 2000, 66, 1147–1151. [Google Scholar]
- Flynne, T.M.; Sanford, R.A.; Ryu, H.; Bethke, C.M.; Levine, A.D.; Ashbolt, N.J.; Santo Domingo, J.W. Functional microbial diversity explains groundwater chemistry in a pristine aquifer. BMC Microbiol. 2013, 13, 146. [Google Scholar]
- Benk, S.A.; Yan, L.; Lehmann, R.; Roth, V.-N.; Schwab, V.F.; Totsche, K.U.; Küsel, K.; Gleixner, G. Fueling diversity in the subsurface: Composition and age of dissolved organic matter in the critical zone. Front. Earth Sci. 2019, 7, 296. [Google Scholar]
- Parisi, V.A.; Brubaker, G.R.; Zenker, M.J.; Prince, R.C.; Gieg, L.M.; da Silva, M.L.B.; Alvarez, P.J.J.; Suflita, J.M. Field metabolomics and laboratory assessments of anaerobic intrinsic bioremediation of hydrocarbons at a petroleum-contaminated site. Microb. Biotechnol. 2009, 2, 202–212. [Google Scholar] [PubMed]
- Agrawal, A.; Gieg, L.M. In situ detection of anaerobic alkane metabolites in subsurface environments. Front. Microbiol. 2013, 4, 140. [Google Scholar]
- Kazy, S.K.; Monier, A.L.; Alvarez, P.J.J. Assessing the correlation between anaerobic toluene degradation activity and bssA concentrations in hydrocarbon-contaminated aquifer material. Biodegradation 2010, 21, 793–800. [Google Scholar] [PubMed]
- Da Silva, M.L.B.; Alvarez, P.J.J. Enhanced anaerobic biodegradation of benzene-toluene-ethylbenzene-xylene-ethanol mixtures in bioaugmented aquifer columns. Appl. Environ. Microbiol. 2004, 70, 4720–4726. [Google Scholar]
- Kimes, N.E.; Callaghan, A.V.; Aktas, D.R.; Smith, W.L.; Sunner, J.; Golding, B.T.; Drozdowska, M.; Hazen, T.C.; Suflita, J.M.; Morris, P.J. Metagenomic analysis and metabolite profiling of deep-sea sediments from the Gulf of Mexico following the Deepwater Horizon oil spill. Front. Microbiol. 2013, 4, 50. [Google Scholar]
- Brow, C.N.; Johnson, R.O.; Johnson, R.L.; Simon, H.M. Assessment of anaerobic toluene biodegradation activity by bssA transcript/gene ratios. Appl. Environ. Microbiol. 2013, 79, 5338–5344. [Google Scholar]
- Beller, H.R.; Kane, S.R.; Legler, T.C.; Alvarez, P.J.J. A real-time polymerase chain reaction method for monitoring anaerobic, hydrocarbon-degrading bacteria based on a catabolic gene. Environ. Sci. Technol. 2002, 36, 3977–3984. [Google Scholar] [CrossRef]
- Oberding, L.K.; Gieg, L.M. Methanogenic paraffin biodegradation: Alkylsuccinate synthase gene quantification and dicarboxylic acid production. Appl. Environ. Microbiol. 2018, 84, e01773-17. [Google Scholar]
- Pilloni, G.; Bayer, A.; Ruth-Anneser, B.; Fillinger, L.; Engel, M.; Griebler, C.; Lueders, T. Dynamics of hydrology and anaerobic hydrocarbon degrader communities in a tar-oil contaminated aquifer. Microorganisms 2019, 7, 46. [Google Scholar]
