Long-Term Helicobacter pylori Infection Switches Gastric Epithelium Reprogramming towards Cancer Stem Cell-Related Differentiation Program in Hp-Activated Gastric Fibroblast-TGFβ Dependent Manner


 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Hp Strain
2.2. Technique of Rat Gastric Fibroblasts Isolation and Their Activation towards Fibroblasts Possessing CAFs Characteristic
2.3. Isolation of Hp-AGF and Normal Gastric Fibroblast (GF)-Conditioned Media
2.4. Long-Term Influence of Supernatants Collected from Hp-AGFs and GFs on Epithelial RGM1 Cells
2.5. RT-PCR Technique
2.6. Influence of TGFβR1 Kinase Activity Receptor Inhibition on Phenotype of Long-Term RGM1 Cells
2.7. Cell Proliferation
2.8. Western Blot
2.9. Determination of the Cells Ability to Release TGFβ1
2.10. Image Acquisition and Immunofluorescence
2.11. Statistical Analyses
3. Results
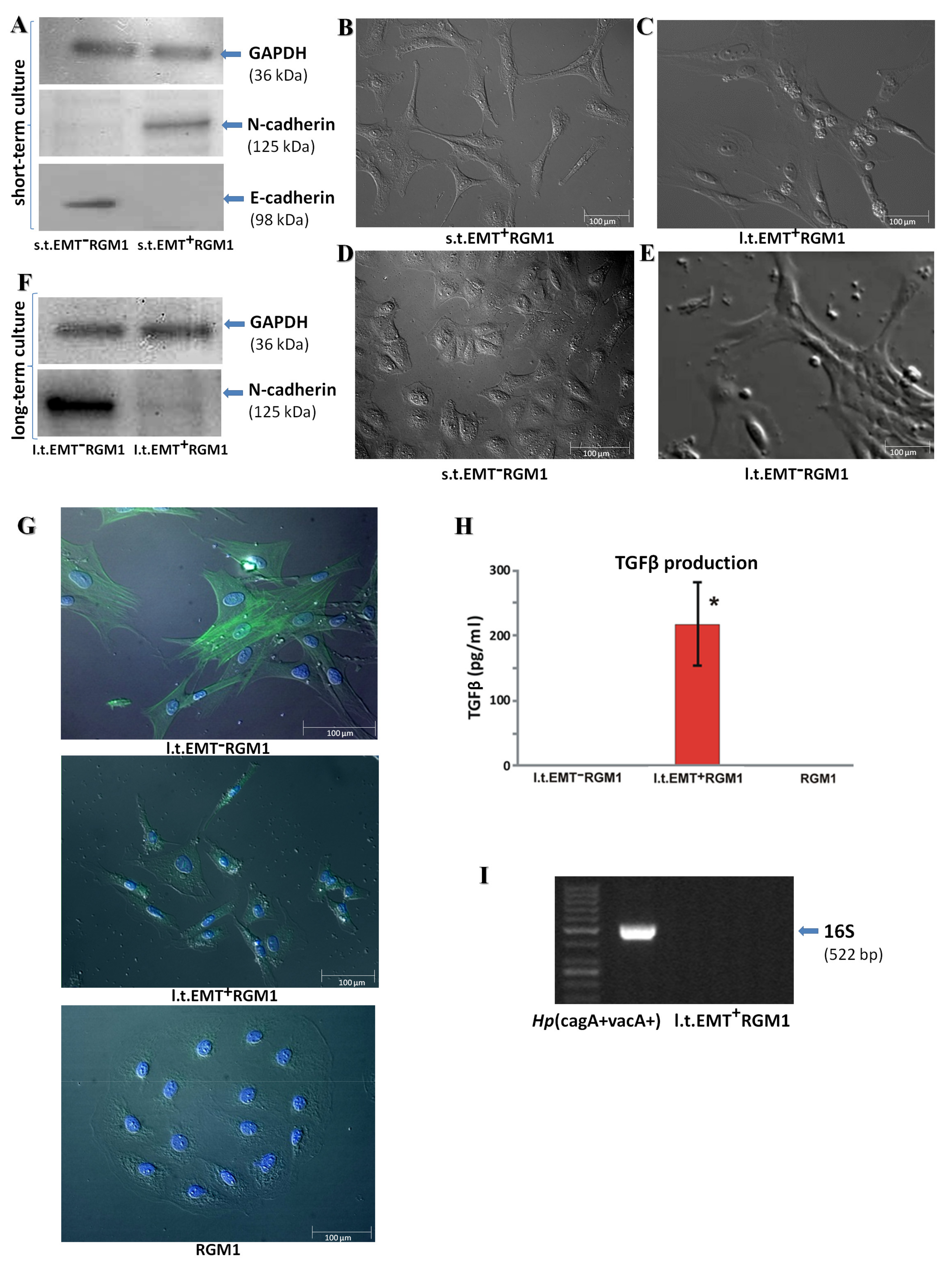
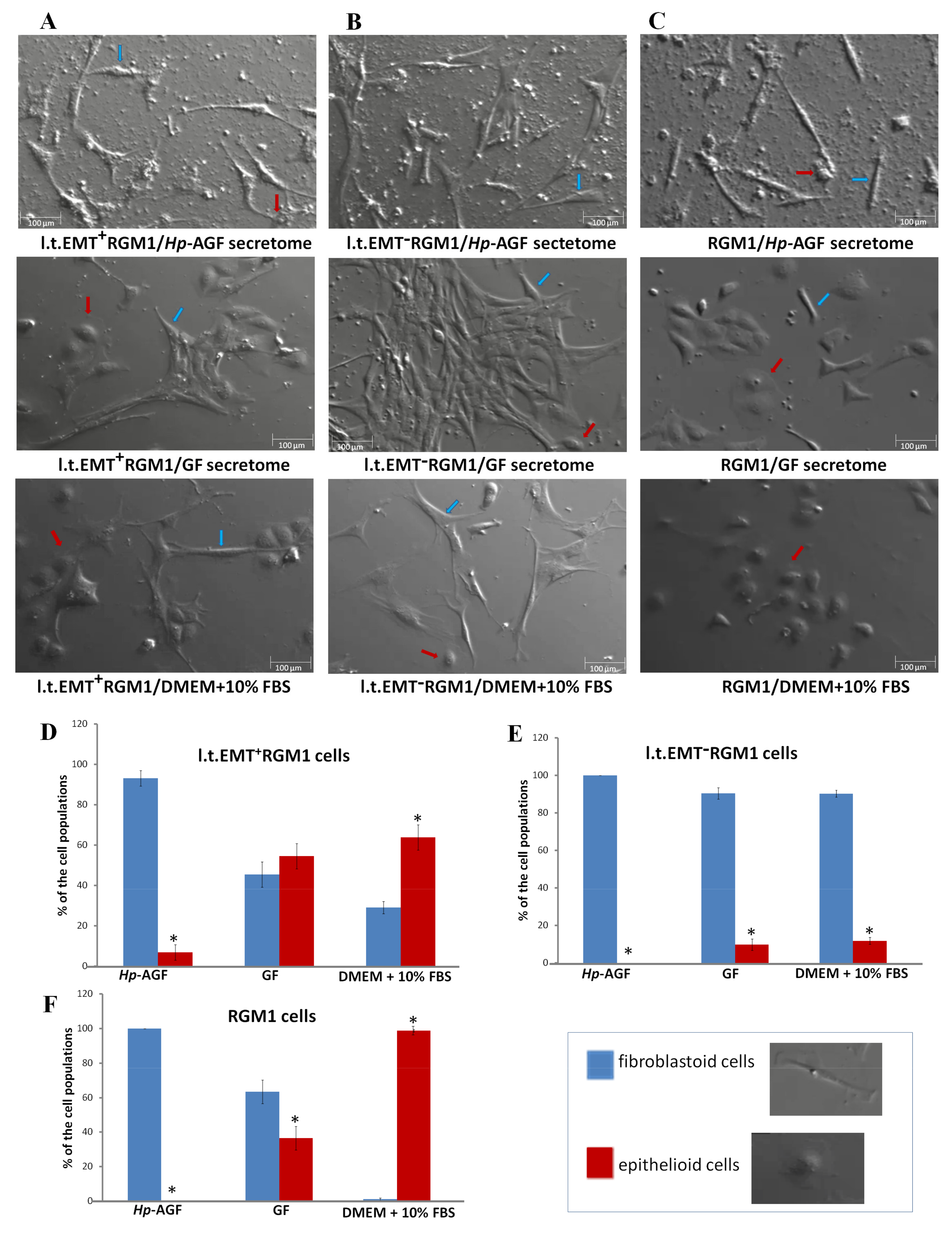
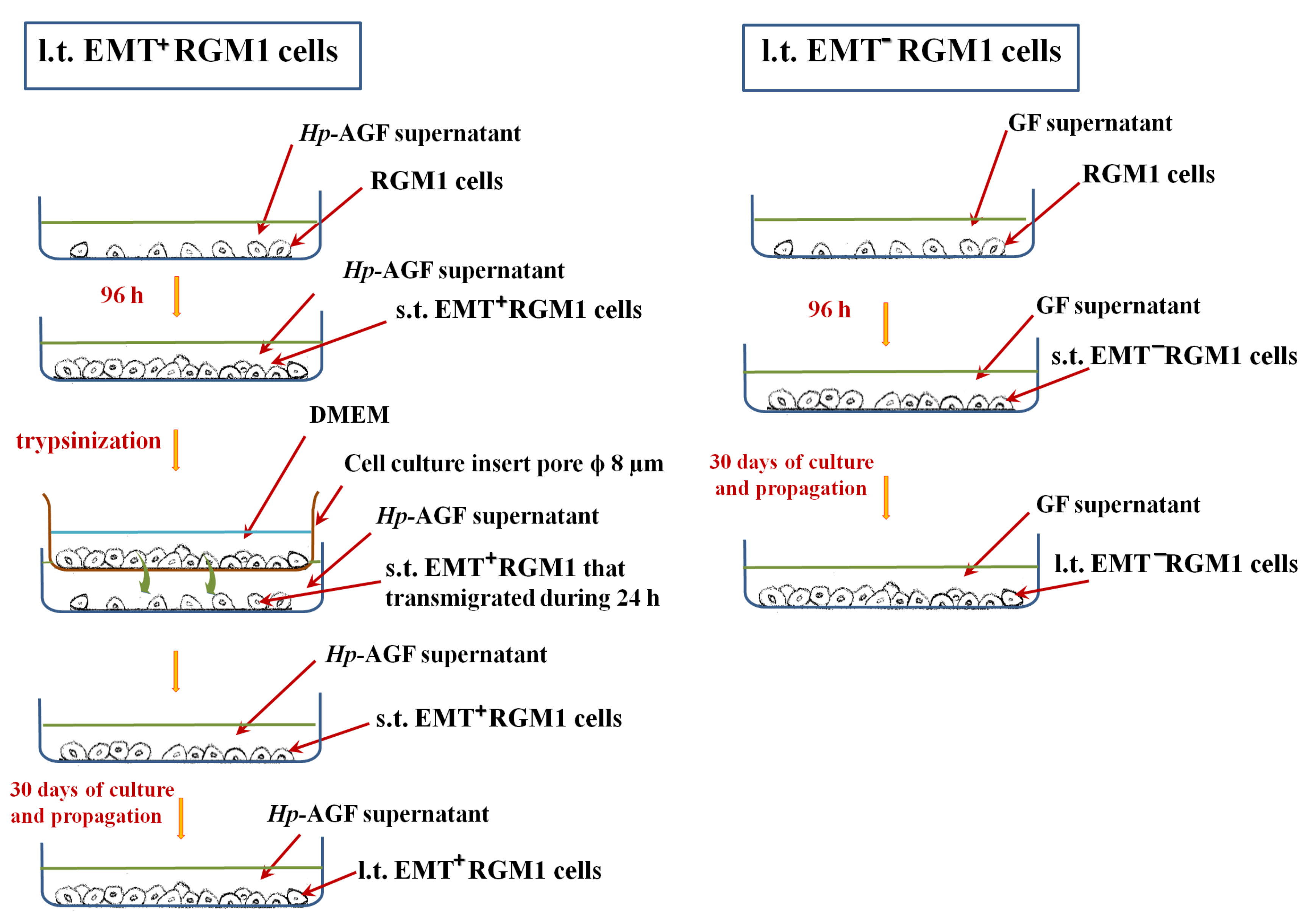
3.1. Long-Term Influence of Hp-AGF Secretome Induces Phenotypical Changes in Pro-Invasive s.t.EMT+RGM1 Cells
3.2. The Phenotypic Plasticity of l.t.EMT+RGM1 Cells
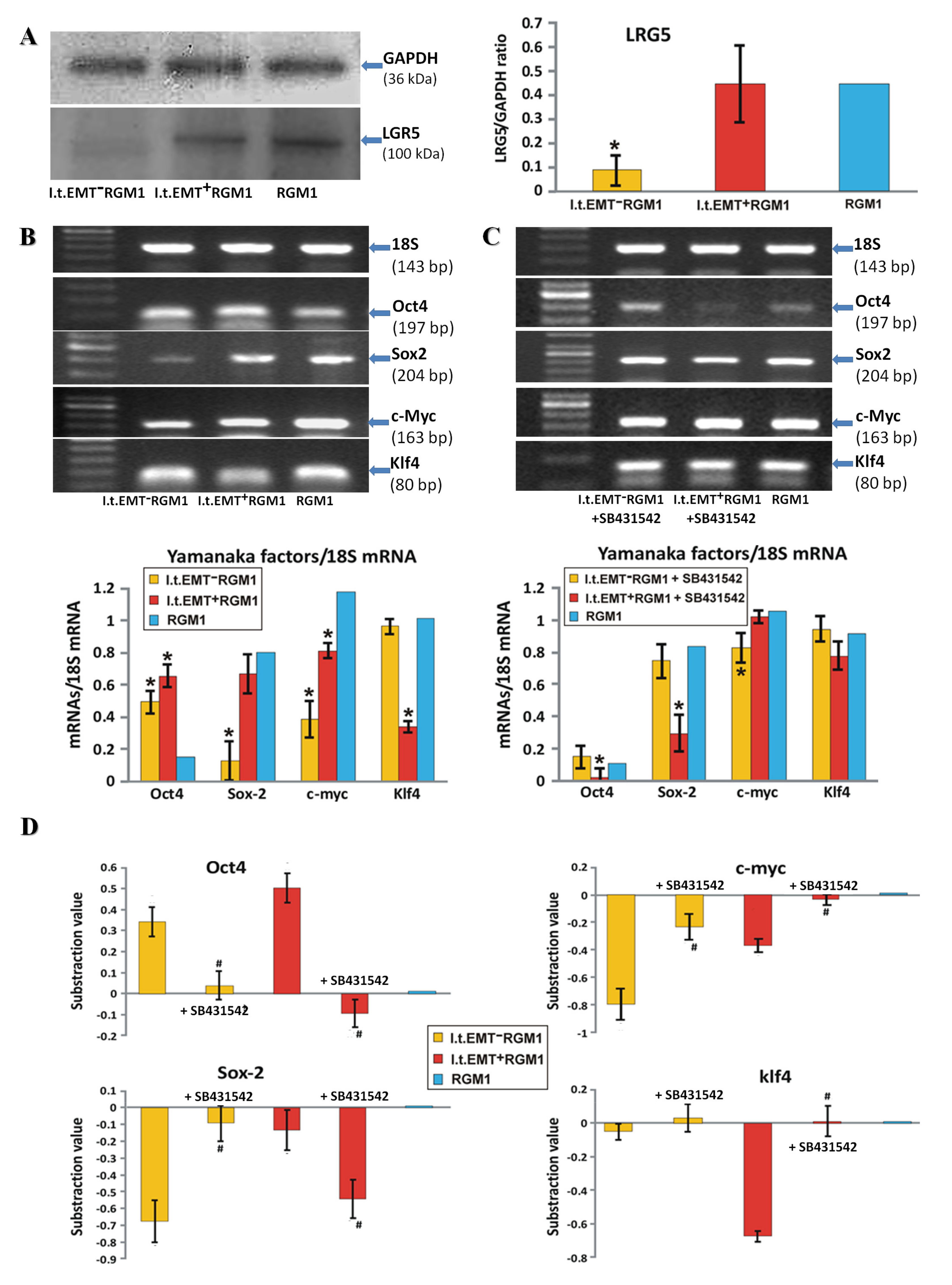
3.3. TGFβR1 Activation Participates in Both Pro-Pluripotent and Pro-Fibrotic RGM1 Reprogramming
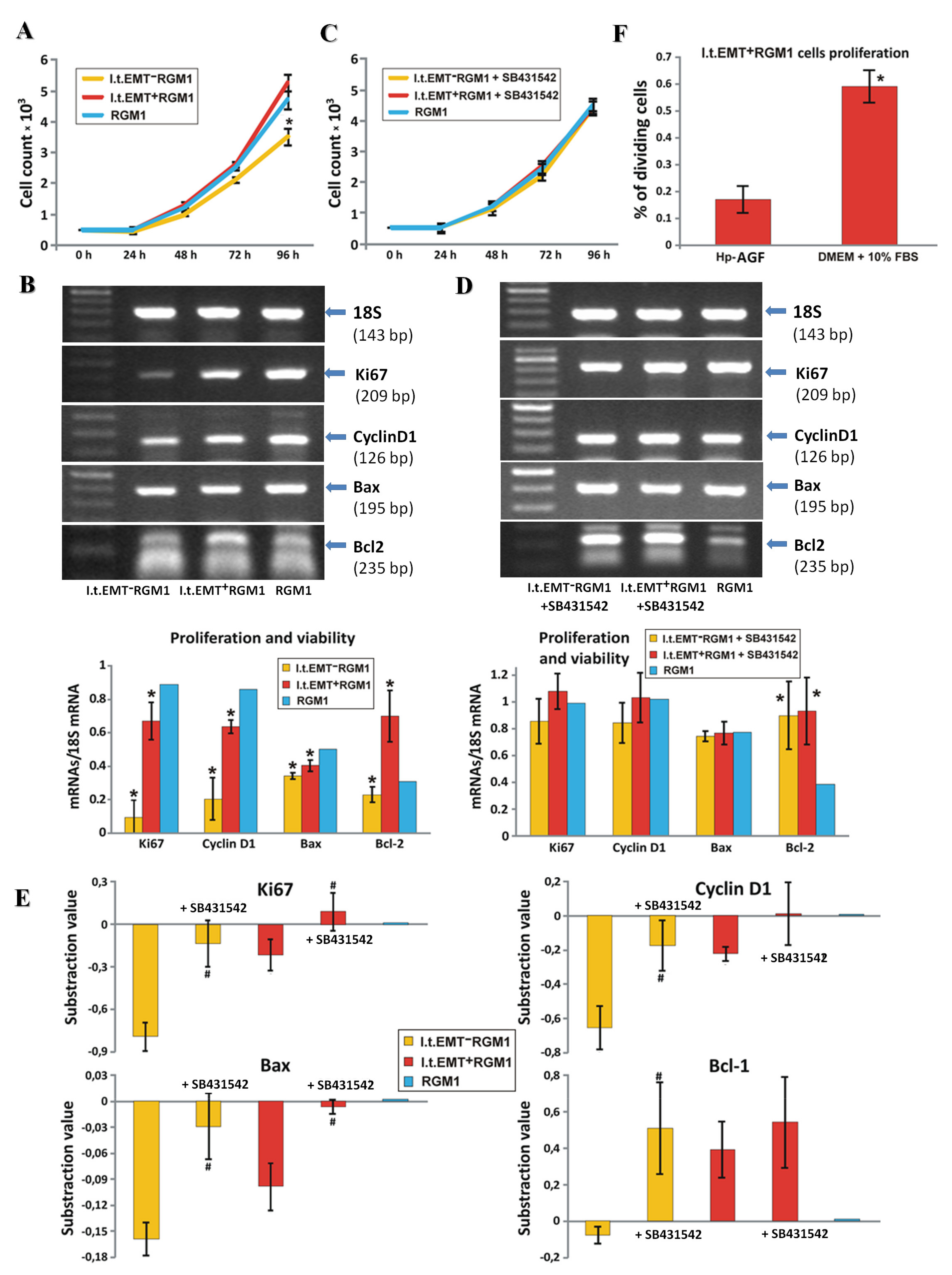
3.4. Hp-AGF Secretome Induces Potentially Pro-Proliferative Phenotype of l.t.EMT+RGM1 Cells
3.5. Hp-AGF Secretome Prompts TGFβ-Dependent Proliferation of l.t.EMT+RGM1 Cells
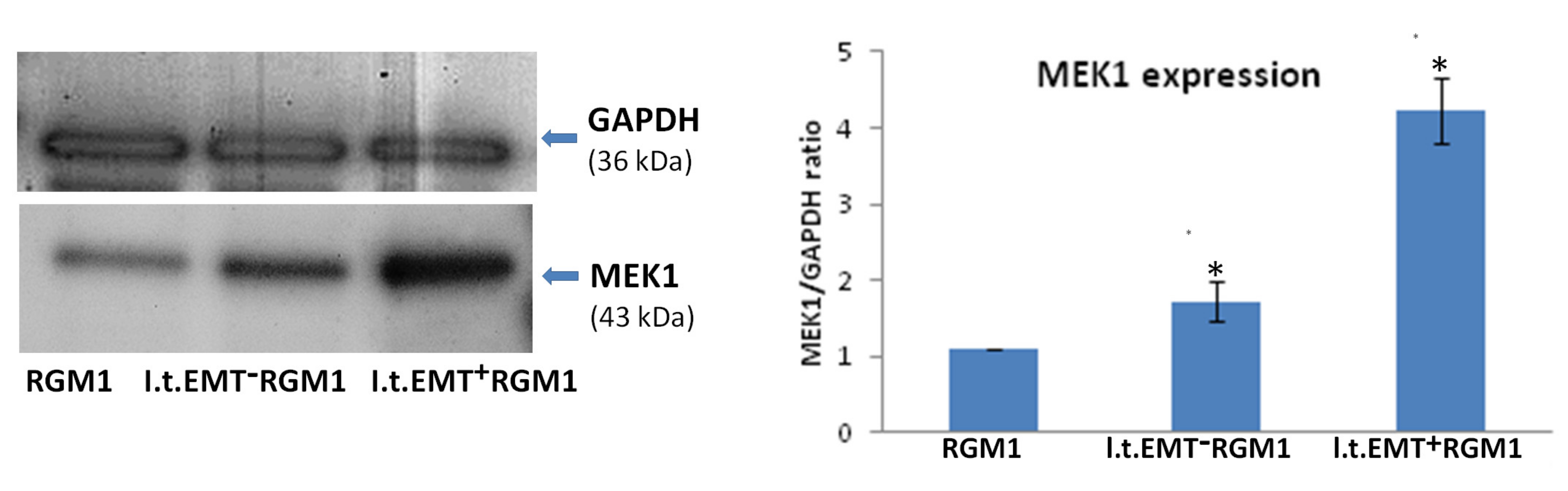
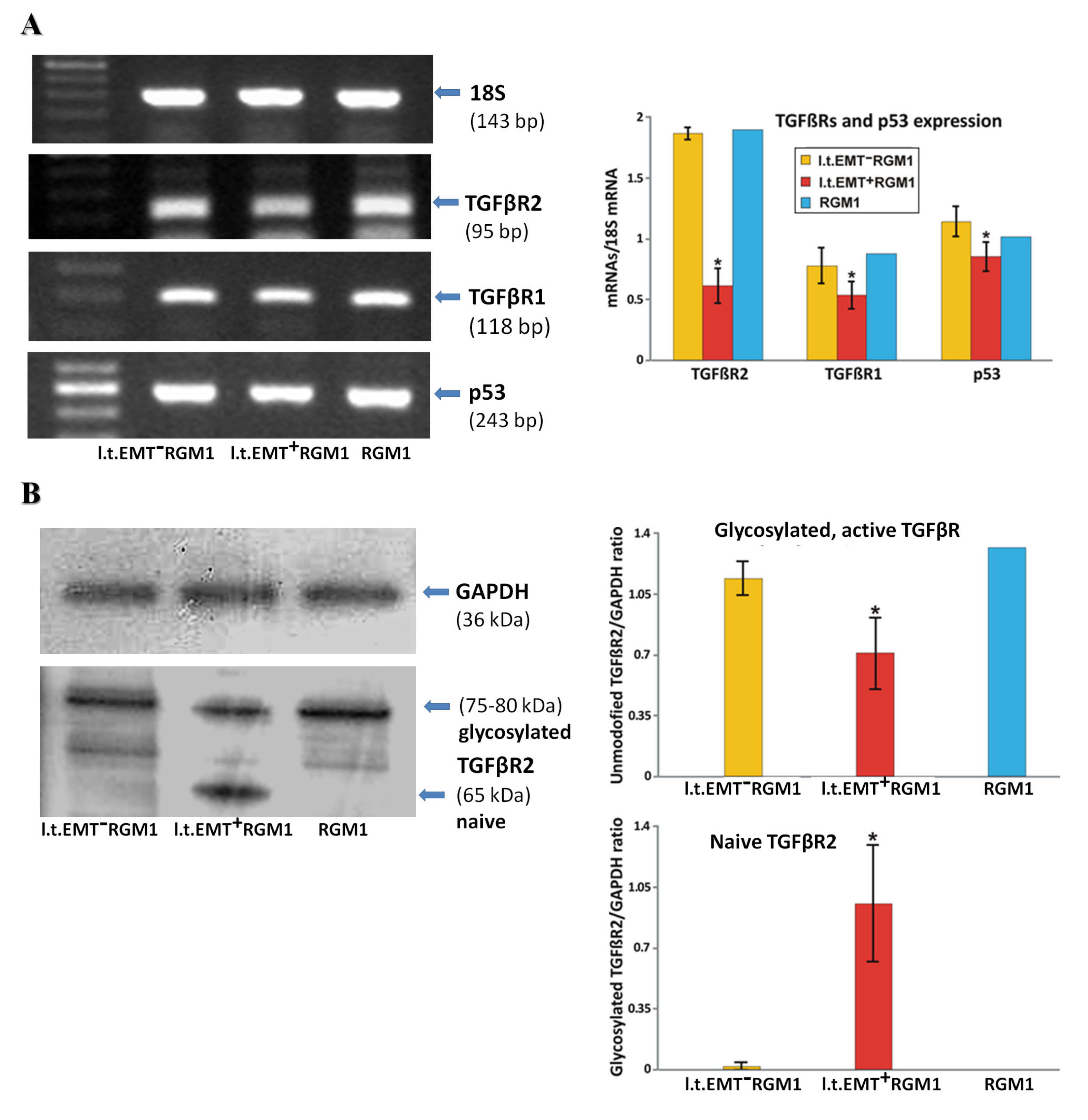
3.6. Interrelations between TGFβR1 and TGFβR2 Activity Underlie Differential Microevolution Pattern of l.t.EMT+RGM1 and l.t.EMT−RGM1 Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dicken, B.J.; Bigam, D.L.; Cass, C.; Mackey, J.R.; Joy, A.A.; Hamilton, S.M. Gastric adenocarcinoma: Review and considerations for future directions. Ann. Surg. 2005, 241, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kong, Y.; Weng, S.; Dong, C.; Zhu, L.; Yang, Z.; Zhong, J.; Yuan, Y. Outcomes of surgery for gastric cancer with distant metastases: A retrospective study from the SEER database. Oncotarget 2017, 8, 4342–4351. [Google Scholar] [CrossRef] [PubMed]
- Apicella, M.; Corso, S.; Giordano, S. Targeted therapies for gastric cancer: Failures and hopes from clinical trias. Oncotarget 2017, 8, 57654–57669. [Google Scholar] [CrossRef] [PubMed]
- Chavarría-Velázquez, C.O.; Torres-Martínez, A.C.; Montaño, L.F.; Rendón-Huerta, E.P. TLR2 Activation induced by H. pylori LPS promotes the differential expression of claudin-4-6-7 and -9 via either STAT3 and ERK1/2 in AGS cells. Immunobiology 2018, 223, 38–48. [Google Scholar] [CrossRef] [PubMed]
- López-Novoa, J.M.; Nieto, M.A. Inflammation and EMT: An alliance towards organ fibrosis and cancer progression. EMBO Mol. Med. 2009, 1, 303–314. [Google Scholar] [CrossRef]
- Posselt, G.; Backert, S.; Wessler, S. The functional interplay of Helicobacter pylori factors with gastric epithelial cells induces a multi-step process in pathogenesis. Cell Commun. Signal. 2013, 11, 77. [Google Scholar] [CrossRef]
- Schneider, S.; Carra, G.; Sahin, U.; Hoy, B.; Rieder, G.; Wessler, S. Complex cellular responses of Helicobacter pylori-colonized gastric adenocarcinoma cells. Infect. Immun. 2011, 79, 2362–2371. [Google Scholar] [CrossRef]
