Microbiome Management by Biological and Chemical Treatments in Maize Is Linked to Plant Health

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Seed Treatments and Sampling Strategy
2.2. DNA Extraction and Amplicon Library Construction
2.3. Bioinformatic Analysis
2.4. Statistical Analysis
2.5. qPCR Analysis of SPA-P69 Gene Copy Numbers
3. Results
3.1. Plant Growth Performance and SPA-P69 Colonization in the Rhizosphere
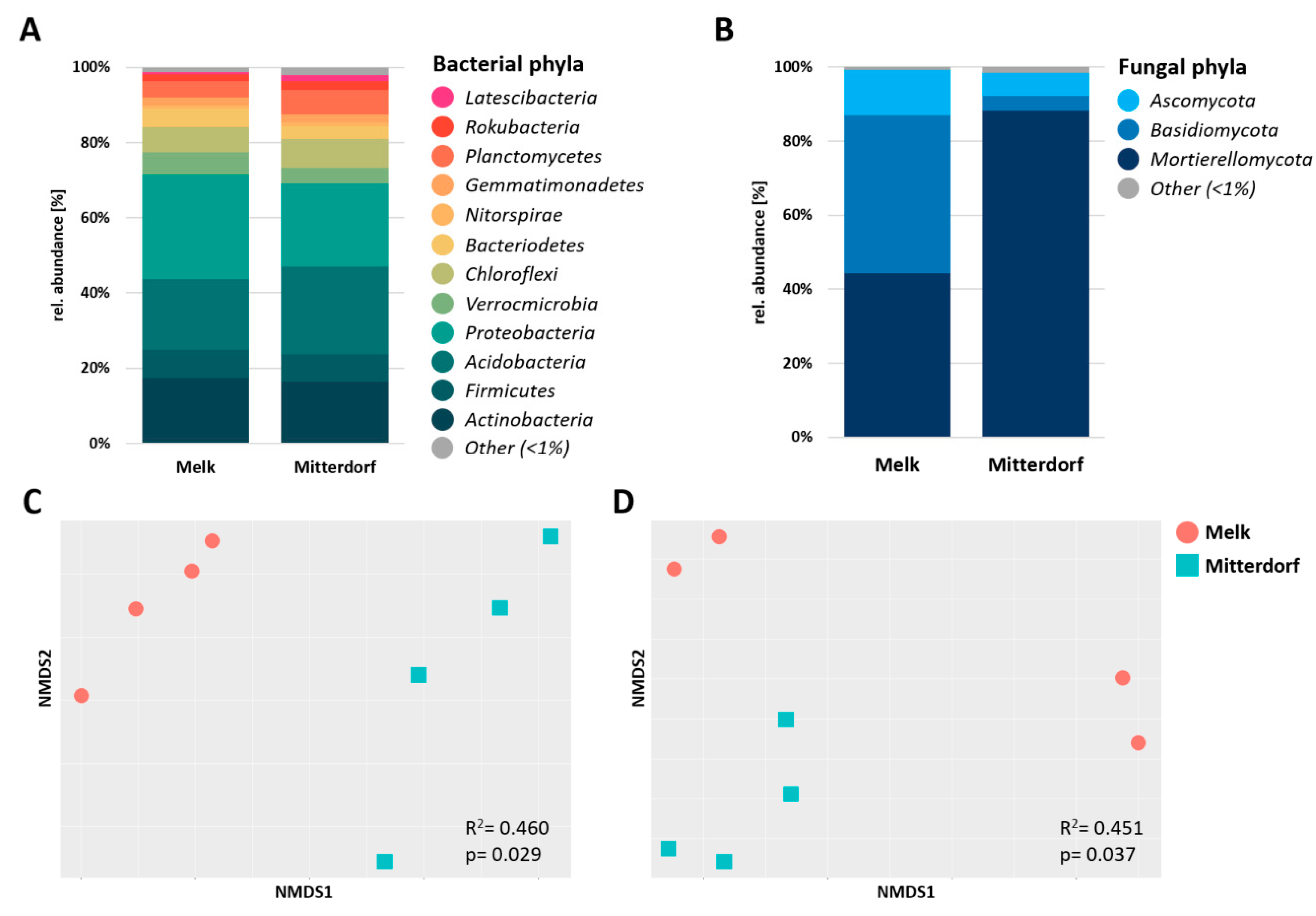
3.2. Microbial Diversity Analysis Comparing the Two Sites
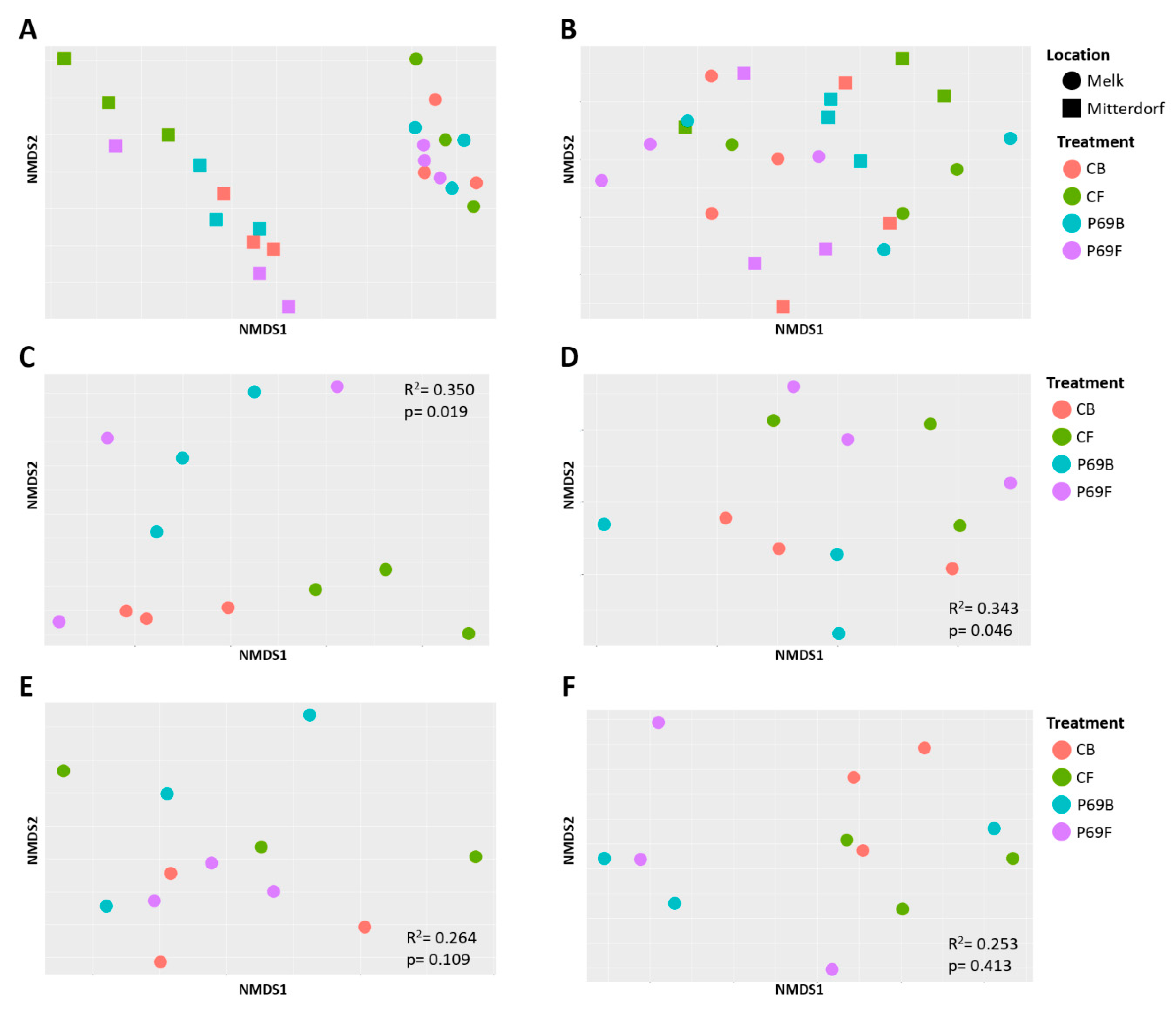
3.3. Influence of Seed Treatments on the Alpha and Beta Microbial Diversity
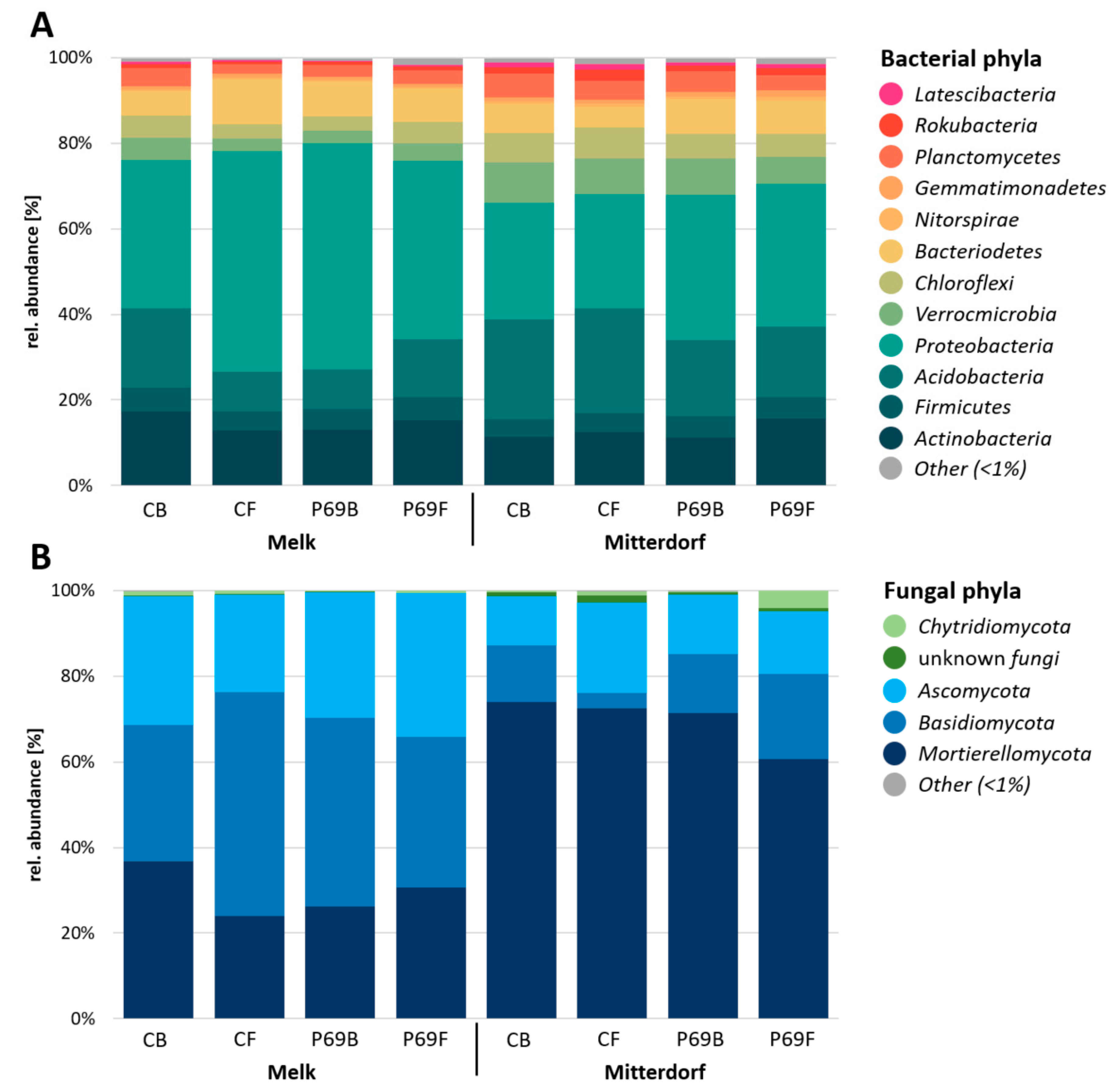
3.4. Microbial Composition in Response to Seed Treatments
3.5. Identification of Responder Taxa in Response to Seed Treatments
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- Vandenkoornhuyse, P.; Quaiser, A.; Duhamel, M.; Van, A.L.; Dufresne, A. The importance of the microbiome of the plant holobiont. New Phytol. 2015, 206, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Cordovez, V.; Dini-Andreote, F.; Carrión, V.J.; Raaijmakers, J.M. Ecology and Evolution of Plant Microbiomes. Annu. Rev. Microbiol. 2019, 73, 69–88. [Google Scholar] [CrossRef] [PubMed]
- Lambers, H.; Mougel, C.; Jaillard, B.; Hinsinger, P. Plant-microbe-soil interactions in the rhizosphere: An evolutionary perspective. Plant Soil 2009, 321, 83–115. [Google Scholar] [CrossRef]
- Mendes, R.; Garbeva, P.; Raaijmakers, J.M. The rhizosphere microbiome: Significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. FEMS Microbiol. Rev. 2013, 37, 634–663. [Google Scholar] [CrossRef]
- Philippot, L.; Raaijmakers, J.M.; Lemanceau, P.; Van Der Putten, W.H. Going back to the roots: The microbial ecology of the rhizosphere. Nat. Rev. Genet. 2013, 11, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Bakker, P.A.H.M.; Doornbos, R.F.; Zamioudis, C.; Berendsen, R.L.; Pieterse, C.M.J. Induced Systemic Resistance and the Rhizosphere Microbiome. Plant Pathol. J. 2013, 29, 136–143. [Google Scholar] [CrossRef]
