Tomato RNA-seq Data Mining Reveals the Taxonomic and Functional Diversity of Root-Associated Microbiota

 ,
,  , , ,
, , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material and Sequencing
2.2. Plant Transcriptome Analysis
2.3. Meta-Transcriptome Filtering and Annotation
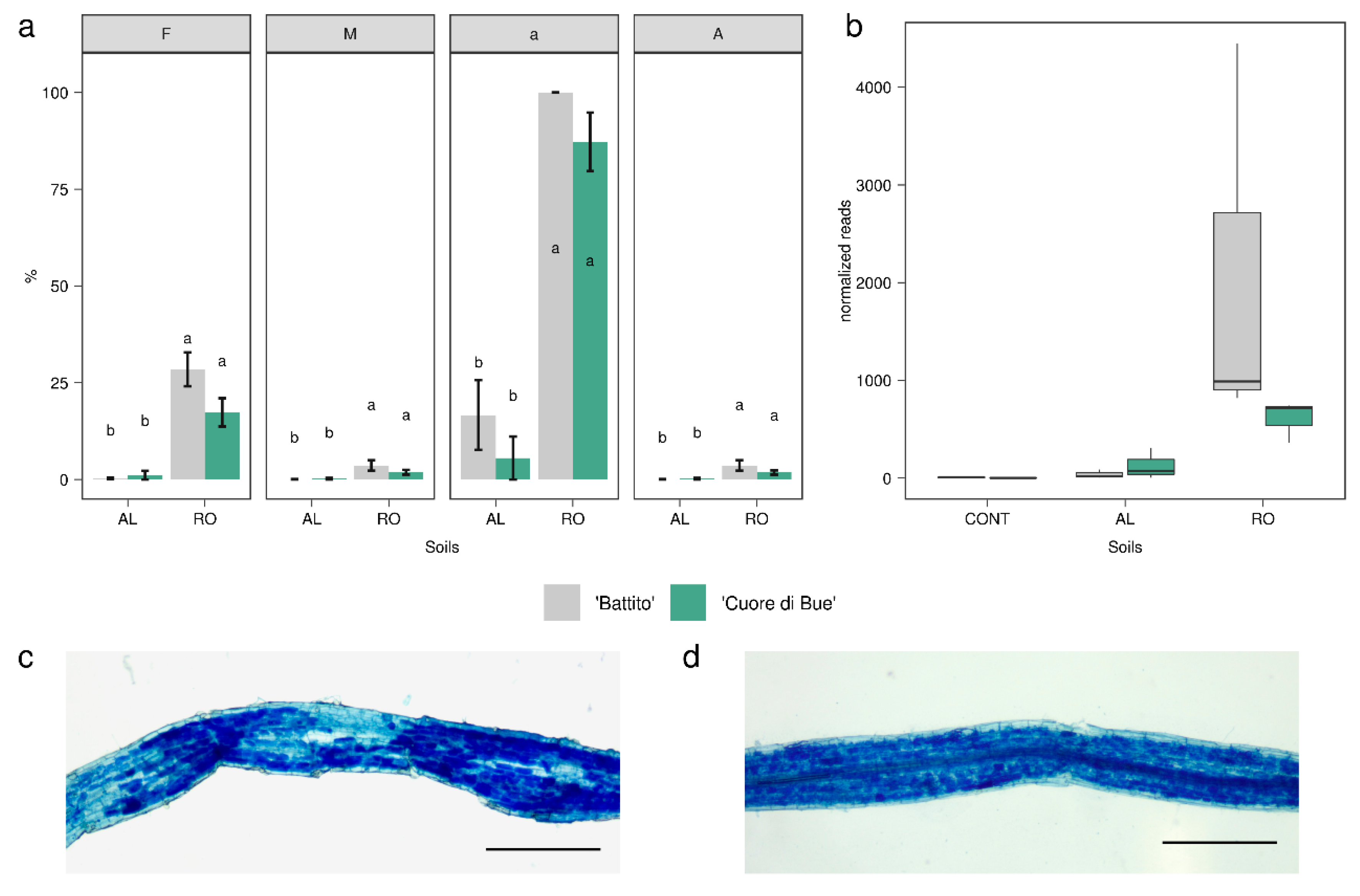
2.4. Analysis of Arbuscular Mycorrhizal Colonization
2.5. Statistical Analysis
3. Results and Discussion
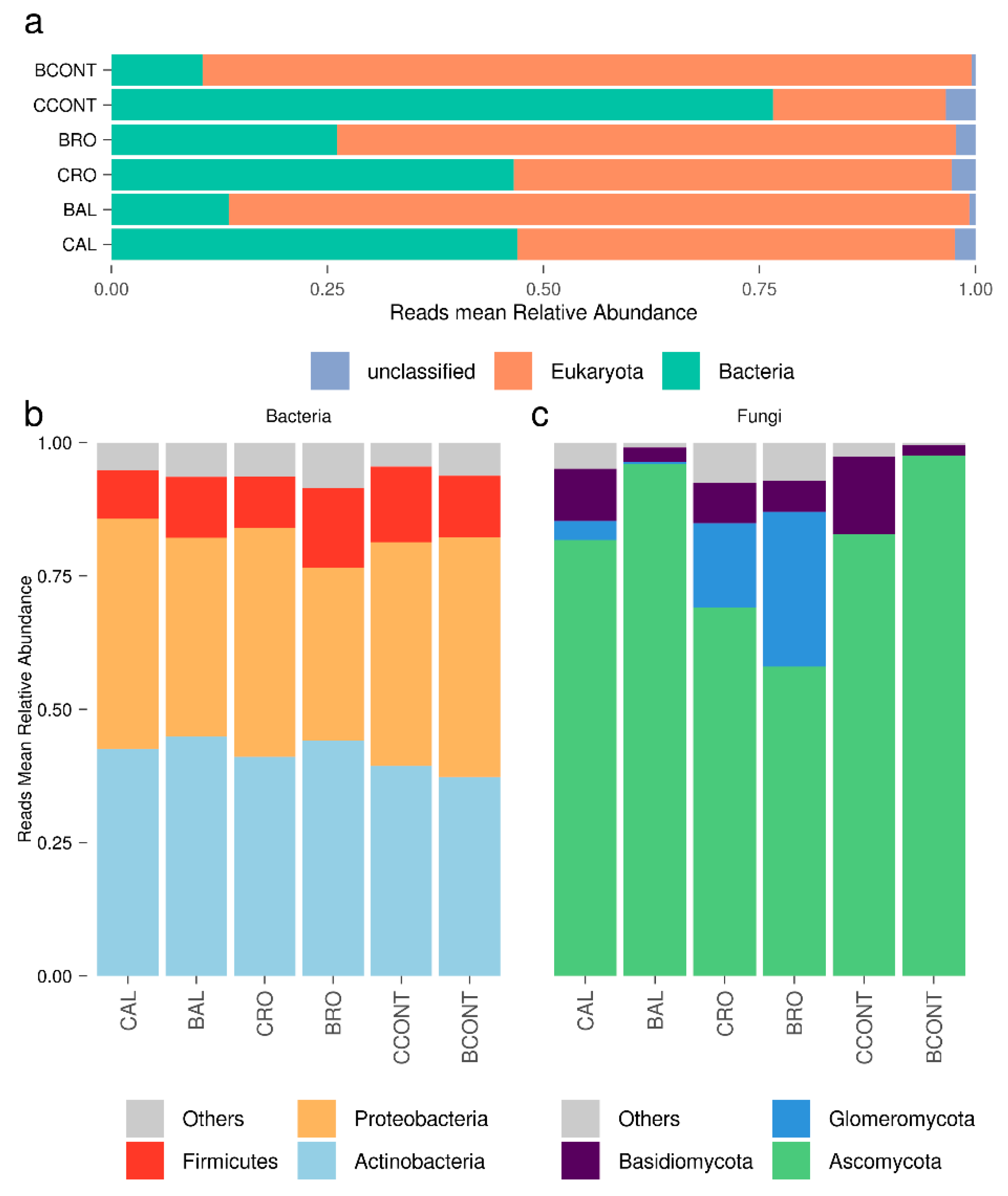
3.1. Reconstructing the Root-Associated Meta-Transcriptome from Host-Targeted RNA-seq Libraries
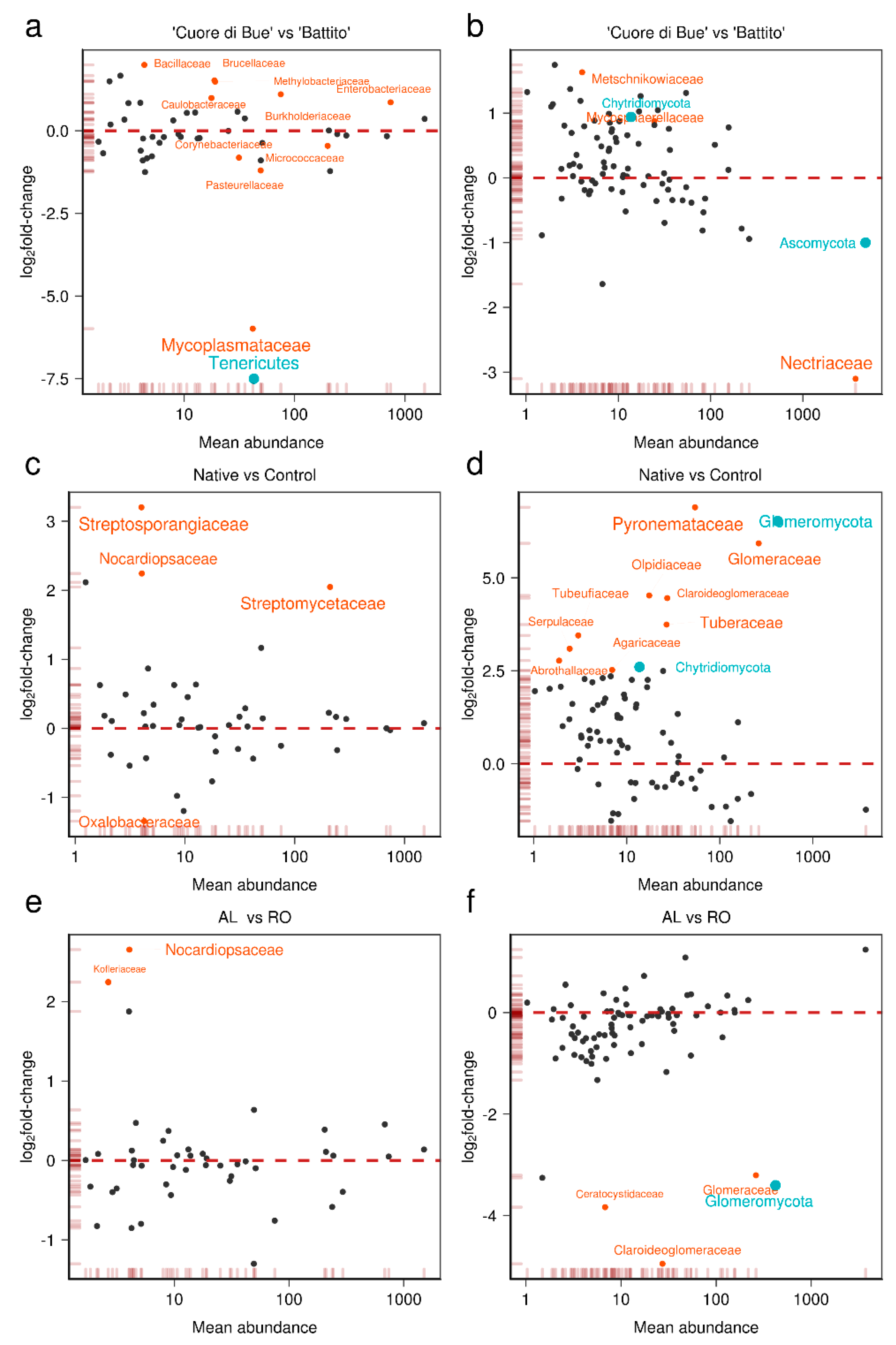
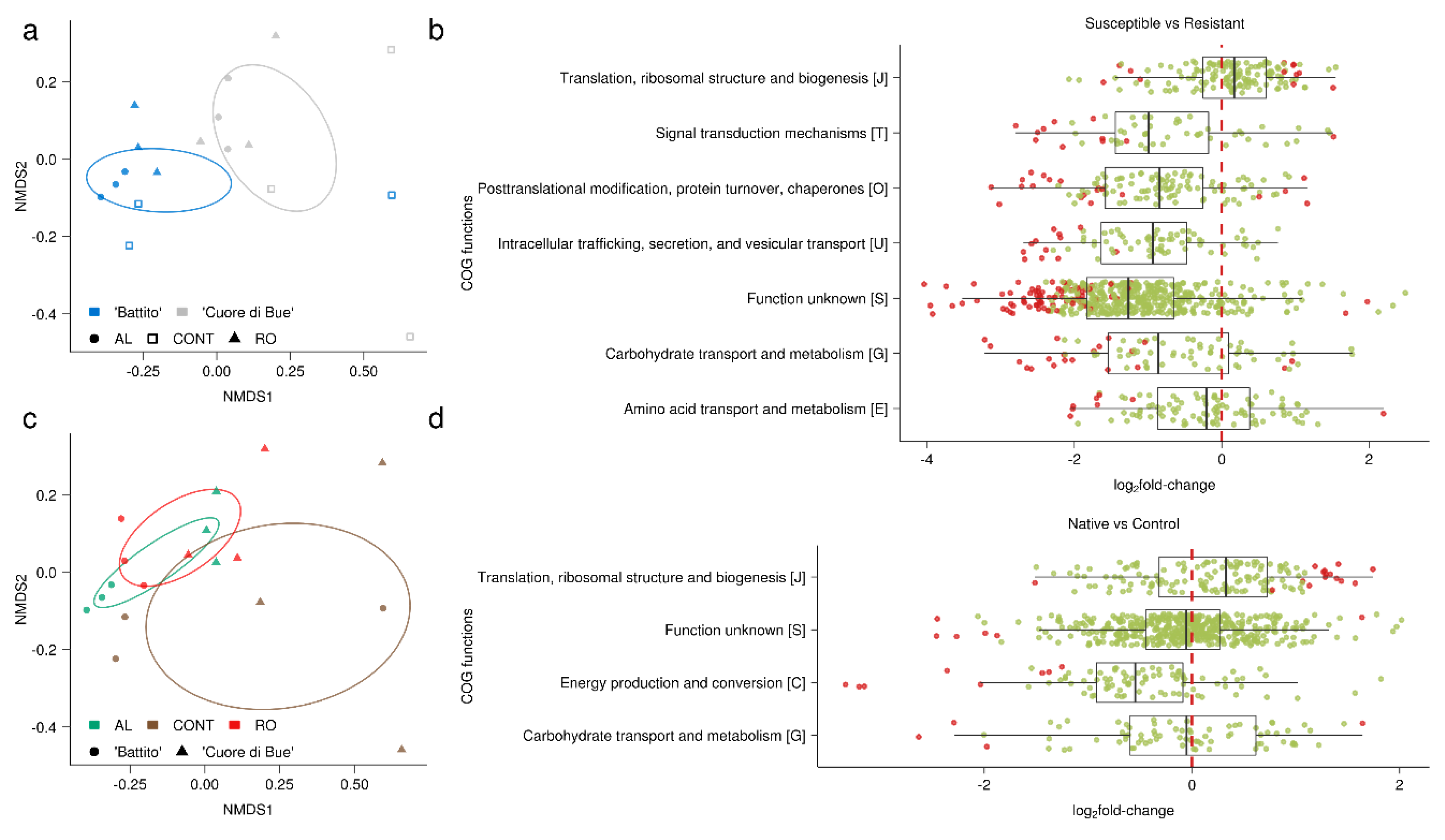
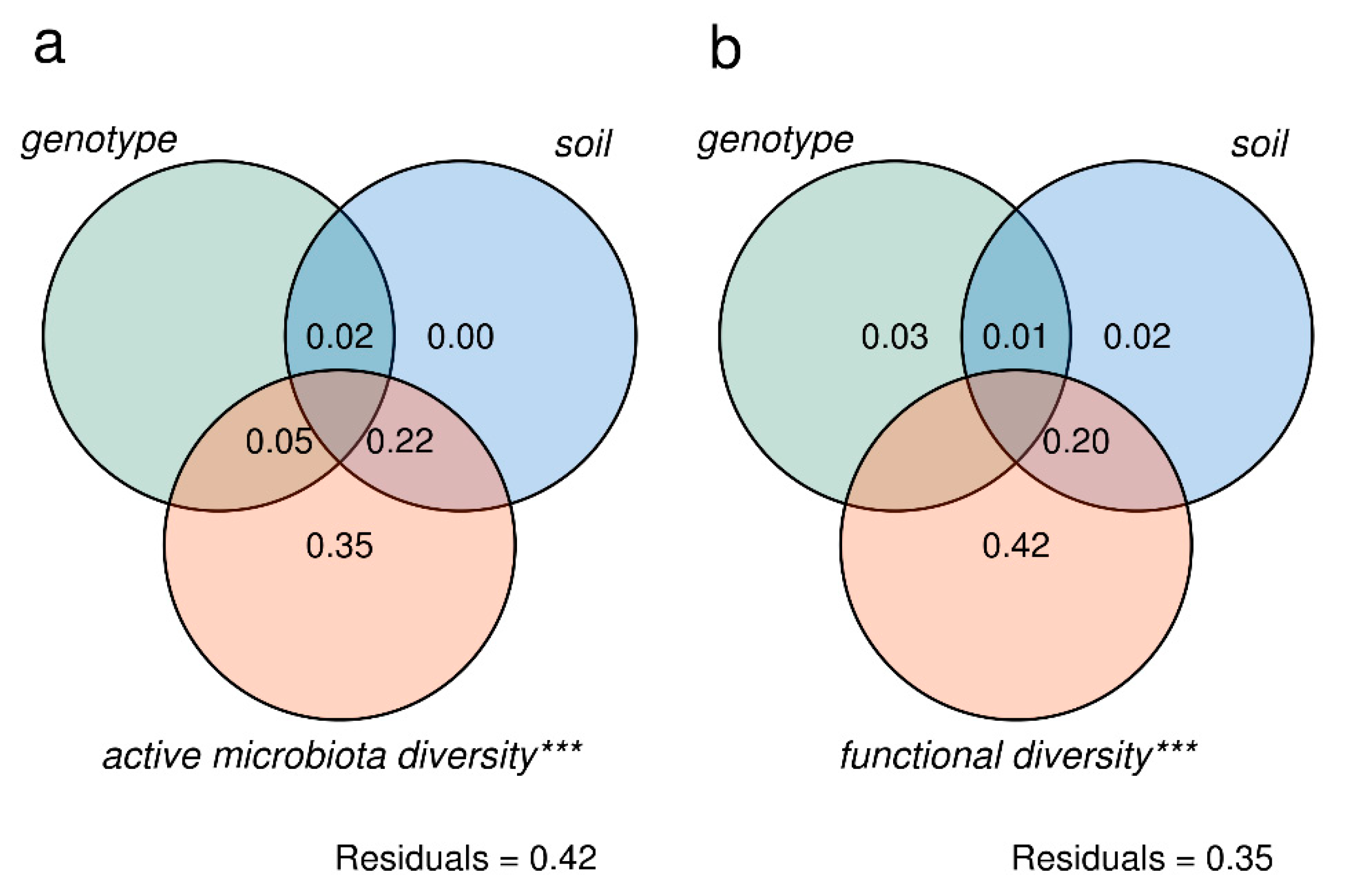
3.2. Tomato Root-Associated Active Microbiota Diversity Is Shaped by Both Soil Type and Host Genotype
3.3. Basal Microbial Metabolisms Are Detected in the Reconstructed Meta-Transcriptome
3.4. Linking the Meta-Transcriptome with the Host Transcriptome
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cowan, D.A.; Ramond, J.-B.; Makhalanyane, T.P.; De Maayer, P. Metagenomics of extreme environments. Curr. Opin. Microbiol. 2015, 25, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.