- Wessler, S.; Backert, S. Molecular mechanisms of epithelial-barrier disruption by Helicobacter pylori. Trends Microbiol. 2008, 16, 397–405. [Google Scholar] [CrossRef]
- Wang, F.; Meng, W.; Wang, B.; Qiao, L. Helicobacter pylori-induced gastric inflammation and gastric cancer. Cancer Lett. 2014, 345, 196–202. [Google Scholar] [CrossRef]
- Ishimoto, T.; Sawayama, H.; Sugihara, H.; Baba, H. Interaction between gastric cancer stem cells and the tumor microenvironment. J. Gastroenterol. 2014, 49, 1111–1120. [Google Scholar] [CrossRef]
- Chung, H.W.; Lim, J.B. Role of the tumor microenvironment in the pathogenesis of gastric carcinoma. World J. Gastroenterol. 2014, 20, 1667–1680. [Google Scholar] [CrossRef] [PubMed]
- Necchi, V.; Candusso, M.; Tava, F.; Luinetti, O.; Ventura, U.; Fiocca, R.; Ricci, V.; Solcia, E. Intracellular, intercellular, and stromal invasion of gastric mucosa, preneoplastic lesions, and cancer by Helicobacter pylori. Gastroenterology 2007, 132, 1009–1023. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.L.; Hsu, W.H.; Kao, M.C.; Chou, C.C.; Lin, C.C.; Liu, C.J.; Weng, B.C.; Kuo, F.C.; Kuo, C.H.; Lin, M.H.; et al. Stromal C-type lectin receptor COLEC12 integrates H. pylori, PGE2-EP2/4 axis and innate immunity in gastric diseases. Sci. Rep. 2018, 8, 3821. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Blobe, G.C. Role of transforming growth factor-beta in hematologic malignancies. Blood 2006, 107, 4589–4596. [Google Scholar] [CrossRef]
- Spender, L.C.; O’Brien, D.I.; Simpson, D.; Dutt, D.; Gregory, C.D.; Allday, M.J.; Clark, L.J.; Inman, G.J. TGF-β induces apoptosis in human B cells via transcriptional regulation of BIK and BCL-XL. Cell Death Differ. 2009, 16, 593–602. [Google Scholar] [CrossRef]
- Shi, Y.; Massague, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Massagué, J. TGFβ in cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef]
- Ham, I.H.; Lee, D.; Hur, H. Role of cancer-associated fibroblast in gastric cancer progression and resistance to treatments. J. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Paraiso, K.H.; Smalley, K.S. Fibroblast-Mediated drug resistance in cancer. Biochem. Pharmacol. 2013, 85, 1033–1041. [Google Scholar] [CrossRef]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Sakai, R. Direct interaction between carcinoma cells and cancer associated fibroblasts for the regulation of cancer invasion. Cancers 2015, 7, 2054–2062. [Google Scholar] [CrossRef] [PubMed]
- Krzysiek-Maczka, G.; Wrobel, T.; Targosz, A.; Szczyrk, U.; Strzalka, M.; Ptak-Belowska, A.; Czyz, J.; Brzozowski, T. Helicobacter pylori-activated gastric fibroblasts induce epithelial-mesenchymal transition of gastric epithelial cells in vitro in a TGF-β-dependent manner. Helicobacter 2019, 24. [Google Scholar] [CrossRef] [PubMed]
- Kuzet, S.E.; Gaggioli, C. Fibroblast activation in cancer: When seed fertilizes soil. Cell Tissue Res. 2016, 365, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Murata, M.; Yamaguchi, T.; Matsuzaki, K.; Okazaki, K. Reversible human TGF-β signal shifting between tumor suppression and fibro-carcinogenesis: Implications of Smad phospho-isoforms for hepatic epithelial-mesenchymal transitions. J. Clin. Med. 2016, 5, 7. [Google Scholar] [CrossRef]