- Berg, G.; Rybakova, D.; Grube, M.; Köberl, M. The plant microbiome explored: Implications for experimental botany. J. Exp. Bot. 2015, 67, 995–1002. [Google Scholar] [CrossRef]
- Lugtenberg, B.; Kamilova, F. Plant-Growth-Promoting Rhizobacteria. Annu. Rev. Microbiol. 2009, 63, 541–556. [Google Scholar] [CrossRef]
- Bennett, J.A.; Klironomos, J. Mechanisms of plant–soil feedback: Interactions among biotic and abiotic drivers. New Phytol. 2018, 222, 91–96. [Google Scholar] [CrossRef]
- Mendes, R.; Kruijt, M.; De Bruijn, I.; Dekkers, E.; Van Der Voort, M.; Schneider, J.H.M.; Piceno, Y.M.; DeSantis, T.Z.; Andersen, G.L.; Bakker, P.A.H.M.; et al. Deciphering the Rhizosphere Microbiome for Disease-Suppressive Bacteria. Science 2011, 332, 1097–1100. [Google Scholar] [CrossRef]
- Bardgett, R.D.; Van Der Putten, W.H. Belowground biodiversity and ecosystem functioning. Nature 2014, 515, 505–511. [Google Scholar] [CrossRef]
- Berg, G.; Köberl, M.; Rybakova, D.; Müller, H.; Grosch, R.; Smalla, K. Plant microbial diversity is suggested as the key to future biocontrol and health trends. FEMS Microbiol. Ecol. 2017, 93, 93. [Google Scholar] [CrossRef]
- Katan, J. Diseases caused by soilborne pathogens: Biology, management and challenges. J. Plant. Pathol. 2017, 99, 305–315. [Google Scholar]
- Oerke, E.-C. Crop losses to pests. J. Agric. Sci. 2006, 144, 31–43. [Google Scholar] [CrossRef]
- De Long, J.R.; Fry, E.L.; Veen, G.F.; Kardol, P. Why are plant–soil feedbacks so unpredictable, and what to do about it? Funct. Ecol. 2019, 33, 118–128. [Google Scholar] [CrossRef]
- Kostenko, O.; Van De Voorde, T.F.J.; Mulder, P.P.J.; Van Der Putten, W.H.; Bezemer, T.M. Legacy effects of aboveground-belowground interactions. Ecol. Lett. 2012, 15, 813–821. [Google Scholar] [CrossRef]