- Müller, J.B.; Ramos, D.T.; Larose, C.; Fernandes, M.; Lazzarin, H.S.C.; Vogel, T.M.; Corseuil, H.X. Combined iron and sulfate reduction biostimulation as a novel approach to enhance BTEX and PAH source-zone biodegradation in biodiesel blend-contaminated groundwater. J. Hazard. Mater. 2017, 326, 229–236. [Google Scholar] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence (5’-3’) | Tm (°C) | Reference |
|---|---|---|---|
| bssA forward | |||
| bssOil | GAA TCC CTG GTT ACA GGT CCA C | 64.1 | This study |
| bssMys | CAA TCC GTG GCA CAA CTG CAT G | 66.3 | This study |
| bssSuf | GAA TAC GTG GAG CGA CCC GCT C | 68.1 | This study |
| bssWin | CAA TCC GTG GCT TCA GGT TCA T | 65 | This study |
| bssA reverse | |||
| bssHitr | TCC TCG TAG CCT TCC CAG TT | 64.6 | This study |
| assA forward | |||
| assOri | CTC CGC CAC GGC CAA CTG | 67.4 | This study |
| assMsd | CTC AGC CAC CGC CAA CTG | 65 | This study |
| assEx | CTC TGC GAC CGC GAA TTG | 63 | This study |
| assSml | TAG CGC CAC GGC CAA CTG | 67 | This study |
| assA reverse | |||
| 8543r | TCG TCR TTG CCC CAY TTN GG | 65.7 | [22] |
| Alkanes | Detected Alkylsuccinates | assA Gene Presence | assA Gene Abundance (copies/L GW) 5 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Well | C6-C10 Alkanes (ppm) | C5 | C5 unsat 1 | C6 | C6 unsat | C7 | C7 unsat | C8 | C8 unsat | C9 | C9 unsat | 2016 | 2017 | 2016 | 2017 | |
| Site A | C02-08 | 5.5 | 2016 | 2016 | 2017 | 2016/2017 | 2016 | 2016 | assA | assA | 1.09 × 107 | 1.17 × 107 | ||||
| C03-14 | 4.4 | 2016 | 2016/2017 | 2016 | 2016 | assA | assA | 2.43 × 108 | 2.86 × 108 | |||||||
| C03-10 | 0.9 | 2016/2017 | 2016 | 2016/2017 | 2016/2017 | 2016/2017 | 2017 | assA | assA | 1.57 × 107 | 2.45 × 107 | |||||
| C01-04 | 0.5 | 2016/2017 | 2016/2017 | 2016/2017 | 2016 | assA | assA | |||||||||
| C03-13 3 | 0.4 | 2017 | 2017 | 2017 | assA | - | 4.64 × 106 | |||||||||
| C01-01 | 0.2 | 2016 | 2016/2017 | 2016/2017 | 2016/2017 | 2016/2017 | 2016/2017 | assA | 3.15 × 107 | |||||||
| C03-12 | 0.1 | 2016 | 2016 | 2017 | assA | assA | 3.81 × 105 | 1.45 × 106 | ||||||||
| C02-06 | <0.1 2 | |||||||||||||||
| C02-07 | <0.1 | assA | 3.27 × 106 | |||||||||||||
| C03-11 | <0.1 | 2016/2017 | 2016/2017 | 2.25 × 106 | 4.34 × 105 | |||||||||||
| MW 07 | <0.1 | 1.86 × 105 | 2.72 × 105 | |||||||||||||
| MW23 | <0.1 | |||||||||||||||
| TRIP BLANK | <0.1 | |||||||||||||||
| Site B | REC-34 | 9.9 | 2016/2017 | 2016/2017 | 2016/2017 | 2016 | assA | assA | 1.15 × 107 | 1.08 × 106 | ||||||
| REC-31 | 5.5 | 2017 | 2016/2017 | 2016/2017 | 2016/2017 | assA | 6.61 × 107 | |||||||||
| ISCO-03-C | 3.1 | 2016 | 2016 | 2016 | 2016 | assA | assA | |||||||||