- Lim, H.; Moon, A. Inflammatory fibroblasts in cancer. Arch. Pharm. Res. 2016, 39, 1021–1031. [Google Scholar] [CrossRef]
- Wang, Y.; Gan, G.; Wang, B.; Wu, J.; Cao, Y.; Zhu, D.; Xu, Y.; Wang, X.; Han, H.; Li, X.; et al. Cancer-Associated fibroblasts promote irradiated cancer cell recovery through autophagy. EBioMedicine 2017, 17, 45–56. [Google Scholar] [CrossRef]
- Wang, F.T.; Sun, W.; Zhang, J.T.; Fan, Y.Z. Cancer-Associated fibroblast regulation of tumor neo-angiogenesis as a therapeutic target in cancer. Oncol. Lett. 2019, 17, 3055–3065. [Google Scholar] [CrossRef]
- Huang, T.X.; Guan, X.Y.; Fu, L. Therapeutic targeting of the crosstalk between cancer-associated fibroblasts and cancer stem cells. Am. J. Cancer Res. 2019, 9, 1889–1904. [Google Scholar]
- Müller, M.; Hermann, P.C.; Liebau, S.; Weidgang, C.; Seufferlein, T.; Kleger, A.; Perkhofer, L. The role of pluripotency factors to drive stemness in gastrointestinal cancer. Stem Cell Res. 2016, 16, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Aponte, P.M.; Caicedo, A. Stemness in cancer: Stem cells, cancer stem cells, and their microenvironment. Stem Cells Int. 2017, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, M.C.; Hollingsworth, R.E.; Hurt, E.M. Cancer stem cell plasticity and tumor hierarchy. World J. Stem Cells 2015, 7, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.S.; Jiang, J.; Liang, X.H.; Tang, T.L. Links between cancer stem cells and epithelial-mesenchymal transition. Onco Targets Ther. 2015, 8, 2973–2980. [Google Scholar] [PubMed]
- Li, C.J.; Chu, P.Y.; Yiang, G.T.; Wu, M.Y. The molecular mechanism of epithelial-mesenchymal transition for breast carcinogenesis. Biomolecules 2019, 9, 476. [Google Scholar] [CrossRef]
- Krzysiek-Maczka, G.; Targosz, A.; Szczyrk, U.; Strzalka, M.; Brzozowski, T.; Ptak-Belowska, A. Involvement of epithelial-mesenchymal transition-inducing transcription factors in the mechanism of Helicobacter pylori-induced fibroblasts activation. J. Physiol. Pharmacol. 2019, 70. [Google Scholar] [CrossRef]
- Krzysiek-Maczka, G.; Targosz, A.; Ptak-Belowska, A.; Korbut, E.; Szczyrk, U.; Strzalka, M.; Brzozowski, T. Molecular alterations in fibroblasts exposed to Helicobacter pylori: A missing link in bacterial inflammation progressing into gastric carcinogenesis? J. Physiol. Pharmacol. 2013, 64, 77–87. [Google Scholar] [PubMed]
- Krzysiek-Maczka, G.; Targosz, A.; Szczyrk, U.; Strzałka, M.; Sliwowski, Z.; Brzozowski, T.; Czyz, J.; Ptak-Belowska, A. Role of Helicobacter pylori infection in cancer-associated fibroblast- induced epithelial-mesenchymal transition in vitro. Helicobacter 2018, 23. [Google Scholar] [CrossRef]
- Kobayashi, I.; Kawano, S.; Tsuji, S.; Matsui, H.; Nakama, A.; Sawaoka, H.; Masuda, E.; Takei, Y.; Kouichi Nagano, K.; Fusamoto, H.; et al. RGM1, a cell line derived from normal gastric mucosa of rat. In Vitro Cell Dev. Biol. Anim. 1996, 32, 259–261. [Google Scholar] [CrossRef]
- Lu, H.J.; Yan, J.; Jin, P.Y.; Zheng, G.H.; Qin, S.M.; Wu, D.M.; Lu, J.; Zheng, Y.L. MicroRNA-152 inhibits tumor cell growth while inducing apoptosis via the transcriptional repression of cathepsin L in gastrointestinal stromal tumor. Cancer Biomark. 2018, 21, 711–722. [Google Scholar] [CrossRef]