- Wightwick, A.M.; Walters, R.; Allinson, G.; Reichman, S.; Menzies, N. Environmental Risks of Fungicides Used in Horticultural Production Systems. In Fungicides; InTech: Reijka, Croatia, 2010; pp. 273–304. [Google Scholar]
- Berg, G. Plant–microbe interactions promoting plant growth and health: Perspectives for controlled use of microorganisms in agriculture. Appl. Microbiol. Biotechnol. 2009, 84, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Carrión, V.J.; Perez-Jaramillo, J.; Cordovez, V.; Tracanna, V.; De Hollander, M.; Ruiz-Buck, D.; Mendes, L.W.; Van Ijcken, W.; Gomez-Exposito, R.; Elsayed, S.S.; et al. Pathogen-induced activation of disease-suppressive functions in the endophytic root microbiome. Science 2019, 366, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Rybakova, D.; Wikström, M.; Birch-Jensen, F.; Postma, J.; Ehlers, R.; Schmuck, M.; Kollmann, R.; Köhl, J.; Berg, G. Verticillium Wilt in Oilseed Rape—the Microbiome is Crucial for Disease Outbreaks as Well as for Efficient Suppression. Plants 2020, 9, 866. [Google Scholar] [CrossRef] [PubMed]
- Alavi, P.; Starcher, M.R.; Zachow, C.; Müller, H.; Berg, G. Root-microbe systems: The effect and mode of interaction of Stress Protecting Agent (SPA) Stenotrophomonas rhizophila DSM14405T. Front. Plant Sci. 2013, 4, 141. [Google Scholar] [CrossRef]
- FAO FAOSTAT. Food and Agriculture Organization of the United Nations. 2018. Available online: http://www.fao.org/faostat/en/ (accessed on 15 April 2020).
- Peiffer, J.A.; Spor, A.; Koren, O.; Jin, Z.; Tringe, S.G.; Dangl, J.L.; Buckler, E.S.; E Ley, R. Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc. Natl. Acad. Sci. USA 2013, 110, 6548–6553. [Google Scholar] [CrossRef] [PubMed]
