Prevalence of Non-B HIV-1 Subtypes in North Italy and Analysis of Transmission Clusters Based on Sequence Data Analysis

 , , ,
, , ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. HIV-1 Subtyping and Sequencing
2.3. Determination of Resistance of HIV-1 Subtypes and CRFs
2.4. Phylogenetic Analyses and Transmission Clusters
2.5. Statistical Analysis
2.6. Sequence Data Availability
3. Results
3.1. Study Population
3.2. HIV-1 Genotyping
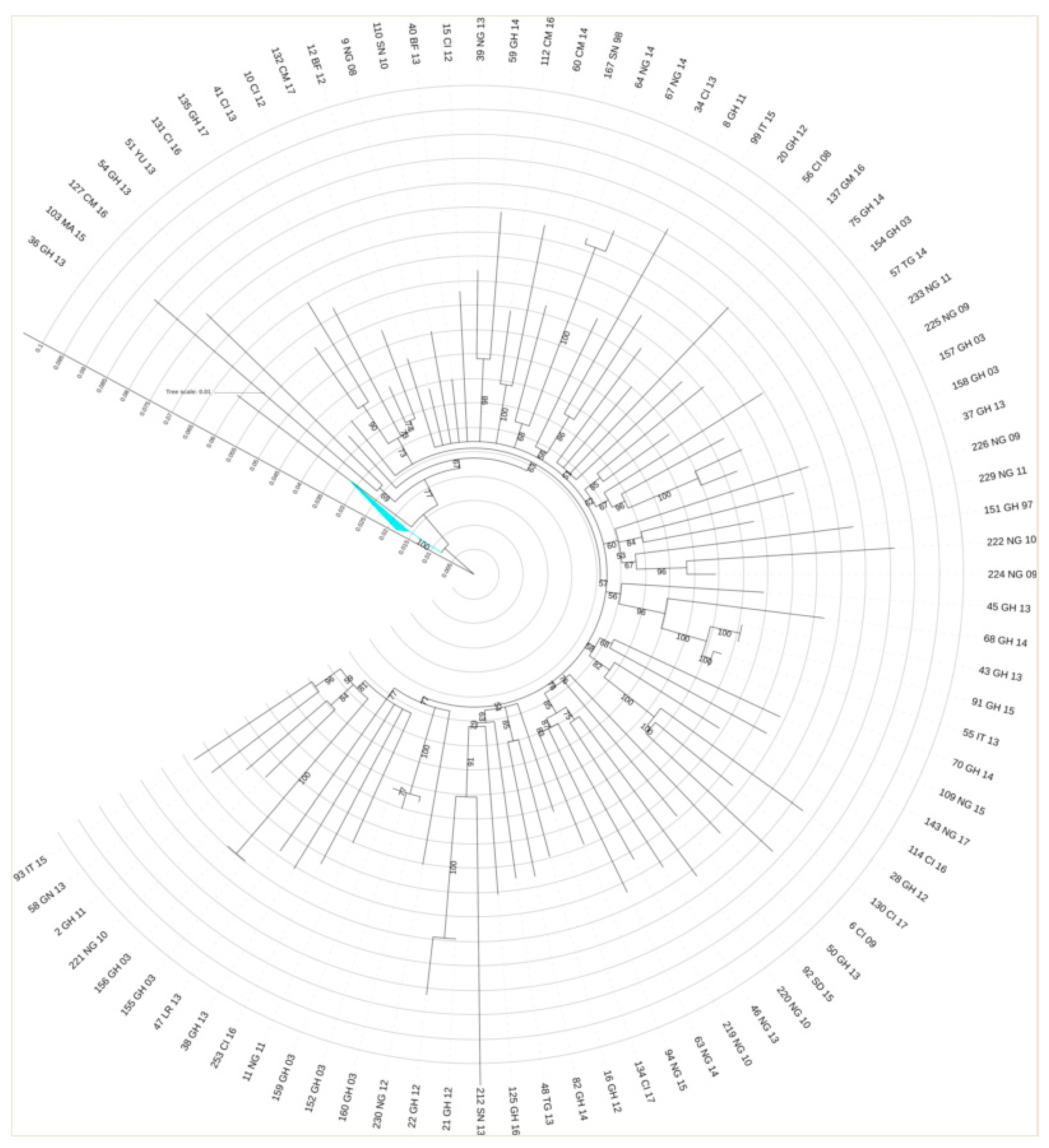
3.3. Transmission Cluster Analysis
3.4. CRF02_AG Transmission Clusters
3.5. F1 Transmission Clusters
3.6. C Transmission Clusters
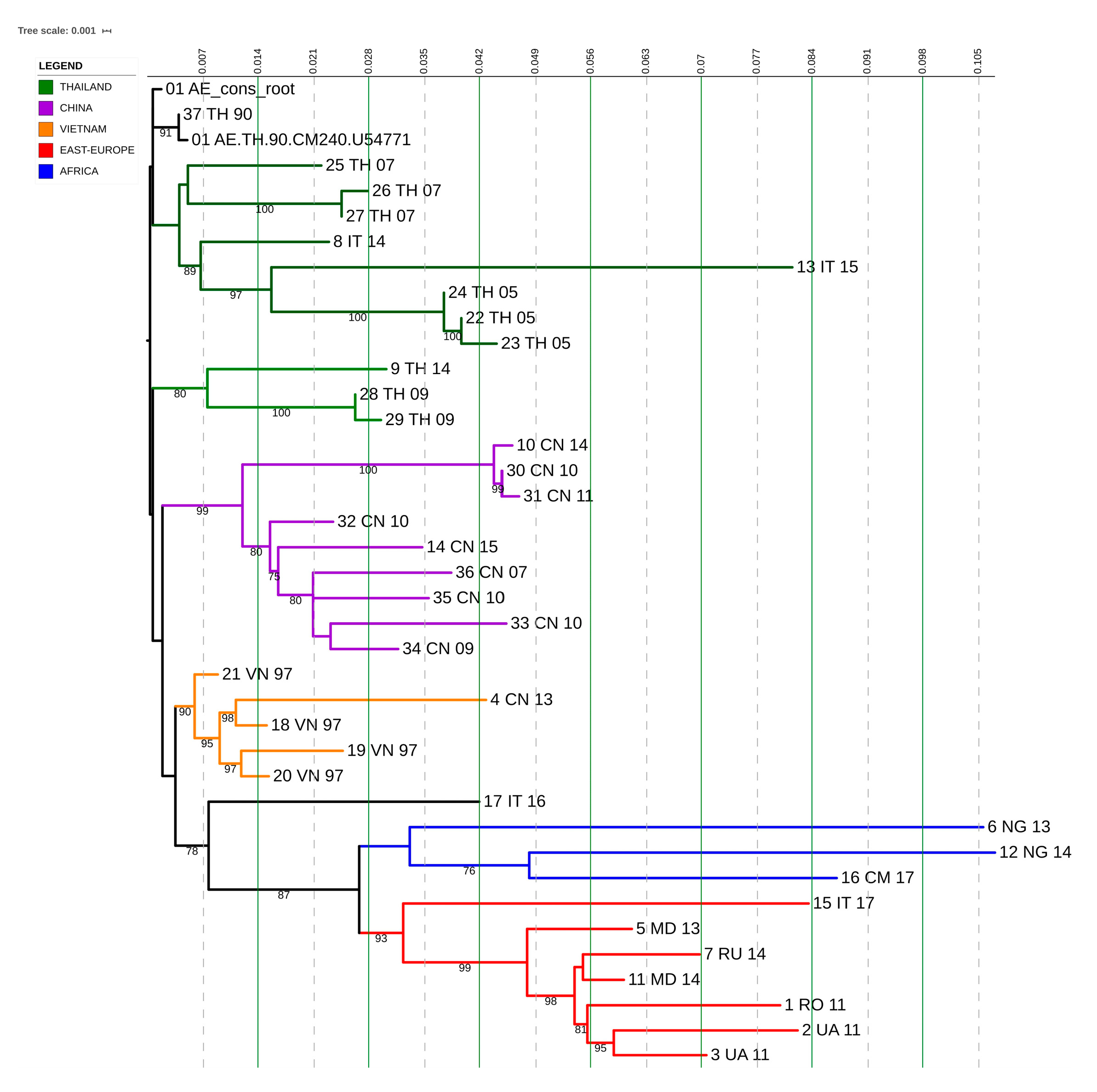
3.7. CRF01_AE Transmission Clusters
3.8. CRF06_cpx and CRF12_BF Transmission Clusters
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Global HIV and AIDS statistics—Fact Sheet 2019; UNAIDS: Geneva, Switzerland, 2019.
- Hemelaar, J. Implications of HIV diversity for the HIV-1 pandemic. J. Infect. 2013, 66, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Aldrich, C.; Hemelaar, J. Global HIV-1 diversity surveillance. Trends Mol. Med. 2012, 18, 691–694. [Google Scholar] [CrossRef] [PubMed]
- Foley, B.T.; Leitner, T.; Paraskevis, D.; Peeters, M. Primate immunodeficiency virus classification and nomenclature review. Infect. Genet. Evol. 2016, 46, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Hemelaar, J.; Elangovan, R.; Yun, J.; Dickson-Tetteh, L.; Fleminger, I.; Kirtley, S.; Williams, B.; Gows-Williams, E.; Ghys, P. WHO-UNAIDS