- Chomczynski, P.; Sacchi, N. Single-Step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Park, J.; Lee, S.Y.; Ooshima, A.; Yang, K.M.; Kang, J.M.; Kim, Y.W.; Kim, S.J. Glucosamine hydrochloride exerts a protective effect against unilateral ureteral obstruction-induced renal fibrosis by attenuating TGF-β signaling. J. Mol. Med. 2013, 91, 1273–1284. [Google Scholar] [CrossRef] [PubMed]
- Marte, B. Tumour heterogeneity. Nature 2013, 501, 327. [Google Scholar] [CrossRef] [PubMed]
- Brungs, D.; Aghmesheh, M.; Vine, K.L.; Becker, T.M.; Carolan, M.G.; Ranson, M. Gastric cancer stem cells: Evidence, potential markers, and clinical implications. J. Gastroenterol. 2016, 51, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Bekaii-Saab, T.; El-Rayes, B. Identifying and targeting cancer stem cells in the treatment of gastric cancer. Cancer 2017, 123, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- May, C.D.; Sphyris, N.; Evans, K.W.; Werden, S.J.; Guo, W.; Mani, S.A. Epithelial-Mesenchymal transition and cancer stem cells: A dangerously dynamic duo in breast cancer progression. Breast Cancer Res. 2011, 13, 202. [Google Scholar] [CrossRef] [PubMed]
- Maugeri-Saccà, M.; Vigneri, P.; De Maria, R. Cancer stem cells and chemosensitivity. Clin. Cancer Res. 2011, 17, 4942–4947. [Google Scholar] [CrossRef] [PubMed]
- Hochedlinger, K.; Yamada, Y.; Beard, C.; Jaenisch, R. Ectopic expression of Oct-4 blocks progenitor-cell differentiation and causes dysplasia in epithelial tissues. Cell 2005, 121, 465–477. [Google Scholar] [CrossRef]
- Al-Marzoqee, F.Y.; Khoder, G.; Al-Awadhi, H.; John, R.; Beg, A.; Vincze, A.; Branicki, F.; Karam, S.M. Upregulation and inhibition of the nuclear translocation of Oct4 during multistep gastric carcinogenesis. Int. J. Oncol. 2012, 41, 1733–1743. [Google Scholar] [CrossRef]
- Chen, Z.; Xu, W.R.; Qian, H.; Zhu, W.; Bu, X.B.; Wang, S.; Yan, Y.M.; Mao, F.; Gu, H.B.; Cao, H.L.; et al. Oct4, a novel marker for human gastric cancer. J. Surg. Oncol. 2009, 99, 414–419. [Google Scholar] [CrossRef]
- Zhang, L.; Guo, X.; Zhang, D.; Fan, Y.; Qin, L.; Dong, S.; Zhang, L. Upregulated miR-132 in Lgr5(+) gastric cancer stem cell-like cells contributes to cisplatin-resistance via SIRT1/CREB/ABCG2 signaling pathway. Mol. Carcinog. 2017, 56, 2022–2034. [Google Scholar] [CrossRef]
- Hoffmann, W. Current status on stem cells and cancers of the gastric epithelium. Int. J. Mol. Sci. 2015, 16, 19153–19169. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, L.; Todaro, M.; De Sousa Melo, F.; Sprick, M.R.; Kemper, K.; Perez Alea, M.; Richel, D.J.; Stassi, G.; Medema, J.P. Single-Cell cloning of colon cancer stem cells reveals a multi-lineage differentiation capacity. Proc. Natl. Acad. Sci. USA 2008, 105, 13427–13432. [Google Scholar] [CrossRef] [PubMed]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT subtype influences epithelial plasticity and mode of cell migration. Dev. Cell 2018, 45, 681–695. [Google Scholar] [CrossRef] [PubMed]
- Aiello, N.M.; Kang, Y. Context-Dependent EMT programs in cancer metastasis. J. Exp. Med. 2019, 216, 1016–1026. [Google Scholar] [CrossRef] [PubMed]
