Gene Cloning, Recombinant Expression, Characterization, and Molecular Modeling of the Glycolytic Enzyme Triosephosphate Isomerase from Fusarium oxysporum

 , , , ,
, , , ,  ,
,  , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Source of Fungal Strain
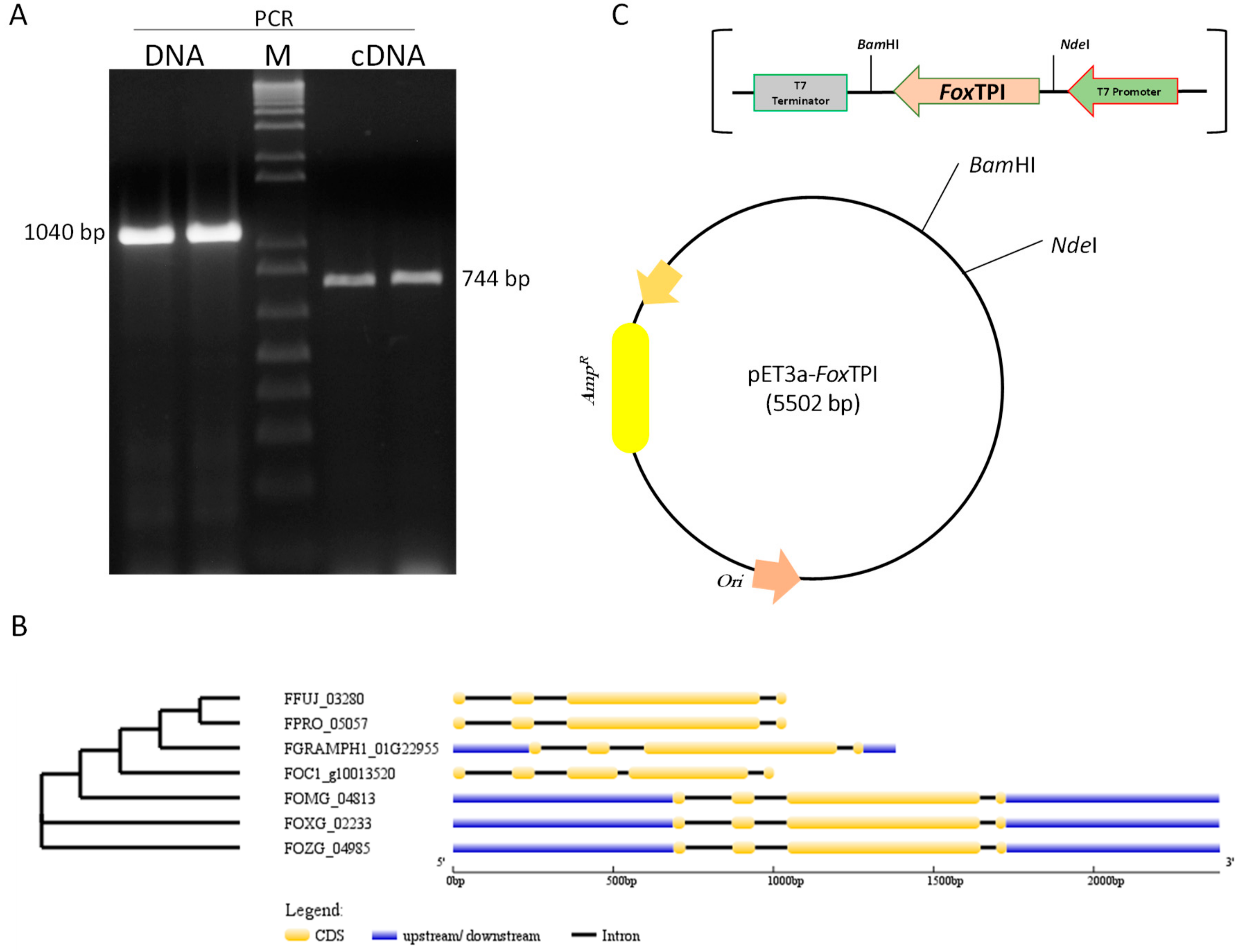
2.2. Nucleic Acids Extraction, RT-PCR, and Cloning
2.3. Aligment of FoxTPI Protein
2.4. Expression and Purification
2.5. Characterization of Functional of FoxTPI
2.5.1. Oligomeric Status of the Protein
2.5.2. Effect of pH on Activity
2.5.3. Effect of Temperature on Activity
2.5.4. Determination of Steady-State Kinetic Parameters
2.6. Evaluation of FoxTPI Protein Stability
2.6.1. Thermal Stability
2.6.2. Stability of FoxTPI in the Presence of Guanidine Hydrochloride (Gdn-HCl)
2.7. Spectroscopic Characterization of the FoxTPI Protein
2.7.1. Circular Dichroism (CD) Analysis
2.7.2. Structural Analysis through Intrinsic Fluorescence
2.8. Bioinformatics and Homology Modeling of FoxTPI
2.9. Assay Inactivation
3. Results and Discussion
3.1. Sequence Analysis and Cloning of FoxTpi Gene
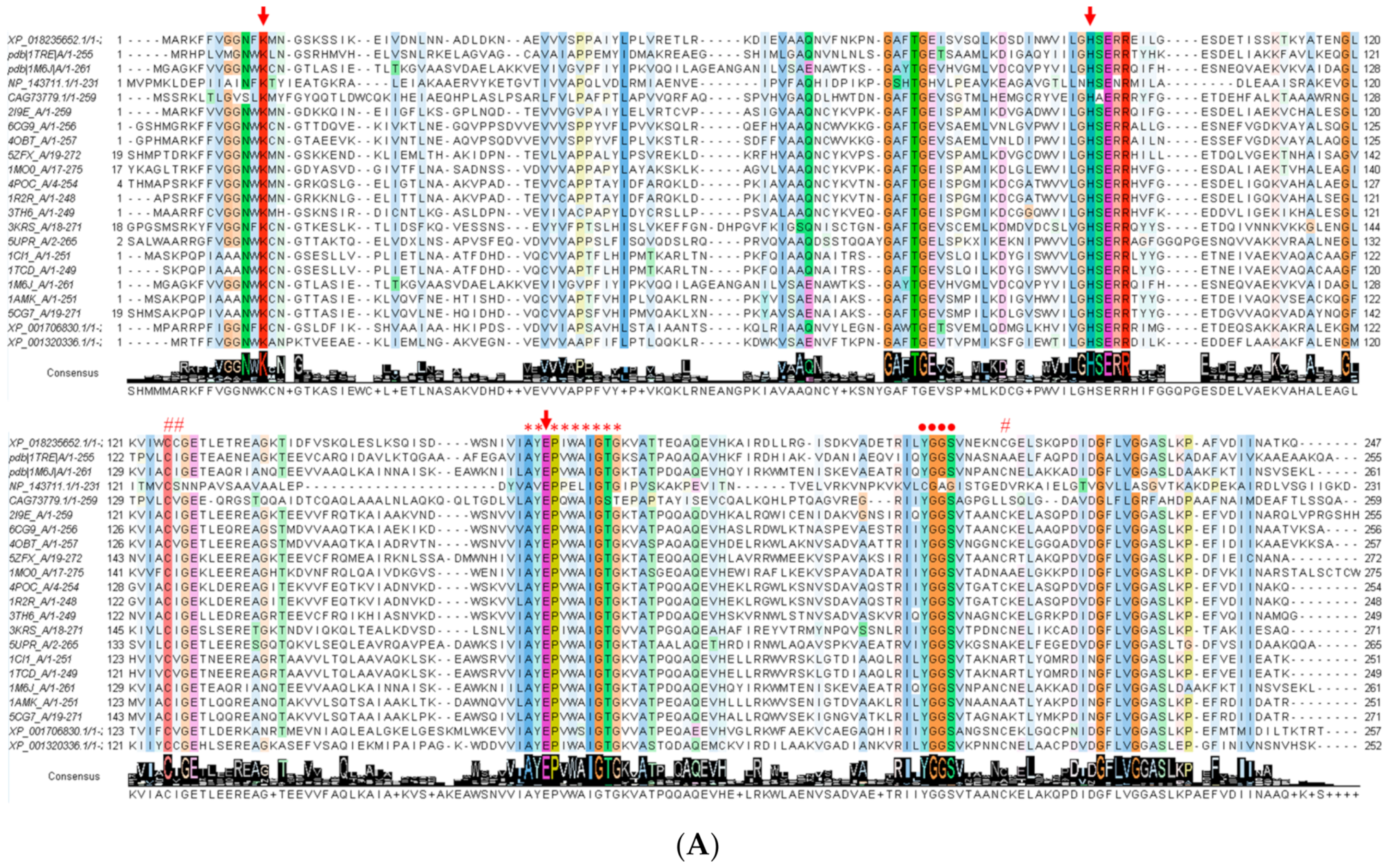
3.2. Alignment of the FoxTPI Protein
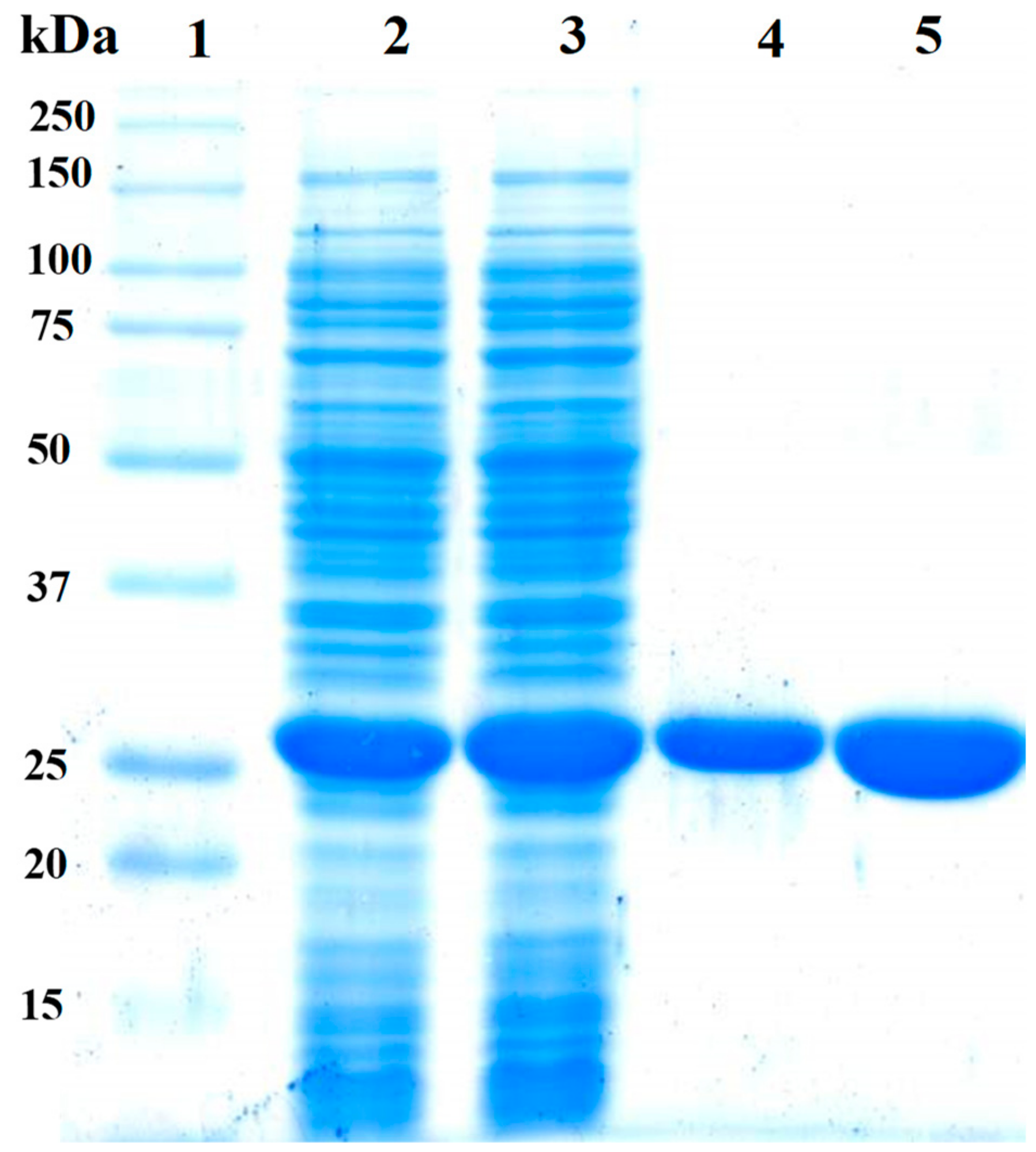
3.3. Expression and Purification of FoxTPI
3.4. Characterization of Functional Parameters of the FoxTPI Protein
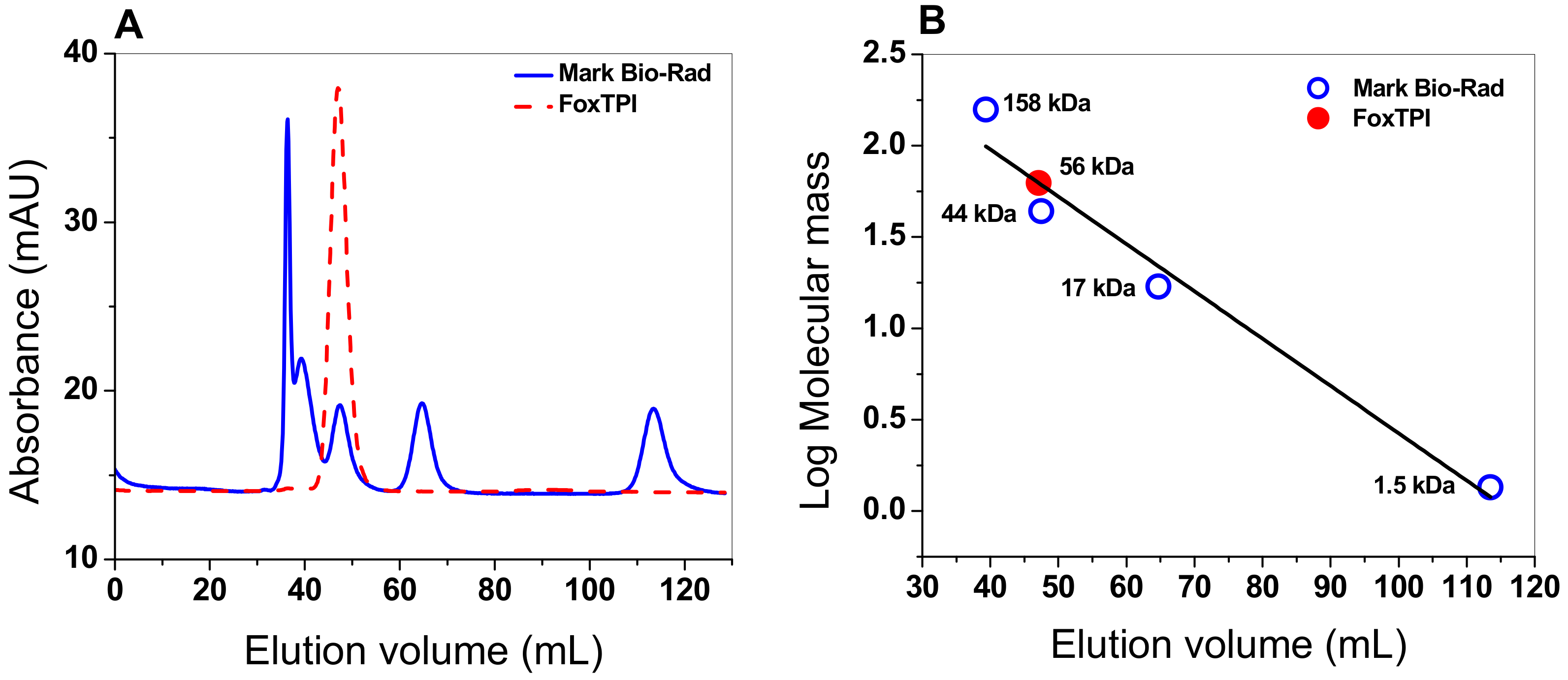
3.4.1. Oligomeric Status of the FoxTPI Protein
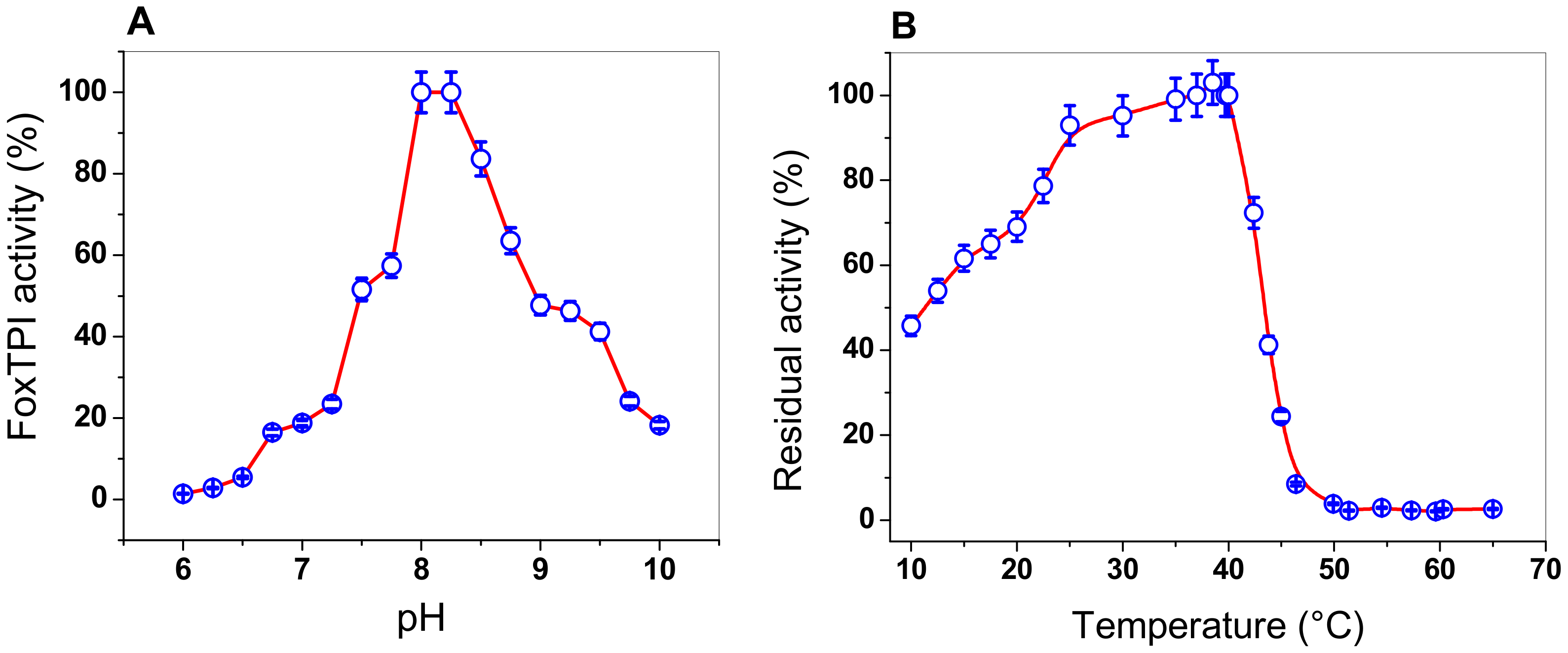
3.4.2. Effect of pH and Temperature on the FoxTPI Activity
3.4.3. Determination of Kinetic Parameters
3.5. Spectroscopic Characterization of the FoxTPI Protein
3.5.1. Circular Dichroism (CD) Analysis
3.5.2. Thermal Stability of the FoxTPI Protein
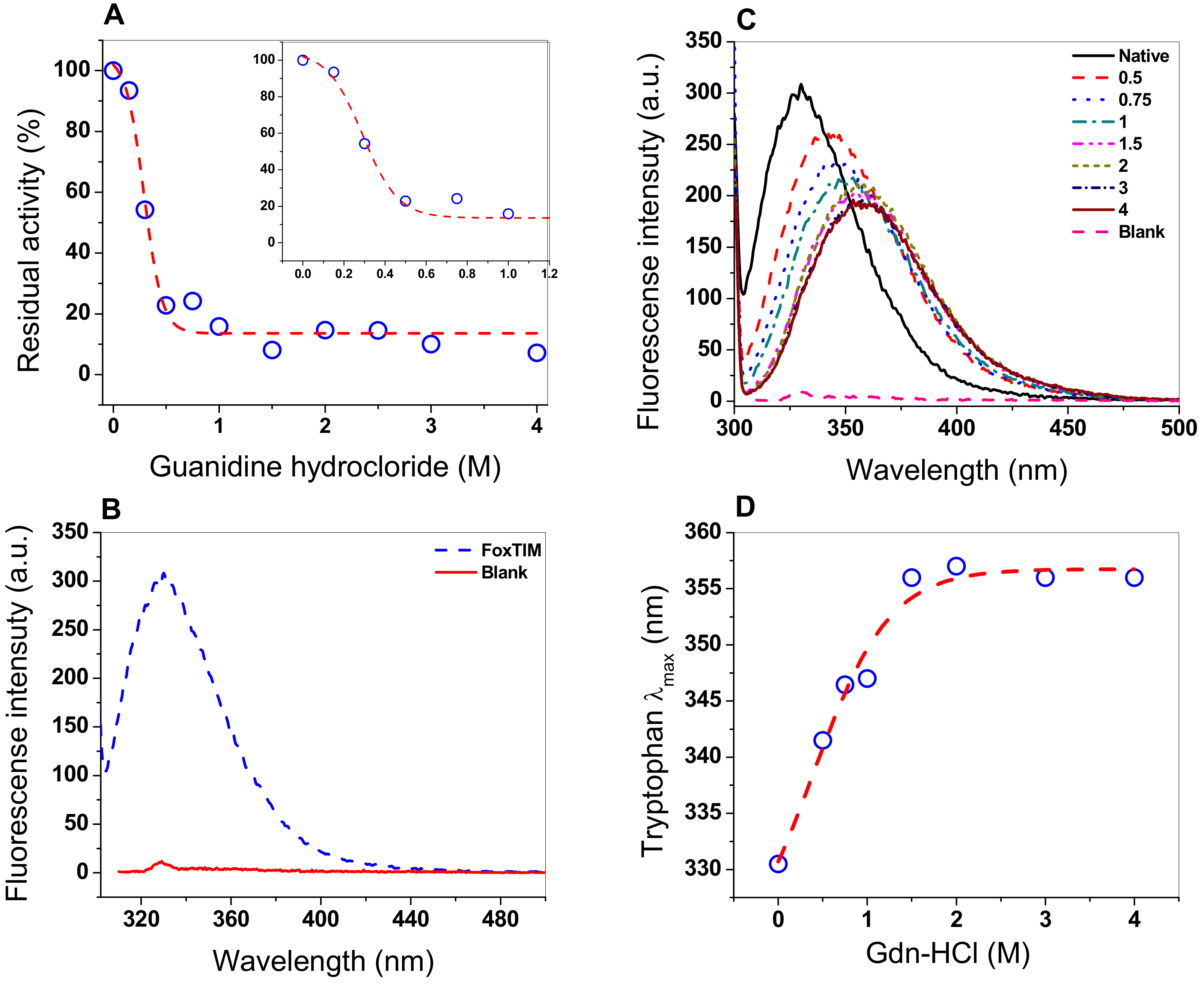
3.6. Assay Stability with Guanidine Hydrochloride (Gdn-HCl)
3.7. Structural Analysis through Intrinsic Fluorescence
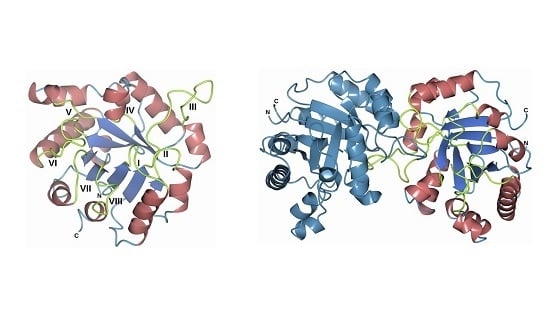
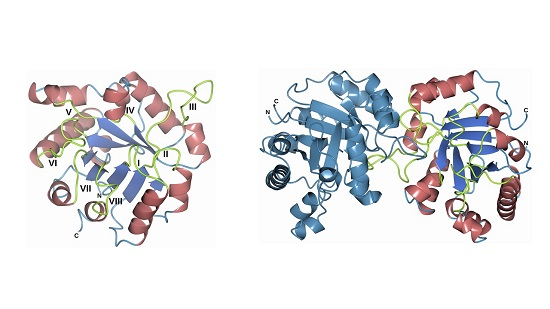
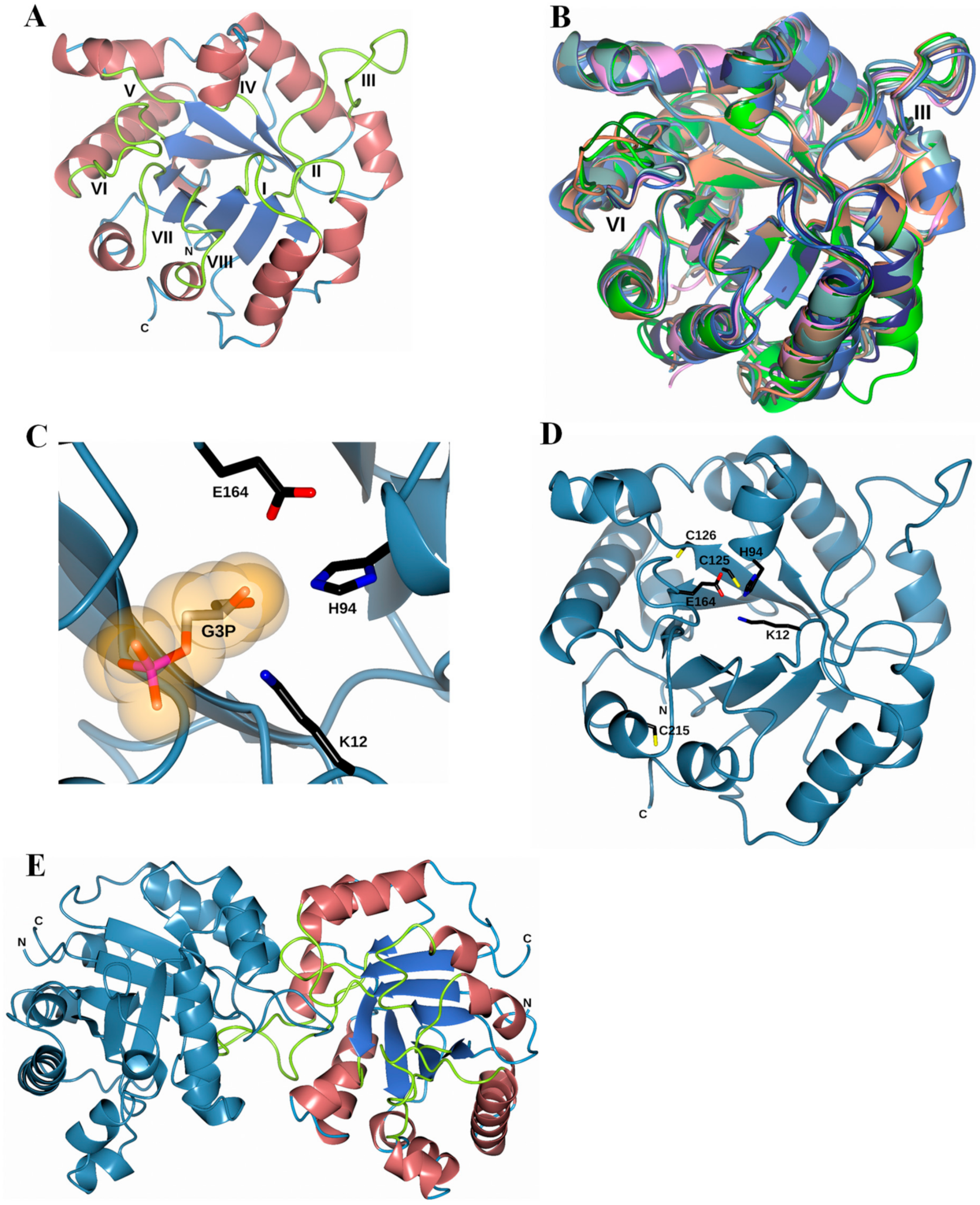
3.8. Homology Modeling of the FoxTPI Protein
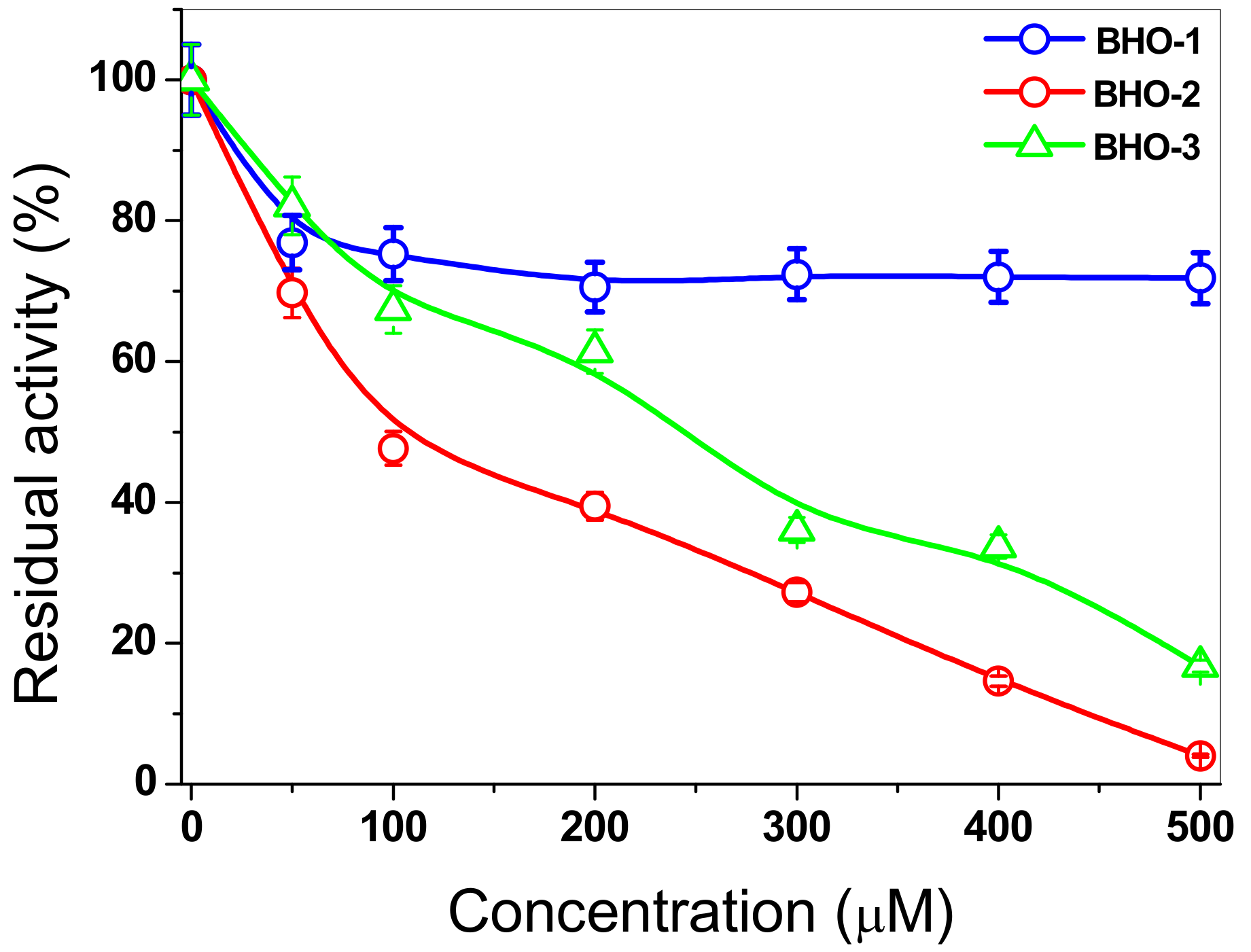
3.9. Inactivation of FoxTPI with Proton-Pump Inhibitor Derivatives
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sharma, L.; Marques, G. Fusarium, an Entomopathogen—A Myth or Reality? Pathogens 2018, 7, 93. [Google Scholar] [CrossRef]