-L.; Gottschick, C.; Bhuju, S.; Masur, C.; Abels, C.; Wagner-Döbler, I. Metatranscriptome Analysis of the Vaginal Microbiota Reveals Potential Mechanisms for Protection against Metronidazole in Bacterial Vaginosis. Msphere 2018, 3, e00262-18. [Google Scholar] [CrossRef] [PubMed]
- Crump, B.C.; Wojahn, J.M.; Tomas, F.; Mueller, R.S. Metatranscriptomics and Amplicon Sequencing Reveal Mutualisms in Seagrass Microbiomes. Front. Microbiol. 2018, 9, 388. [Google Scholar] [CrossRef] [PubMed]
- Saminathan, T.; García, M.; Ghimire, B.; Lopez, C.; Bodunrin, A.; Nimmakayala, P.; Abburi, V.L.; Levi, A.; Balagurusamy, N.; Reddy, U.K. Metagenomic and Metatranscriptomic Analyses of Diverse Watermelon Cultivars Reveal the Role of Fruit Associated Microbiome in Carbohydrate Metabolism and Ripening of Mature Fruits. Front. Plant Sci. 2018, 9, 4. [Google Scholar] [CrossRef]
- Cox, J.W.; Ballweg, R.A.; Taft, D.H.; Velayutham, P.; Haslam, D.B.; Porollo, A. A fast and robust protocol for metataxonomic analysis using RNAseq data. Microbiome 2017, 5, 7. [Google Scholar] [CrossRef]
- Mateus, I.D.; Masclaux, F.G.; Aletti, C.; Rojas, E.C.; Savary, R.; Dupuis, C.; Sanders, I.R. Dual RNA-seq reveals large-scale non-conserved genotype × genotype-specific genetic reprograming and molecular crosstalk in the mycorrhizal symbiosis. ISME J. 2019, 13, 1226–1238. [Google Scholar] [CrossRef]