- Zhi, Y.; Mou, Z.; Chen, J.; He, Y.; Dong, H.; Fu, X.; Wu, Y. B7H1 expression and epithelial- to-mesenchymal transition phenotypes on colorectal cancer stem-like cells. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Chang, Y.W.; Su, Y.J.; Hsiao, M.; Wei, K.C.; Lin, W.H.; Liang, C.L.; Chen, S.C.; Lee, J.L. Diverse targets of β-catenin during the epithelial-mesenchymal transition define cancer stem cells and predict disease relapse. Cancer Res. 2015, 75, 3398–3410. [Google Scholar] [CrossRef]
- Garg, M. Urothelial cancer stem cells and epithelial plasticity: Current concepts and therapeutic implications in bladder cancer. Cancer Metastasis Rev. 2015, 34, 691–701. [Google Scholar] [CrossRef]
- Unternaehrer, J.J.; Zhao, R.; Kim, K.; Cesana, M.; Powers, J.T.; Ratanasirintrawoot, S.; Onder, T.; Shibue, T.; Weinberg, R.A.; Daley, G.Q. The epithelial-mesenchymal transition factor SNAIL paradoxically enhances reprogramming. Stem Cell Rep. 2014, 3, 691–698. [Google Scholar] [CrossRef]
- Comes, S.; Gagliardi, M.; Laprano, N.; Fico, A.; Cimmino, A.; Palamidessi, A.; De Cesare, D.; De Falco, S.; Angelini, A.; Scita, G.; et al. L-Proline induces a mesenchymal-like invasive program in embryonic stem cells by remodeling H3K9 and H3K36 methylation. Stem Cell Rep. 2013, 1, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, E.; Hermann, P.C.; Mueller, M.T.; Huber, S.; Balic, A.; Miranda-Lorenzo, I.; Zagorac, S.; Alcala, S.; Rodriguez-Arabaolaza, I.; Carlos Ramirez, J. Nodal/Activin signaling drives self-renewal and tumorigenicity of pancreatic cancer stem cells and provides a target for combined drug therapy. Cell Stem Cell 2011, 9, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Sainz, B., Jr.; Alcala, S.; Garcia, S.; Sanchez-Ripoll, Y.; Azevedo, M.M.; Cioffi, M.; Tatari, M.; Miranda-Lorenzo, I.; Hidalgo, M.; Gomez-Lopez, G.; et al. Microenvironmental hCAP-18/LL-37 promotes pancreatic ductal adenocarcinoma by activating its cancer stem cell compartment. Gut 2015, 64, 1921–1935. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.C.; Sancho, P.; Canamero, M.; Martinelli, P.; Madriles, F.; Michl, P.; Gress, T.; De Pascual, R.; Gandia, L.; Guerra, C.; et al. Nicotine promotes initiation and progression of KRAS-induced pancreatic cancer via Gata6-dependent dedifferentiation of acinar cells in mice. Gastroenterology 2014, 147. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.C.; Trabulo, S.M.; Sainz, B., Jr.; Balic, A.; Garcia, E.; Hahn, S.A.; Vandana, M.; Sahoo, S.K.; Tunici, P.; Bakker, A.; et al. Multimodal treatment eliminates cancer stem cells and leads to long-term survival in primary human pancreatic cancer tissue xenografts. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Mueller, M.T.; Hermann, P.C.; Witthauer, J.; Rubio-Viqueira, B.; Leicht, S.F.; Huber, S.; Ellwart, J.W.; Mustafa, M.; Bartenstein, P.; D’Haese, J.G.; et al. Combined targeted treatment to eliminate tumorigenic cancer stem cells in human pancreatic cancer. Gastroenterology 2009, 137, 1102–1113. [Google Scholar] [CrossRef]
- Radisky, D.C.; LaBarge, M.A. Epithelial-Mesenchymal transition and the stem cell phenotype. Cell Stem Cell 2008, 2, 511–512. [Google Scholar] [CrossRef]
- Scheel, C.; Weinberg, R.A. Phenotypic plasticity and epithelial-mesenchymal transitions in cancer and normal stem cells? Int. J. Cancer 2011, 129, 2310–2314. [Google Scholar] [CrossRef]