- Walters, W.A.; Jin, Z.; Youngblut, N.; Wallace, J.G.; Sutter, J.; Zhang, W.; González-Peña, A.; Peiffer, J.; Koren, O.; Shi, Q.; et al. Large-scale replicated field study of maize rhizosphere identifies heritable microbes. Proc. Natl. Acad. Sci. USA 2018, 115, 7368–7373. [Google Scholar] [CrossRef] [PubMed]
- Raaijmakers, J.M.; Paulitz, T.; Steinberg, C.; Alabouvette, C.; Moënne-Loccoz, Y. The rhizosphere: A playground and battlefield for soilborne pathogens and beneficial microorganisms. Plant Soil 2008, 321, 341–361. [Google Scholar] [CrossRef]
- Schmidt, C.S.; Alavi, M.; Cardinale, M.; Müller, H.; Berg, G. Stenotrophomonas rhizophila DSM14405T promotes plant growth probably by altering fungal communities in the rhizosphere. Biol. Fertil. Soils 2012, 48, 947–960. [Google Scholar] [CrossRef]
- Cernava, T.; Chen, X.; Krug, L.; Li, H.; Yang, M.; Berg, G. The tea leaf microbiome shows specific responses to chemical pesticides and biocontrol applications. Sci. Total Environ. 2019, 667, 33–40. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2010, 108, 4516–4522. [Google Scholar] [CrossRef]
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 2015, 18, 1403–1414. [Google Scholar] [CrossRef]
- White, T.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: San Diego, CA, USA, 1990; pp. 315–322. [Google Scholar]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; A Johnson, A.J.; Holmes, S. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Kõljalg, U.; Nilsson, R.H.; Abarenkov, K.; Tedersoo, L.; Taylor, A.F.S.; Bahram, M.; Bates, S.T.; Bruns, T.D.; Bengtsson-Palme, J.; Callaghan, T.M.; et al. Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 2013, 22, 5271–5277. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, R.H.; Larsson, K.-H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Glöckner, F.O.; Tedersoo, L.; et al. The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2019, 47, D259–D264. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahe, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Lugtenberg, B.J.; Dekkers, L.C. What makes Pseudomonas bacteria rhizosphere competent? Environ. Microbiol. 1999, 1, 9–13. [Google Scholar] [CrossRef]