- Laurence, M.H.; Walsh, J.L.; Shuttleworth, L.A.; Robinson, D.M.; Johansen, R.M.; Petrovic, T.; Vu, T.T.H.; Burgess, L.W.; Summerell, B.A.; Liew, E.C.Y. Six novel species of Fusarium from natural ecosystems in Australia. Fungal Divers. 2016, 77, 349–366. [Google Scholar] [CrossRef]
- Al-Hatmi, A.M.; Meis, J.F.; de Hoog, G.S. Fusarium: Molecular Diversity and Intrinsic Drug Resistance. PLoS Pathog. 2016, 12, e1005464. [Google Scholar] [CrossRef]
- Dean, R.; Van Kan, J.A.L.; Pretorius, Z.A.; Hammond-Kosack, K.E.; Di Pietro, A.; Spanu, P.D.; Rudd, J.J.; Dickman, M.; Kahmann, R.; Ellis, J.; et al. The top 10 fungal pathogens in molecular plant pathology. Mol. Plant. Pathol. 2012, 13, 414–430. [Google Scholar] [CrossRef]
- Buruchara, R.A.; Camacho, L. Common bean reaction to Fusarium oxysporum f. sp. phaseoli, the cause of severe vascular wilt in Central Africa. J. Phytopathol. 2000, 148, 39–45. [Google Scholar] [CrossRef]
- Antoniou, A.; Tsolakidou, M.D.; Stringlis, I.A.; Pantelides, I.S. Rhizosphere Microbiome Recruited from a Suppressive Compost Improves Plant Fitness and Increases Protection against Vascular Wilt Pathogens of Tomato. Front. Plant. Sci. 2017, 8, 2022. [Google Scholar] [CrossRef] [PubMed]
- Ortoneda, M.; Guarro, J.; Madrid, M.P.; Caracuel, Z.; Roncero, M.I.; Mayayo, E.; Di Pietro, A. Fusarium oxysporum as a Multihost Model for the Genetic Dissection of Fungal Virulence in Plants and Mammals. Infect. Immun. 2004, 72, 1760–1766. [Google Scholar] [CrossRef] [PubMed]
- Moussa, T.A.A.; Al-Zahrani, H.S.; Kadasa, N.M.S.; Ahmed, S.A.; de Hoog, G.S.; Al-Hatmi, A.M.S. Two new species of the Fusarium fujikuroi species complex isolated from the natural environment. Antonie van Leeuwenhoek 2017, 110, 819–832. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.E.; Dignani, M.C.; Anaissie, E.J. Taxonomy, biology, and clinical aspects of Fusarium species. Clin. Microbiol. Rev. 1994, 7, 479–504. [Google Scholar] [CrossRef]
- Nucci, M.; Anaissie, E. Fusarium Infections in Immunocompromised Patients. Clin. Microbiol. Rev. 2007, 20, 695–704. [Google Scholar] [CrossRef]
- Al-Hatmi, A.M.S.; Hagen, F.; Menken, S.B.J.; Meis, J.F.; de Hoog, G.S. Global molecular epidemiology and genetic diversity of Fusarium, a significant emerging group of human opportunists. Emerg. Microbes Infect. 2016b, 5, 1958–2015. [Google Scholar] [CrossRef]
- Wang, L.; Li, C.; Zhang, Y.; Qiao, C.; Ye, Y. Synthesis and biological evaluation of benzofuroxan derivatives as fungicides against phytopathogenic fungi. J. Agric. Food Chem. 2013, 61, 8632–8640. [Google Scholar] [CrossRef] [PubMed]