- Kovalchuk, A.; Zeng, Z.; Ghimire, R.P.; Kivimäenpää, M.; Raffaello, T.; Liu, M.; Mukrimin, M.; Kasanen, R.; Sun, H.; Julkunen-Tiitto, R.; et al. Dual RNA-seq analysis provides new insights into interactions between Norway spruce and necrotrophic pathogen Heterobasidion annosum s.l. BMC Plant Biol. 2019, 19, 2. [Google Scholar] [CrossRef]
- Young, E.; Carey, M.; Meharg, A.A.; Meharg, C. Microbiome and ecotypic adaption of Holcus lanatus (L.) to extremes of its soil pH range, investigated through transcriptome sequencing. Microbiome 2018, 6, 48. [Google Scholar] [CrossRef]
- Celaj, A.; Markle, J.; Danska, J.; Parkinson, J. Comparison of assembly algorithms for improving rate of metatranscriptomic functional annotation. Microbiome 2014, 2, 39. [Google Scholar] [CrossRef]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef]
- Chialva, M.; Salvioli di Fossalunga, A.; Daghino, S.; Ghignone, S.; Bagnaresi, P.; Chiapello, M.; Novero, M.; Spadaro, D.; Perotto, S.; Bonfante, P. Native soils with their microbiotas elicit a state of alert in tomato plants. New Phytol. 2018, 220, 1296–1308. [Google Scholar] [CrossRef]