- Adam, E.; Groenenboom, A.E.; Kurm, V.; Rajewska, M.; Schmidt, R.; Tyc, O.; Weidner, S.; Berg, G.; De Boer, W.; Salles, J.F. Controlling the Microbiome: Microhabitat Adjustments for Successful Biocontrol Strategies in Soil and Human Gut. Front. Microbiol. 2016, 7, 1079. [Google Scholar] [CrossRef]
- Berg, G.; Smalla, K. Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere. FEMS Microbiol. Ecol. 2009, 68, 1–13. [Google Scholar] [CrossRef]
- Costa, R.; Götz, M.; Mrotzek, N.; Lottmann, J.; Berg, G.; Smalla, K.; Götz, M. Effects of site and plant species on rhizosphere community structure as revealed by molecular analysis of microbial guilds. FEMS Microbiol. Ecol. 2006, 56, 236–249. [Google Scholar] [CrossRef]
- Fan, K.; Weisenhorn, P.; Gilbert, J.A.; Chu, H. Wheat rhizosphere harbors a less complex and more stable microbial co-occurrence pattern than bulk soil. Soil Boil. Biochem. 2018, 125, 251–260. [Google Scholar] [CrossRef]
- Nettles, R.; Watkins, J.; Ricks, K.; Boyer, M.; Licht, M.; Atwood, L.W.; Peoples, M.; Smith, R.G.; Mortensen, D.A.; Koide, R.T. Influence of pesticide seed treatments on rhizosphere fungal and bacterial communities and leaf fungal endophyte communities in maize and soybean. Appl. Soil Ecol. 2016, 102, 61–69. [Google Scholar] [CrossRef]
- Fournier, B.; Dos Santos, S.P.; Gustavsen, J.A.; Imfeld, G.; Lamy, F.; Mitchell, E.A.D.; Mota, M.; Noll, D.; Planchamp, C.; Heger, T. Impact of a synthetic fungicide (fosetyl-Al and propamocarb-hydrochloride) and a biopesticide (Clonostachys rosea) on soil bacterial, fungal, and protist communities. Sci. Total Environ. 2020, 738, 139635. [Google Scholar] [CrossRef]