- Etzerodt, T.; Wetterhorn, K.; Dionisio, G.; Rayment, I. Functional characterization of a soluble NADPH-cytochrome P450 reductase from Fusarium graminearum. Protein Expr. Purif. 2017, 138, 69–75. [Google Scholar] [CrossRef]
- Lv, P.; Chen, Y.; Zhao, Z.; Shi, T.; Wu, X.; Xue, J.; Li, Q.X.; Hua, R. Design, Synthesis, and Antifungal Activities of 3-Acyl Thiotetronic Acid Derivatives: New Fatty Acid Synthase Inhibitors. J. Agric. Food. Chem. 2018, 66, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Hemphill, A. Drug target identification in protozoan parasites. Expert Opin. Drug Discov. 2016, 11, 815–824. [Google Scholar] [CrossRef]
- Liu, X.; Sun, J.; Wu, H. Glycolysis-related proteins are broad spectrum vaccine candidates against aquacultural pathogens. Vaccine 2017, 35, 3813–3816. [Google Scholar] [CrossRef]
- Enríquez, F.S.; Rodríuez, R.A.; Hernández, A.G.; Oria, H.J.; Gutiérrez, C.P.; Pérez, H.G.; de la Mora, D.L.M.I.; Castillo, V.A.; García, T.I.; Méndez, S.T.; et al. Determining the molecular mechanism of inactivation by chemical modification of triosephosphate isomerase from the human parasite Giardia lamblia: A study for antiparasitic drug design. Proteins 2011, 79, 2711–2724. [Google Scholar] [CrossRef]
- Hernández, O.B.; Navarrete, V.G.; Nava, Z.C.; Castillo, V.A.; Méndez, S.T.; Torres, A.A.; Gómez, M.S.; Marcial, Q.J.; Ponce, M.M.; Rufino-González, Y.; et al. Novel giardicidal compounds bearing proton pump inhibitor scaffold proceeding through triosephosphate isomerase inactivation. Sci. Rep. 2017, 7, 7810. [Google Scholar] [CrossRef]
- Wierenga, R.K.; Kapetaniou, E.G.; Venkatesan, R. Triosephosphate isomerase: A highly evolved biocatalyst. Cell. Mol. Life Sci. 2010, 67, 3961–3982. [Google Scholar] [CrossRef]
- Zinsser, V.L.; Farnell, E.; Dunne, D.W.; Timson, D.J. Triosephosphate isomerase from the blood fluke Schistosoma mansoni: Biochemical characterisation of a potential drug and vaccine target. FEBS Lett. 2013, 587, 3422–3427. [Google Scholar] [CrossRef]