| ISCO-04-C 4 | 2.8 | 2016 | 2016 | 2016 | assA | 2.61 × 106 | - | |||||||||
| REC-24 | 2.8 | 2016/2017 | 2016/2017 | 2016/2017 | assA | assA | 1.69 × 107 | |||||||||
| REC-11 4 | 2.7 | 2016 | 2016 | assA | 2.21 × 107 | - | ||||||||||
| REC-12 | 2.2 | 2016 | 2016 | assA | assA | 3.00 × 107 | 1.34 × 107 | |||||||||
| ISO-49 | 1.3 | 2017 | 2017 | 2017 | 2017 | assA | assA | 1.76 × 107 | 1.03 × 107 | |||||||
| S14-7R | 1.2 | 2016/2017 | 2016/2017 | 2016/2017 | 2017 | assA | assA | 3.44 × 106 | 9.80 × 107 | |||||||
| REC-26 | 0.2 | assA | assA | 3.02 × 109 | 1.45 × 109 | |||||||||||
| ISCO-03-B 4 | <0.1 | 2016 | assA | 4.64 × 106 | - | |||||||||||
| S14-49B | <0.1 | 1.36 × 106 | 1.42 × 106 | |||||||||||||
| Trip Blank | <0.1 | |||||||||||||||
| Monoaromatic Hydrocarbon | Benzylsuccinates | bssA Gene Presence | bssA Gene Abundance (copies/L GW) 4 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Well | Toluene (ppm) | Ethylbenzene (ppm) | Xylenes (ppm) | BzSucc | EBSucc | MeBzSucc | 2016 | 2017 | 2016 | 2017 | |
| Site A | C02-08 | 0.072 | 0.861 | 1.71 | 2016/2017 | bssA | bssA | ||||
| C03-14 | 0.0976 | 1.8 | 0.0102 | 2016/2017 | bssA | ||||||
| C03-10 | 0.128 | 1.26 | 0.178 | bssA | bssA | 4.48 × 109 | 3.18 × 109 | ||||
| C01-04 | 0.0153 | 0.0912 | 0.0081 | bssA | bssA | 3.90 × 107 | 1.13 × 108 | ||||
| C03-13 2 | <0.0003 | 0.013 | 0.0102 | ||||||||
| C01-01 | 0.0014 | <0.0005 | 0.001 | bssA | 1.09 × 108 | ||||||
| C03-12 | <0.0003 | 0.0018 | 0.0015 | bssA | bssA | 3.84 × 1010 | 4.08 × 109 | ||||
| C02-06 | <0.0003 | <0.0005 | <0.0005 | ||||||||
| C02-07 | <0.0003 | <0.0005 | <0.0005 | bssA | |||||||
| C03-11 | <0.0003 | <0.0005 | <0.0005 | bssA | bssA | ||||||
| MW07 | <0.0003 | <0.0005 | 0.0102 | bssA | |||||||
| MW23 | <0.0003 | <0.0005 | 0.0102 | bssA | bssA | ||||||
| TRIP BLANK | <0.0003 1 | <0.0005 1 | <0.0005 | ||||||||
| Site B | REC-34 | 3.83 | 0.325 | 6.11 | 2016/2017 | 2016 | 2016/2017 | bssA | bssA | 2.89 × 106 | 1.06 × 107 |
| REC-31 | 0.867 | 0.394 | 0.979 | 6.17 × 108 | |||||||
| ISCO-03-C | 0.557 | 0.0051 | 0.0051 | bssA | bssA | 3.30 × 106 | 6.29 × 108 | ||||
| ISCO-04-C 3 | 0.92 | 0.0323 | 0.0323 | ||||||||
| REC-24 | 0.377 | 0.297 | 0.675 | bssA | bssA | ||||||
| REC-113 | 0.732 | 0.0408 | 4.96 | bssA | 1.05 × 106 | - | |||||
| ISO-49 | 0.87 | 0.223 | 4.46 | 2016 | 2016 | bssA | bssA | 1.24 × 107 | 4.37 × 108 | ||
| REC-12 | 0.0299 | 0.0489 | 0.0432 | bssA | 3.51 × 106 | 3.11 × 107 | |||||
| S14-7R | 0.125 | 0.0365 | 0.0754 | 2016/2017 | bssA | bssA | 1.15 × 106 | 1.59 × 108 | |||
| ISCO-03-B 3 | 0.0713 | 0.108 | 0.0454 | bssA | 1.44 × 106 | - | |||||