- Kalia, A.; Gosal, S.K. Effect of pesticide application on soil microorganisms. Arch. Agron. Soil Sci. 2011, 57, 569–596. [Google Scholar] [CrossRef]
- Sułowicz, S.; Cycoń, M.; Piotrowska-Seget, Z. Non-target impact of fungicide tetraconazole on microbial communities in soils with different agricultural management. Ecotoxicology 2016, 25, 1047–1060. [Google Scholar] [CrossRef] [PubMed]
- Vasanthakumari, M.M.; Shridhar, J.; Madhura, R.J.; Nandhitha, M.; Kasthuri, C.; Janardhana, B.; Nataraja, K.N.; Ravikanth, G.; Shaanker, R.U. Role of endophytes in early seedling growth of plants: A test using systemic fungicide seed treatment. Plant Physiol. Rep. 2018, 24, 86–95. [Google Scholar] [CrossRef]
- Naether, A.; Foesel, B.U.; Naegele, V.; Wüst, P.K.; Weinert, J.; Bonkowski, M.; Alt, F.; Oelmann, Y.; Polle, A.; Lohaus, G.; et al. Environmental Factors Affect Acidobacterial Communities below the Subgroup Level in Grassland and Forest Soils. Appl. Environ. Microbiol. 2012, 78, 7398–7406. [Google Scholar] [CrossRef]
- Orlando, V.; Chitambar, J.J.; Dong, K.; Chizhov, V.N.; Mollov, D.; Bert, W.; Subbotin, S.A. Molecular and morphological characterisation of Xiphinema americanum-group species (Nematoda: Dorylaimida) from California, USA, and other regions, and co-evolution of bacteria from the genus Candidatus Xiphinematobacter with nematodes. Nematology 2016, 18, 1015–1043. [Google Scholar] [CrossRef]
- Vandekerckhove, T.; Willems, A.; Gillis, M.; Coomans, A. Occurrence of novel verrucomicrobial species, endosymbiotic and associated with parthenogenesis in Xiphinema americanum-group species (Nematoda, Longidoridae). Int. J. Syst. Evol. Microbiol. 2000, 50, 2197–2205. [Google Scholar] [CrossRef]
- Al-Shammari, T.A.; Bahkali, A.H.; Elgorban, A.M.; El-Kahky, M.T.; Al-Sum, B.A. The use of Trichoderma longibrachiatum and Mortierella alpina against root-knot nematode, Meloidogyne javanica on tomato. J. Pure Appl. Microbiol 2013, 7, 199–207. [Google Scholar]