- Kumar, K.; Bhargava, P.; Roy, U. Cloning, overexpression and characterization of Leishmania donovani triosephosphate isomerase. Exp. Parasitol. 2012, 130, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Vilgalys, R.; Hester, M. Rapid genetic identification and mapping of enzymatically amplified ribosomal DNA from several Cryptococcus species. J. Bacteriol. 1990, 172, 4239–4246. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Russel, D.W. Rapid Isolation of Yeast DNA. In Molecular Cloning, a Laboratory Manual; Sambrook, J., Russel, D.W., Eds.; Cold Spring Harbor Laboratory: New York, NY, USA, 2001; pp. 631–632. [Google Scholar]
- Stajich, J.E.; Harris, T.; Brunk, B.P.; Brestelli, J.; Fischer, S.; Harb, O.S.; Kissinger, J.C.; Li, W.; Nayak, V.; Pinney, D.F.; et al. FungiDB: An integrated functional genomics database for fungi. Nucleic Acids Res. 2012, 40, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2 a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef]
- Gómez, M.S.; Terrón, H.J.; de la Mora, M.I.; García, T.I.; López, V.G.; Reyes, V.H.; Oria, H.J. Cloning, expression, purification and characterization of his-tagged human glucose-6-phosphate dehydrogenase: A simplified method for protein yield. Protein J. 2013, 32, 585–592. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–6855. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar]
- Gómez, M.S.; Terrón, H.J.; De la Mora, D.I.; González, V.A.; Marcial, Q.J.; García, T.I.; Vanoye, C.A.; López, V.G.; Hernández, A.G.; Oria, H.J.; et al. The stability of G6PD is affected by mutations with different clinical phenotypes. Int. J. Mol. Sci. 2014, 15, 21179–21201. [Google Scholar] [CrossRef]
- Gómez, M.S.; Marcial, Q.J.; Vanoye, C.A.; Enríquez, F.S.; De la Mora, D.I.; González, V.A.; García, T.I.; Martínez, R.V.; Sierra, P.E.; Lazcano, P.F.; et al. Mutations of Glucose-6-Phosphate Dehydrogenase Durham, Santa-Maria and A+ Variants Are Associated with Loss Functional and Structural Stability of the Protein. Int. J. Mol. Sci. 2015, 16, 28657–28668. [Google Scholar] [CrossRef]
- Gómez, M.S.; Marcial, Q.J.; Vanoye, C.A.; Serrano, P.H.; González, V.A.; Martínez, R.V.; Hernández, O.B.; Sierra, P.E.; Castillo, R.R.A.; Cuevas, C.M.; et al. Functional and Biochemical Characterization of Three Recombinant Human Glucose-6-Phosphate Dehydrogenase Mutants: Zacatecas, Vanua-Lava and Viangchan. Int. J. Mol. Sci. 2016, 17, 787. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, N.E.J.; Ortega, C.D.; Serrano, P.H.; González, V.A.; Vanoye, C.A.; Hernández, O.B.; Sierra, P.E.; Hernández, P.J.; Rodríguez, B.E.; Arreguin, E.R.; et al. Biochemical Analysis of Two Single Mutants that Give Rise to a Polymorphic G6PD A-Double Mutant. Int. J. Mol. Sci. 2017, 18, 2244. [Google Scholar] [CrossRef] [PubMed]