- Giannoni, E.; Bianchini, F.; Masieri, L.; Serni, S.; Torre, E.; Calorini, L.; Chiarugi, P. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res. 2010, 70, 6945–6956. [Google Scholar] [CrossRef]
- Honma, N.; Genda, T.; Matsuda, Y.; Yamagiwa, S.; Takamura, M.; Ichida, T.; Aoyagi, Y. MEK/ERK signaling is a critical mediator for integrin-induced cell scattering in highly metastatic hepatocellular carcinoma cells. Lab. Investig. 2006, 86, 687–696. [Google Scholar] [CrossRef][Green Version]
- Cowley, S.; Paterson, H.; Kemp, P.; Marshall, C.J. Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell 1994, 77, 841–852. [Google Scholar] [CrossRef]
- Mansour, S.J.; Matten, W.T.; Hermann, A.S.; Candia, J.M.; Rong, S.; Fukasawa, K.; Vande Woude, G.F.; Ahn, N.G. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science 1994, 265, 966–970. [Google Scholar] [CrossRef] [PubMed]
- Montesano, R.; Soriano, J.V.; Hosseini, G.; Pepper, M.S.; Schramek, H. Constitutively active mitogen-activated protein kinase kinase MEK1 disrupts morphogenesis and induces an invasive phenotype in Madin-Darby canine kidney epithelial cells. Cell Growth Differ. 1999, 10, 317–332. [Google Scholar] [PubMed]
- Pinkas, J.; Leder, P. MEK1 signaling mediates transformation and metastasis of EpH4 mammary epithelial cells independent of an epithelial to mesenchymal transition. Cancer Res. 2002, 62, 4781–4790. [Google Scholar]
- Voisin, L.; Julien, C.; Duhamel, S.; Gopalbhai, K.; Claveau, I.; Saba-El-Leil, M.K.; Rodrigue-Gervais, I.G.; Gaboury, L.; Lamarre, D.; Basik, M.; et al. Activation of MEK1 or MEK2 isoform is sufficient to fully transform intestinal epithelial cells and induce the formation of metastatic tumors. BMC Cancer 2008, 17, 337. [Google Scholar] [CrossRef]
- Xu, Z.; Shen, M.X.; Ma, D.Z.; Wang, L.W.; Zha, X.L. TGF-β1-promoted epithelial- to–mesenchymal transformation and cell adhesion contribute to TGF-β 1-enhanced cell migration in SMMC-7721 cells. Cell Res. 2003, 13, 343–350. [Google Scholar] [CrossRef]
- Nantajit, D.; Lin, D.; Li, J.J. The network of epithelial-mesenchymal transition: Potential new targets for tumor resistance. J. Cancer Res. Clin. Oncol. 2015, 141, 1697–1713. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-β -induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef]
- Attisano, L.; Wrana, J.L. Signal integration in TGF-β, WNT, and Hippo pathways. F1000Prime Rep. 2013, 5, 172013. [Google Scholar] [CrossRef]
- Morris, S.M.; Carter, K.T.; Baek, J.Y.; Koszarek, A.; Yeh, M.M.; Knoblaugh, S.E.; Grady, W.M. TGF-β signaling alters the pattern of liver tumorigenesis induced by Pten inactivation. Oncogene 2015, 34, 3273–3282. [Google Scholar] [CrossRef] [PubMed]
- Horie, Y.; Suzuki, A.; Kataoka, E.; Sasaki, T.; Hamada, K.; Sasaki, J.; Mizuno, K.; Hasegawa, G.; Kishimoto, H.; Iizuka, M.; et al. Hepatocyte-Specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J. Clin. Investig. 2004, 113, 1774–1783. [Google Scholar] [CrossRef] [PubMed]
- Ikushima, H.; Miyazono, K. TGFβ signalling: A complex web in cancer progression. Nat. Rev. Cancer 2010, 10, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Akhurst, R.J. Differentiation plasticity regulated by TGF-beta family proteins in development and disease. Nat. Cell Biol. 2007, 9, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Shuang, L.; Chen, S.; Zeng, J. TGF-β signaling: A complex role in tumorigenesis. Mol. Med. Rep. 2018, 17, 699–704. [Google Scholar]