- Eroshin, V.; Dedyukhina, E. Effect of lipids from Mortierella hygrophila on plant resistance to phytopathogens. World J. Microbiol. Biotechnol. 2002, 18, 165–167. [Google Scholar] [CrossRef]
- Zhang, K.; Bonito, G.; Hsu, C.-M.; Hameed, K.; Vilgalys, R.; Liao, H.-L. Mortierella elongata Increases Plant Biomass among Non-Leguminous Crop Species. Agronomy 2020, 10, 754. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Table | PGPR Strain | Fungicide |
|---|---|---|
| CB | No | No |
| CF | No | YES |
| P69B | Stenotrophomonas rhizophila SPA-P69 | No |
| P69F | Stenotrophomonas rhizophila SPA-P69 | YES |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kusstatscher, P.; Wicaksono, W.A.; Thenappan, D.P.; Adam, E.; Müller, H.; Berg, G. Microbiome Management by Biological and Chemical Treatments in Maize Is Linked to Plant Health. Microorganisms 2020, 8, 1506. https://doi.org/10.3390/microorganisms8101506
Kusstatscher P, Wicaksono WA, Thenappan DP, Adam E, Müller H, Berg G. Microbiome Management by Biological and Chemical Treatments in Maize Is Linked to Plant Health. Microorganisms. 2020; 8(10):1506. https://doi.org/10.3390/microorganisms8101506
Chicago/Turabian StyleKusstatscher, Peter, Wisnu Adi Wicaksono, Dhivya P. Thenappan, Eveline Adam, Henry Müller, and Gabriele Berg. 2020. "Microbiome Management by Biological and Chemical Treatments in Maize Is Linked to Plant Health" Microorganisms 8, no. 10: 1506. https://doi.org/10.3390/microorganisms8101506
APA StyleKusstatscher, P., Wicaksono, W. A., Thenappan, D. P., Adam, E., Müller, H., & Berg, G. (2020). Microbiome Management by Biological and Chemical Treatments in Maize Is Linked to Plant Health. Microorganisms, 8(10), 1506. https://doi.org/10.3390/microorganisms8101506