- Cortés, M.Y.Y.; Vanoye, C.A.; Castillo, R.R.A.; Serrano, P.H.; González, V.A.; Ortega, C.D.; Hernández, O.B.; Moreno, V.L.M.; Prada, G.D.; Sierra, P.E.; et al. Cloning and biochemical characterization of three glucose-6-phosphate dehydrogenase mutants presents in the Mexican population. Int. J. Biol. Macromol. 2018, 119, 926–936. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Schulte-Sasse, M.; Pardo-Ávila, F.; Pulido-Mayoral, N.O.; Vázquez-Lobo, A.; Costas, M.; García-Hernández, E.; Rodríguez-Romero, A.; Fernández-Velasco, D.A. Structural, thermodynamic and catalytic characterization of an ancestral triosephosphate isomerase reveal early evolutionary coupling between monomer association and function. FEBS J. 2019, 286, 882–900. [Google Scholar] [CrossRef]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins 2009, 77, 114–122. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardsona, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, D66, 486–501. [Google Scholar] [CrossRef]
- Mukherjee, S.; Roychowdhury, A.; Dutta, D.; Das, A.K. Crystal structures of triosephosphate isomerase from methicillin resistant Staphylococcus aureus MRSA252 provide structural insights into novel modes of ligand binding and unique conformations of catalytic loop. Biochimie 2012, 94, 2532–2544. [Google Scholar] [CrossRef]
- McNicholas, S.; Potterton, E.; Wilson, K.S.; Nobleb, M.E.M. Presenting your structures: The CCP4mg molecular-graphics software. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 386–394. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the ExPASy server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- Laskowski, R.A.; Hutchinson, E.G.; Michie, A.D.; Wallace, A.C.; Jones, M.L.; Thornton, J.M. PDBsum: A Web-based database of summaries and analyses of all PDB structures. Trends Biochem. Sci. 1997, 22, 488–490. [Google Scholar] [CrossRef]
- González, M.E.; Zubillaga, R.A.; Saavedra, E.; Chánez, C.M.E.; Pérez, M.R.; Hernández, A.A. Conserved cysteine 126 in triosephosphate isomerase is required not for enzymatic activity but for proper folding and stability. Biochemistry 2004, 43, 3255–3263. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, R.A.; Hernández, S.A.; del Pozo, Y.L.; Kornhauser, A.; Fernández, V.D.A. Structure and inactivation of triosephosphate isomerase from Entamoeba histolytica. J. Mol. Biol. 2002, 322, 669–675. [Google Scholar] [CrossRef]