- Chialva, M.; Zhou, Y.; Spadaro, D.; Bonfante, P. Not only priming: Soil microbiota may protect tomato from root pathogens. Plant Signal. Behav. 2018, 13, e1464855. [Google Scholar] [CrossRef] [PubMed]
- Poli, A.; Lazzari, A.; Prigione, V.; Voyron, S.; Spadaro, D.; Varese, G.C. Influence of plant genotype on the cultivable fungi associated to tomato rhizosphere and roots in different soils. Fungal Biol. 2016, 120, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Voyron, S.; Ercole, E.; Ghignone, S.; Poli, A.; Spadaro, D.; Bonfante, P.; Girlanda, M. Plant Genotype Control over the Recruitment of the Tomato Fungal Microbiota. In Proceedings of the Ecology of Soil Microorganisms, Prague, Czech Republic, 29 November–3 December 2015; p. 175. [Google Scholar]
- Leinonen, R.; Sugawara, H.; Shumway, M. International Nucleotide Sequence Database Collaboration The Sequence Read Archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Tabata, S.; Hirakawa, H.; Asamizu, E.; Shirasawa, K.; Isobe, S.; Kaneko, T.; Nakamura, Y.; Shibata, D.; Aoki, K.; et al. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 2012, 485, 635–641. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Kopylova, E.; Noe, L.; Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef]
- Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018, 46, D8–D13. [CrossRef] [PubMed]
- Pongor, L.S.; Vera, R.; Ligeti, B. Fast and Sensitive Alignment of Microbial Whole Genome Sequencing Reads to Large Sequence Datasets on a Desktop PC: Application to Metagenomic Datasets and Pathogen Identification. PLoS ONE 2014, 9, e103441. [Google Scholar] [CrossRef] [PubMed]
- Dick, J.M. Calculation of the relative metastabilities of proteins using the CHNOSZ software package. Geochem. Trans. 2008, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Trouvelot, A.; Kough, J.; Gianinazzi-Pearson, V. Mesure du taux de mycorhization VA d’un système radiculaire. Recherche de méthodes d’estimation ayant une signification fonctionnelle. In Physiological and Genetical Aspects of Mycorrhizae, Proceedings of the 1st European Symposium on Mycorrhizae, Dijon, France, 1–5 July 1985; Gianinazzi-Pearson, V., Gianinazzi, S., Eds.; Institut National de la Recherche Agronomique: Paris, France, 1986; pp. 217–221. [Google Scholar]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. R package Version 2.5-6. 2019. Available online: https://CRAN.R-project.org/package=vegan (accessed on 23 December 2019).