| REC-26 | 0.0523 | 0.0421 | 0.403 | bssA | 4.53 × 106 | 1.38 × 108 | |||||
| S14-49B | <0.0003 | <0.0005 | <0.0005 | bssA | |||||||
| Trip Blank | <0.0003 | <0.0005 | <0.0005 | ||||||||
| Targeted FAE Gene | Reported Detection Limit (copies/L or g) | Highest Reported Sample Abundance (copies/L or g) | Targeted Electron-Accepting Conditions | Cited Primer Reference | Reference |
|---|---|---|---|---|---|
| assA | ~log 5 | ~log 9 | Sulfate-reducers and methanogenic consortia | This Study | |
| not reported | ~log 6 | Methanogenic paraffin degrading consortium | Oberding and Gieg [62] | ||
| ~log 3 | ~log 8 | Sulfate-reducers and methanogenic consortia | Aitken et al. [40] | ||
| bssA | ~log 5 | ~log 10 | Nitrate- and sulfate-reducers | This Study | |
| ~log 2 | ~log 8 | Sulfate-reducers | Winderl et al. [29] | Pilloni et al. [63] | |
| ~log 4 | ~log 8 | Nitrate- and sulfate-reducers | Winderl et al. [19] non-qPCR primers 7772f/8546r | Müller et al. [64] | |
| ~log 4 | ~log 4 | Sulfate-reducers and methanogenic consortia | Beller et al. [30] | Oka et al. [32] | |
| ~log 2 | ~log 3 | Nitrate-reducers | Beller et al. [61] | Oka et al. [32] | |
| ~log 1 | ~log 8 | Nitrate-reducers | Beller et al. [61] | Kazy et al. [57] | |
| ~log 3 | ~log 8 | Sulfate-reducers | Beller et al. [30] | ||
| ~log 3 | ~log 7 | Deltaproteobacterial “F1” | Winderl et al. [29] | ||
| ~log 2 | ~log 8 | Nitrate-reducers | Beller et al. [61] | Da Silva and Alvarez [58] | |
| ~log 3 | ~log 8 | Nitrate-reducers | Beller et al. [61] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kharey, G.; Scheffer, G.; Gieg, L.M. Combined Use of Diagnostic Fumarate Addition Metabolites and Genes Provides Evidence for Anaerobic Hydrocarbon Biodegradation in Contaminated Groundwater. Microorganisms 2020, 8, 1532. https://doi.org/10.3390/microorganisms8101532
Kharey G, Scheffer G, Gieg LM. Combined Use of Diagnostic Fumarate Addition Metabolites and Genes Provides Evidence for Anaerobic Hydrocarbon Biodegradation in Contaminated Groundwater. Microorganisms. 2020; 8(10):1532. https://doi.org/10.3390/microorganisms8101532
Chicago/Turabian StyleKharey, Gurpreet, Gabrielle Scheffer, and Lisa M. Gieg. 2020. "Combined Use of Diagnostic Fumarate Addition Metabolites and Genes Provides Evidence for Anaerobic Hydrocarbon Biodegradation in Contaminated Groundwater" Microorganisms 8, no. 10: 1532. https://doi.org/10.3390/microorganisms8101532
APA StyleKharey, G., Scheffer, G., & Gieg, L. M. (2020). Combined Use of Diagnostic Fumarate Addition Metabolites and Genes Provides Evidence for Anaerobic Hydrocarbon Biodegradation in Contaminated Groundwater. Microorganisms, 8(10), 1532. https://doi.org/10.3390/microorganisms8101532