- Bourguignon, S.C.; Meirelles, M.N.; Pacheco, R.S.; De Simone, S.G. Purification and Partial characterization of Trypanosoma cruzi triosephosphate isomerase. Memórias do Instituto Oswaldo Cruz 1998, 93, 219–224. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ogungbe, I.V.; Setzer, W.N. Comparative Molecular Docking of Antitrypanosomal Natural Products into Multiple Trypanosoma brucei Drug Targets. Molecules 2009, 14, 1513–1536. [Google Scholar] [CrossRef] [PubMed]
- García, T.I.; De la Mora, D.L.M.I.; Hernández, A.G.; Molina, O.D.; Caballero, S.S.; Olivos, G.A.; Nava, G.; López, V.G.; Enríquez, F.S. First characterization of a microsporidial triosephosphate isomerase and the biochemical mechanisms of its inactivation to propose a new druggable target. Sci. Rep. 2018, 8, 8591. [Google Scholar] [CrossRef]
- Bao, S.; Chen, D.; Yu, S.; Chen, H.; Tan, L.; Hu, M.; Qiu, X.; Song, C.; Ding, C. Characterization of triosephosphate isomerase from Mycoplasma gallisepticum. FEMS Microbiol. Lett. 2015, 362, fnv140. [Google Scholar] [CrossRef][Green Version]
- Mora, I.D.L.M.D.L.; Torres, L.A.; Enríquez, F.S.; Méndez, S.T.; Castillo, V.A.; Gómez, M.S.; López, V.G.; Marcial, Q.J.; Torres, A.A.; García, T.I.; et al. Structural effects of protein aging: Terminal marking by deamidation in human triosephosphate isomerase. PLoS ONE 2015, 10, e0123379. [Google Scholar] [CrossRef]
- López, C.L.M.; Jiménez, S.P.; Baruch, T.N.; Trasviña, A.C.H.; Díaz, Q.C.; Lara, G.S.; Winkler, R.; Brieba, L.G. Structural Basis for Redox Regulation of Cytoplasmic and Chloroplastic Triosephosphate Isomerases from Arabidopsis thaliana. Front. Plant. Sci. 2016, 7, 1817. [Google Scholar] [CrossRef]
- Reyes, V.H.; Hernández, A.G.; López, V.G.; Cabrera, N.; Pérez, M.R.; de Gómez, P.M.T.; Gómez, P.A. Factors that control the reactivity of the interface cysteine of triosephosphate isomerase from Trypanosoma brucei and Trypanosoma cruzi. Biochemistry 2001, 40, 3134–3140. [Google Scholar] [CrossRef]