- McMurdie, P.J.; Holmes, S. Waste Not, Want Not: Why Rarefying Microbiome Data Is Inadmissible. PLoS Comput. Biol. 2014, 10, e1003531. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Fox, J.; Weisberg, S. An R Companion to Applied Regression, 3rd eds.; Sage: Thousand Oaks, CA, USA, 2019. [Google Scholar]
- Levene, H. Robust tests for equality of variances. In Contributions to Probability and Statistics: Essays in Honor of Harold Hotelling; Olkin, I., Ed.; Stanford University Press: Palo Alto, CA, USA, 1960; pp. 278–292. [Google Scholar]
- Kruskal, W.H. A Nonparametric test for the Several Sample Problem. Ann. Math. Stat. 1952, 23, 525–540. [Google Scholar] [CrossRef]
- Tukey, J.W. Comparing Individual Means in the Analysis of Variance. Biometrics 1949, 5, 99–114. [Google Scholar] [CrossRef]
- De Mendiburu, F. Agricolae: Statistical Procedures for Agricultural Research. R Package Version 1.3-1. 2019. Available online: https://CRAN.R-project.org/package=agricolae (accessed on 23 December 2019).
- Dunn, O.J. Multiple Comparisons Using Rank Sums. Technometrics 1964, 6, 241–252. [Google Scholar] [CrossRef]
- Ogle, D.H.; Wheeler, P.; Dinno, A. FSA: Fisheries Stock Analysis. R Package Version 0.8.26. 2019. Available online: https://github.com/droglenc/FSA (accessed on 23 December 2019).
- Derksen, S.; Keselman, H.J. Backward, forward and stepwise automated subset selection algorithms: Frequency of obtaining authentic and noise variables. Br. J. Math. Stat. Psychol. 1992, 45, 265–282. [Google Scholar] [CrossRef]
- Dray, S.; Blanchet, G.; Borcard, D.; Guenard, G.; Jombart, T.; Larocque, G.; Legendre, P.; Madi, N.; Wagner, H.H. Adespatial: Multivariate Multiscale Spatial Analysis. R package Version 0.3-7. 2019. Available online: https://CRAN.R-project.org/package=adespatial (accessed on 23 December 2019).
- Xiong, X.; Frank, D.N.; Robertson, C.E.; Hung, S.S.; Markle, J.; Canty, A.J.; McCoy, K.D.; Macpherson, A.J.; Poussier, P.; Danska, J.S.; et al. Generation and Analysis of a Mouse Intestinal Metatranscriptome through Illumina Based RNA-Sequencing. PLoS ONE 2012, 7, e36009. [Google Scholar] [CrossRef] [PubMed]
- Westreich, S.T.; Korf, I.; Mills, D.A.; Lemay, D.G. SAMSA: A comprehensive metatranscriptome analysis pipeline. BMC Bioinform. 2016, 17, 399. [Google Scholar] [CrossRef] [PubMed]
- Danielsen, L.; Thürmer, A.; Meinicke, P.; Buée, M.; Morin, E.; Martin, F.; Pilate, G.; Daniel, R.; Polle, A.; Reich, M. Fungal soil communities in a young transgenic poplar plantation form a rich reservoir for fungal root communities. Ecol. Evol. 2012, 2, 1935–1948. [Google Scholar] [CrossRef]
- Palaniyandi, S.A.; Yang, S.H.; Zhang, L.; Suh, J.-W. Effects of actinobacteria on plant disease suppression and growth promotion. Appl. Microbiol. Biotechnol. 2013, 97, 9621–9636. [Google Scholar] [CrossRef]
- Poomthongdee, N.; Duangmal, K.; Pathom-aree, W. Acidophilic actinomycetes from rhizosphere soil: Diversity and properties beneficial to plants. J. Antibiot. 2015, 68, 106–114. [Google Scholar] [CrossRef]
- Detheridge, A.P.; Brand, G.; Fychan, R.; Crotty, F.V.; Sanderson, R.; Griffith, G.W.; Marley, C.L. The legacy effect of cover crops on soil fungal populations in a cereal rotation. Agric. Ecosyst. Environ. 2016, 228, 49–61. [Google Scholar] [CrossRef]
- Bulgarelli, D.; Rott, M.; Schlaeppi, K.; Ver Loren van Themaat, E.; Ahmadinejad, N.; Assenza, F.; Rauf, P.; Huettel, B.; Reinhardt, R.; Schmelzer, E.; et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 2012, 488, 91–95. [Google Scholar] [CrossRef]