- Cordenonsi, M.; Dupont, S.; Maretto, S.; Insinga, A.; Imbriano, C.; Piccolo, S. Links between tumor suppressors: p53 is required for TGF-β gene responses by cooperating with Smads. Cell 2003, 113, 301–314. [Google Scholar] [CrossRef]
- Kalo, E.; Buganim, Y.; Shapira, K.E.; Besserglick, H.; Goldfinger, N.; Weisz, L.; Stambolsky, P.; Henis, Y.I.; Rotter, V. Mutant p53 attenuates the Smad-dependent transforming growth factor β1 (TGF-β1) signaling pathway by repressing the expression of TGF-β receptor type II. Mol. Cell. Biol. 2007, 27, 8228–8242. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Y.; Liu, Y.; Wang, N.; Qi, Y.; Du, J. Krüppel-Like factor 4 transcriptionally regulates TGF-β1 and contributes to cardiac myofibroblast differentiation. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Sun, H.; Peng, Z.; Tang, H.; Xie, D.; Jia, Z.; Zhong, L.; Zhao, S.; Ma, Z.; Gao, Y.; Zeng, L.; et al. Loss of KLF4 and consequential downregulation of Smad7 exacerbate oncogenic TGF-β signaling in and promote progression of hepatocellular carcinoma. Oncotarget 2017, 36, 2957–2968. [Google Scholar] [CrossRef]
- He, M.; Zheng, B.; Zhang, Y.; Zhang, X.H.; Wang, C.; Yang, Z.; Sun, Y.; Wu, X.L.; Wen, J.K. KLF4 mediates the link between TGF-β1-induced gene transcription and H3 acetylation in vascular smooth muscle cells. FASEB J. 2015, 29, 4059–4070. [Google Scholar] [CrossRef]
- Podolec, J.; Baran, J.; Siedlinski, M.; Urbanczyk, M.; Krupinski, M.; Bartus, K.; Niewiara, L.; Podolec, M.; Guzik, T.; Tomkiewicz-Pajak, L.; et al. Serum rantes, transforming growth factor-β1 and interleukin-6 levels correlate with cardiac muscle fibrosis in patients with aortic valve stenosis. J. Physiol. Pharmacol. 2018, 69. [Google Scholar] [CrossRef]
- Wei, D.; Gong, W.; Kanai, M.; Schlunk, C.; Wang, L.; Yao, J.C.; Wu, T.T.; Huang, S.; Xie, K. Drastic down-regulation of Krüppel-like factor 4 expression is critical in human gastric cancer development and progression. Cancer Res. 2005, 65, 2746–2754. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Kanai, M.; Huang, S.; Xie, K. Emerging role of KLF4 in human gastrointestinal cancer. Carcinogenesis 2006, 1, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Li, H.X.; Han, M.; Bernier, M.; Zheng, B.; Sun, S.G.; Su, M.; Zhang, R.; Fu, J.R.; Wen, J.K. Krüppel-Like factor 4 promotes differentiation by transforming growth factor-beta receptor-mediated Smad and p38 MAPK signaling in vascular smooth muscle cells. J. Biol. Chem. 2010, 285, 17846–17856. [Google Scholar] [CrossRef]
- Kim, M.; Kim, S.H.; Lim, J.W.; Kim, H. Lycopene induces apoptosis by inhibiting nuclear translocation of β-catenin in gastric cancer cells. J. Physiol. Pharmacol. 2019, 70. [Google Scholar] [CrossRef]
- Hashimoto, I.; Nagata, T.; Sekine, S.; Moriyama, M.; Shibuya, K.; Hojo, S.; Matsui, K.; Yoshioka, I.; Okumura, T.; Hori, T.; et al. Prognostic significance of KLF4 expression in gastric cancer. Oncol. Lett. 2017, 13, 819–826. [Google Scholar] [CrossRef]
- Cordenonsi, M.; Montagner, M.; Adorno, M.; Zacchigna, L.; Martello, G.; Mamidi, A.; Soligo, S.; Dupont, S.; Piccolo, S. Integration of TGF-β and Ras/MAPK signaling through p53 phosphorylation. Science 2007, 315, 840–843. [Google Scholar] [CrossRef]