- Williams, J.C.; Zeelen, J.P.; Neubauer, G.; Vriend, G.; Backmann, J.; Michels, P.A.; Lambeir, A.M.; Wierenga, R.K. Structural and mutagenesis studies of leishmania triosephosphate isomerase: A point mutation can convert a mesophilic enzyme into a superstable enzyme without losing catalytic power. Protein Eng. 1999, 12, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, D.; Prakash, S.; Gupta, K.; Balaram, P. Mass spectrometric analysis of dimer-disrupting mutations in Plasmodium triosephosphate isomerase. Anal. Biochem. 2016, 500, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Lara, G.S.; Estrella, P.; Portillo, C.; Cruces, M.E.; Jimenez, S.P.; Fattori, J.; Migliorini, F.A.C.; Lopez, H.M.; Diaz, Q.C.; Lopez, C.M.; et al. Substrate-Induced Dimerization of Engineered Monomeric Variants of Triosephosphate Isomerase from Trichomonas vaginalis. PLoS ONE 2015, 10, e0141747. [Google Scholar] [CrossRef]
- Alvarez, M.; Zeelen, J.P.; Mainfroid, V.; Rentier, D.F.; Martial, J.A.; Wyns, L.; Wierenga, R.K.; Maes, D. Triose-phosphate isomerase (TIM) of the psychrophilic bacterium Vibrio marinus. Kinetic and structural properties. J. Biol. Chem. 1998, 273, 2199–2206. [Google Scholar] [CrossRef]
- Chu, C.H.; Lai, Y.J.; Huang, H.; Sun, Y.J. Kinetic and structural properties of triosephosphate isomerase from Helicobacter pylori. Proteins 2008, 71, 396–406. [Google Scholar] [CrossRef]
- Zhai, X.; Amyes, T.L.; Wierenga, R.K.; Loria, J.P.; Richard, J.P. Structural mutations that probe the interactions between the catalytic and dianion activation sites of triosephosphate isomerase. Biochemistry 2013, 52, 5928–5940. [Google Scholar] [CrossRef][Green Version]
- Landa, A.; Rojo, D.A.; Jiménez, L.; Fernández, V.D.A. Sequencing, expression and properties of triosephosphate isomerase from Entamoeba histolytica. Eur. J. Biochem. 1997, 247, 348–355. [Google Scholar] [CrossRef]
- Son, J.; Kim, S.; Kim, S.E.; Lee, H.; Lee, M.R.; Hwang, K.Y. Structural Analysis of an Epitope Candidate of Triosephosphate Isomerase in Opisthorchis viverrini. Sci. Rep. 2018, 10, 15075. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism | kcat (GAP) (min−1) | Km (GAP) (mM) | kcat/Km (GAP) (min−1 mM−1) | Reference |
|---|---|---|---|---|
| Fusarium oxysporum | 2.9 × 105 | 0.47 | 6.0 × 105 | [This study] |
| Encephalitozoon intestinalis | 1.0 × 105 | 0.83 | 2.0 × 106 | [48] |
| Saccharomyces cerevisiae | 2.8 × 105 | 1.10 | 4.3 × 106 | [44] |
| Homo sapiens | 1.8 × 105 | 0.74 | 4.2 × 106 | [50] |
| Giardia lamblia | 4.6 × 105 | 0.78 | 9.8 × 106 | [17] |
| Arabidopsis thaliana | 1.5 × 105 | 0.48 | 5.3 × 106 | [51] |