- Lundberg, D.S.; Lebeis, S.L.; Paredes, S.H.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; Del Rio, T.G.; et al. Defining the core Arabidopsis thaliana root microbiome. Nature 2012, 488, 86–90. [Google Scholar] [CrossRef]
- Alabouvette, C.; Olivain, C.; L–Haridon, F.; Aimé, S.; Steinberg, C. Using strains of Fusarium oxysporum to control Fusarium wilts: Dream or reality? In Novel Biotechnologies for Biocontrol Agent Enhancement and Management; Vurro, M., Gressel, J., Eds.; NATO Security through Science Series; Springer: Dordrecht, The Netherlands, 2007; pp. 157–177. ISBN 978-1-4020-5799-1. [Google Scholar]
- Green, S.J.; Inbar, E.; Michel, F.C.; Hadar, Y.; Minz, D. Succession of Bacterial Communities during Early Plant Development: Transition from Seed to Root and Effect of Compost Amendment. Appl. Environ. Microbiol. 2006, 72, 3975–3983. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mendes, R.; Kruijt, M.; De Bruijn, I.; Dekkers, E.; Van der Voort, M.; Schneider, J.H.M.; Piceno, Y.M.; DeSantis, T.Z.; Andersen, G.L.; Bakker, P.A.H.M.; et al. Deciphering the Rhizosphere Microbiome for Disease-Suppressive Bacteria. Science 2011, 332, 1097–1100. [Google Scholar] [CrossRef] [PubMed]
- Rosendahl, S. Communities, populations and individuals of arbuscular mycorrhizal fungi. New Phytol. 2008, 178, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Öpik, M.; Moora, M.; Liira, J.; Zobel, M. Composition of root-colonizing arbuscular mycorrhizal fungal communities in different ecosystems around the globe. J. Ecol. 2006, 94, 778–790. [Google Scholar] [CrossRef]
- Teixeira, P.J.P.; Colaianni, N.R.; Fitzpatrick, C.R.; Dangl, J.L. Beyond pathogens: Microbiota interactions with the plant immune system. Curr. Opin. Microbiol. 2019, 49, 7–17. [Google Scholar] [CrossRef]
- Del Giorgio, P.A.; Cole, J.J. Bacterial Growth Efficiency in Natural Aquatic Systems. Annu. Rev. Ecol. Syst. 1998, 29, 503–541. [Google Scholar] [CrossRef]
- Leimena, M.M.; Ramiro-Garcia, J.; Davids, M.; Van den Bogert, B.; Smidt, H.; Smid, E.J.; Boekhorst, J.; Zoetendal, E.G.; Schaap, P.J.; Kleerebezem, M. A comprehensive metatranscriptome analysis pipeline and its validation using human small intestine microbiota datasets. BMC Genom. 2013, 14, 530. [Google Scholar] [CrossRef]
- Liao, W.; Ren, J.; Wang, K.; Wang, S.; Zeng, F.; Wang, Y.; Sun, F. Alignment-free Transcriptomic and Metatranscriptomic Comparison Using Sequencing Signatures with Variable Length Markov Chains. Sci. Rep. 2016, 6, 1–15. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Y.-X.; Zhang, N.; Hu, B.; Jin, T.; Xu, H.; Qin, Y.; Yan, P.; Zhang, X.; Guo, X.; et al. NRT1.1B is associated with root microbiota composition and nitrogen use in field-grown rice. Nat. Biotechnol. 2019, 37, 676–684. [Google Scholar] [CrossRef]
- Zgadzaj, R.; Garrido-Oter, R.; Jensen, D.B.; Koprivova, A.; Schulze-Lefert, P.; Radutoiu, S. Root nodule symbiosis in Lotus japonicus drives the establishment of distinctive rhizosphere, root, and nodule bacterial communities. Proc. Natl. Acad. Sci. USA 2016, 113, E7996–E8005. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SRA 1 Run | SRA 1 Sample Accession | Raw | Filtered | Unique Filtered Reads (%) | Mapped on NCBI (%) | Bacteria | Eukaryotes | Mapped % (eggNOG) |
|---|---|---|---|---|---|---|---|---|---|
| BAL_R1 | SRR6368019 | SRS2751529 | 11,382,297 | 184,182 | 88.88 | 27.12 | 4952 | 16,074 | 33.41 |
| BAL_R2 | SRR6368032 | SRS2751512 | 20,308,970 | 391,019 | 93.74 | 25.88 | 8662 | 34,437 | 34.28 |
| BAL_R3 | SRR6368031 | SRS2751523 | 12,614,238 | 187,812 | 95.77 | 41.37 | 3141 | 54,951 | 59.10 |
| BRO_R1 | SRR6368030 | SRS2751514 | 14,986,370 | 230,155 | 95.61 | 15.59 | 2909 | 7937 | 23.69 |