- Elston, R.; Inman, G.J. Crosstalk between p53 and TGF-β Signalling. J. Signal Transduct. 2012, 294097, 1–10. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence | Size of PCR Product (bp) | Annealing Temp. (°C) |
|---|---|---|---|
| 18S | Forward 5′-GTTGGTTTTGATCTGATAAATGC-3′ Reverse 5′-CATTAAATCAGTTATGGTTCCTTTG-3′ | 143 | 60 |
| Bax | Forward 5′-CTGCCAACCCACCCTGGTCT-3′ Reverse 5′-TGGCAGCTGACATGTTTTCTG-3′ | 195 | 55 |
| BCL2 | Forward 5′-ACTGAGTACCTGAACCGGCATC-3′ Reverse 5′-GGAGAAATCAAACAGAGGTCGC-3′ | 108 | 60 |
| c-Myc | Forward 5′-CCACACAGCCCACTGGTCCT-3′ Reverse 5′-GGCTGGAGCATTTGCGGTTGTT-3′ | 163 | 60 |
| Cyclin D1 | Forward 5′-TGCTTGGGAAGTTGTGTTGG-3′ Reverse 5′-AATGCCATCACGGTCCCTAC-3′ | 126 | 60 |
| c-Myc | Forward 5′-CCACACAGCCCACTGGTCCT-3′ Reverse 5′-GGCTGGAGCATTTGCGGTTGTT-3′ | 163 | 60 |
| Ki-67 | Forward 5′-AACCAGGACTTTGTGCTCTGTAA-3′ Reverse 5′-CTCTTTTGGCTTCCATTTCTTC-3′ | 209 | 60 |
| Klf4 | Forward 5′-TTCTCCACGTTCGCGTCCGG-3′ Reverse 5′-TCTCGCCAACGGTTAGTCGGGG-3′ | 80 | 60 |
| Oct4 | Forward 5′-GGAGGGATGGCATACTGTGGACCT-3′ Reverse 5′-TCCTGGGACTCCTCGGGACTAGG-3′ | 197 | 60 |
| p53 | Forward 5′-AGTGAAGGGACTAGCATTGTC-3′ Reverse 5′-GGATGCCCGTGCTGCCGAGGAG-3′ | 243 | 60 |
| Sox2 | Forward 5′-AGAACCCCAAGATGCACAAC-3′ Reverse 5′-CTCCGGGAAGCGTGTACTTA-3′ | 204 | 60 |
| TGFβ rec 2 | Forward 5′-TGTGGCAGAGCGCTTCAGT-3′ Reverse 5′-TGTTCAGGGAGCCGTCTTCT-3′ | 95 | 60 |
| TGFβ rec 1 | Forward 5′-GCAGACTGGACCAGCAATGAC-3′ Reverse 5′-CTGCAATCAGGATCACTGCAA-3′ | 118 | 60 |
| Hp 16S | Forward 5′-GTCAAGAGATCAGCCTATGTCC-3′ Reverse 5′-TGGCAATCAGCGTCAGGTAATG-3′ | 522 | 54 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krzysiek-Maczka, G.; Targosz, A.; Szczyrk, U.; Wrobel, T.; Strzalka, M.; Brzozowski, T.; Czyz, J.; Ptak-Belowska, A. Long-Term Helicobacter pylori Infection Switches Gastric Epithelium Reprogramming towards Cancer Stem Cell-Related Differentiation Program in Hp-Activated Gastric Fibroblast-TGFβ Dependent Manner. Microorganisms 2020, 8, 1519. https://doi.org/10.3390/microorganisms8101519
Krzysiek-Maczka G, Targosz A, Szczyrk U, Wrobel T, Strzalka M, Brzozowski T, Czyz J, Ptak-Belowska A. Long-Term Helicobacter pylori Infection Switches Gastric Epithelium Reprogramming towards Cancer Stem Cell-Related Differentiation Program in Hp-Activated Gastric Fibroblast-TGFβ Dependent Manner. Microorganisms. 2020; 8(10):1519. https://doi.org/10.3390/microorganisms8101519
Chicago/Turabian StyleKrzysiek-Maczka, Gracjana, Aneta Targosz, Urszula Szczyrk, Tomasz Wrobel, Malgorzata Strzalka, Tomasz Brzozowski, Jaroslaw Czyz, and Agata Ptak-Belowska. 2020. "Long-Term Helicobacter pylori Infection Switches Gastric Epithelium Reprogramming towards Cancer Stem Cell-Related Differentiation Program in Hp-Activated Gastric Fibroblast-TGFβ Dependent Manner" Microorganisms 8, no. 10: 1519. https://doi.org/10.3390/microorganisms8101519
APA StyleKrzysiek-Maczka, G., Targosz, A., Szczyrk, U., Wrobel, T., Strzalka, M., Brzozowski, T., Czyz, J., & Ptak-Belowska, A. (2020). Long-Term Helicobacter pylori Infection Switches Gastric Epithelium Reprogramming towards Cancer Stem Cell-Related Differentiation Program in Hp-Activated Gastric Fibroblast-TGFβ Dependent Manner. Microorganisms, 8(10), 1519. https://doi.org/10.3390/microorganisms8101519