| Trypanozoma cruzi | 2.7 × 105 | 0.43 | 1.0 × 107 | [52] |
| Leishmania mexicana | 2.5 × 105 | 0.41 | 1.0 × 107 | [53] |
| Plasmodium falciparum | 2.5 × 105 | 0.35 | 7.1 × 105 | [54] |
| Trichomonas vaginalis | 7.9 × 104 | 0.23 | 3.5 × 105 | [55] |
| Vibrio marinus | 4.2 × 105 | 1.9 | 2.2 × 105 | [56] |
| Helicobacter pilory | 8.8 × 104 | 3.5 | 2.6 × 104 | [57] |
| Chicken muscle | 1.9 × 105 | 0.29 | 1.1 × 107 | [58] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández-Ochoa, B.; Gómez-Manzo, S.; Alcaraz-Carmona, E.; Serrano-Posada, H.; Centeno-Leija, S.; Arreguin-Espinosa, R.; Cuevas-Cruz, M.; González-Valdez, A.; Mendoza-Espinoza, J.A.; Acosta Ramos, M.; et al. Gene Cloning, Recombinant Expression, Characterization, and Molecular Modeling of the Glycolytic Enzyme Triosephosphate Isomerase from Fusarium oxysporum. Microorganisms 2020, 8, 40. https://doi.org/10.3390/microorganisms8010040
Hernández-Ochoa B, Gómez-Manzo S, Alcaraz-Carmona E, Serrano-Posada H, Centeno-Leija S, Arreguin-Espinosa R, Cuevas-Cruz M, González-Valdez A, Mendoza-Espinoza JA, Acosta Ramos M, et al. Gene Cloning, Recombinant Expression, Characterization, and Molecular Modeling of the Glycolytic Enzyme Triosephosphate Isomerase from Fusarium oxysporum. Microorganisms. 2020; 8(1):40. https://doi.org/10.3390/microorganisms8010040
Chicago/Turabian StyleHernández-Ochoa, Beatriz, Saúl Gómez-Manzo, Erick Alcaraz-Carmona, Hugo Serrano-Posada, Sara Centeno-Leija, Roberto Arreguin-Espinosa, Miguel Cuevas-Cruz, Abigail González-Valdez, José Alberto Mendoza-Espinoza, Marcelo Acosta Ramos, and et al. 2020. "Gene Cloning, Recombinant Expression, Characterization, and Molecular Modeling of the Glycolytic Enzyme Triosephosphate Isomerase from Fusarium oxysporum" Microorganisms 8, no. 1: 40. https://doi.org/10.3390/microorganisms8010040
APA StyleHernández-Ochoa, B., Gómez-Manzo, S., Alcaraz-Carmona, E., Serrano-Posada, H., Centeno-Leija, S., Arreguin-Espinosa, R., Cuevas-Cruz, M., González-Valdez, A., Mendoza-Espinoza, J. A., Acosta Ramos, M., Cortés-Maldonado, L., Montiel-González, A. M., Pérez de la Cruz, V., Rocha-Ramírez, L. M., Marcial-Quino, J., & Sierra-Palacios, E. (2020). Gene Cloning, Recombinant Expression, Characterization, and Molecular Modeling of the Glycolytic Enzyme Triosephosphate Isomerase from Fusarium oxysporum. Microorganisms, 8(1), 40. https://doi.org/10.3390/microorganisms8010040