| BRO_R2 | SRR6368029 | SRS2751516 | 15,367,923 | 207,560 | 95.43 | 17.80 | 2853 | 8265 | 18.92 |
| BRO_R3 | SRR6368036 | SRS2751513 | 17,906,164 | 484,731 | 95.56 | 10.99 | 5185 | 13,803 | 29.70 |
| BCONT_R1 | SRR6368035 | SRS2751519 | 12,893,929 | 93,736 | 85.68 | 19.14 | 5114 | 676 | 29.46 |
| BCONT_R2 | SRR6368034 | SRS2751517 | 23,428,531 | 382,171 | 84.86 | 42.85 | 4751 | 98,337 | 44.00 |
| BCONT_R3 | SRR6368033 | SRS2751518 | 13,161,547 | 117,290 | 91.33 | 34.91 | 2982 | 8861 | 23.11 |
| CAL_R1 | SRR6368022 | SRS2751525 | 13,376,685 | 342,990 | 88.63 | 8.59 | 4691 | 7049 | 36.23 |
| CAL_R2 | SRR6368021 | SRS2751526 | 12,343,880 | 317,136 | 81.39 | 13.11 | 9361 | 7628 | 34.98 |
| CAL_R3 | SRR6368024 | SRS2751522 | 18,637,725 | 380,329 | 89.13 | 9.16 | 5928 | 6830 | 32.38 |
| CRO_R1 | SRR6368023 | SRS2751524 | 13,977,334 | 208,810 | 90.82 | 10.80 | 4651 | 3801 | 31.17 |
| CRO_R2 | SRR6368026 | SRS2751530 | 18,315,136 | 534,056 | 89.60 | 10.61 | 9998 | 13,518 | 34.78 |
| CRO_R3 | SRR6368025 | SRS2751520 | 27,710,101 | 581,439 | 91.36 | 12.77 | 8804 | 8195 | 18.68 |
| CCONT_R1 | SRR6368028 | SRS2751515 | 17,006,711 | 138,440 | 89.62 | 17.90 | 3071 | 820 | 13.90 |
| CCONT_R2 | SRR6368027 | SRS2751521 | 29,267,531 | 246,552 | 92.28 | 22.81 | 11,392 | 2488 | 22.10 |
| CCONT_R3 | SRR6368020 | SRS2751527 | 16,244,284 | 98,253 | 94.61 | 16.58 | 2804 | 1210 | 21.72 |
| Source | Df | SS | MS | F | R2 | p | Explained Variance (%) |
|---|---|---|---|---|---|---|---|
| Bacteria (family) | |||||||
| Genotype | 1 | 0.10739 | 0.107388 | 4.5390 | 0.20102 | 0.0032 | 20.10 |
| Soil | 2 | 0.05908 | 0.029542 | 1.2487 | 0.11060 | 0.2511 | 11.06 |
| Genotype × Soil | 2 | 0.0838 | 0.041915 | 1.7716 | 0.15692 | 0.0762 | 15.69 |
| Residual | 12 | 0.28391 | 0.023659 | 0.53145 | 53.15 | ||
| Total | 17 | 0.53421 | 1 | 100 | |||
| Fungi (family) | |||||||
| Genotype | 1 | 0.69477 | 0.69477 | 7.6321 | 0.28835 | 0.0004 | 28.83 |
| Soil | 2 | 0.48786 | 0.24393 | 2.6796 | 0.20247 | 0.0294 | 20.25 |
| Genotype × Soil | 2 | 0.13449 | 0.06724 | 0.7387 | 0.05581 | 0.6368 | 5.58 |
| Residual | 12 | 1.09239 | 0.09103 | 0.45337 | 45.34 | ||
| Total | 17 | 2.40951 | 1 | 100 | |||
| COG genes | |||||||
| Genotype | 1 | 0.3782 | 0.3781 | 2.7336 | 0.11915 | 0.0253 | 11.92 |
| Soil | 2 | 0.7818 | 0.39089 | 2.8254 | 0.24630 | 0.0091 | 24.63 |
| Genotype × Soil | 2 | 0.3539 | 0.17696 | 1.2791 | 0.11151 | 0.2334 | 11.15 |
| Residual | 12 | 1.6601 | 0.13835 | 0.52304 | 52.30 | ||
| Total | 17 | 3.1740 | 1 | 100 | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chialva, M.; Ghignone, S.; Novero, M.; Hozzein, W.N.; Lanfranco, L.; Bonfante, P. Tomato RNA-seq Data Mining Reveals the Taxonomic and Functional Diversity of Root-Associated Microbiota. Microorganisms 2020, 8, 38. https://doi.org/10.3390/microorganisms8010038
Chialva M, Ghignone S, Novero M, Hozzein WN, Lanfranco L, Bonfante P. Tomato RNA-seq Data Mining Reveals the Taxonomic and Functional Diversity of Root-Associated Microbiota. Microorganisms. 2020; 8(1):38. https://doi.org/10.3390/microorganisms8010038
Chicago/Turabian StyleChialva, Matteo, Stefano Ghignone, Mara Novero, Wael N. Hozzein, Luisa Lanfranco, and Paola Bonfante. 2020. "Tomato RNA-seq Data Mining Reveals the Taxonomic and Functional Diversity of Root-Associated Microbiota" Microorganisms 8, no. 1: 38. https://doi.org/10.3390/microorganisms8010038
APA StyleChialva, M., Ghignone, S., Novero, M., Hozzein, W. N., Lanfranco, L., & Bonfante, P. (2020). Tomato RNA-seq Data Mining Reveals the Taxonomic and Functional Diversity of Root-Associated Microbiota. Microorganisms, 8(1), 38. https://doi.org/10.3390/microorganisms8010038