Network for HIV isolation characterization. Global and regional molecular epidemiology of HIV-1, 1990–2015: A systematic review, global survey, and trend analysis. Lancet Infect. Dis. 2018, 19, 143–155. [Google Scholar] [CrossRef]
- Delatorre, E.; Mir, D.; Bello, G. Spatiotemporal dynamics of the HIV-1 subtype G epidemic in West and Central Africa. PLoS ONE 2014, 9, e98908. [Google Scholar] [CrossRef] [PubMed]
- Easterbrook, P.J.; Smith, M.; Mullen, J.; O’Shea, S.; Christie, I.; de Ruiter, A.; Tatt, I.D.; Geretti, A.M.; Zuckerman, M. Impact of HIV-1 viral subtype on disease progression and response to antiretroviral therapy. J. Int. AIDS Soc. 2010, 13, 4. [Google Scholar] [CrossRef]
- Kiwanuka, N.; Laeyendecker, O.; Robb, M.; Kigozi, G.; Arroyo, M.; McCutchan, F.; Eller, L.A.; Eller, M.; Makumbi, F.; Birx, D.; et al. Effect of human immunodeficiency virus Type 1 (HIV-1) subtype on disease progression in persons from Rakai, Uganda, with incident HIV-1 infection. J. Infect. Dis. 2008, 197, 707–713. [Google Scholar] [CrossRef]
- Baeten, J.M.; Chohan, B.; Lavreys, L.; Chohan, V.; McClelland, R.S.; Certain, L.; Mandaliya, K.; Jaoko, W.; Overbaugh, J. HIV-1 subtype D infection is associated with faster disease progression than subtype A in spite of similar plasma HIV-1 loads. J. Infect. Dis. 2007, 195, 1177–1180. [Google Scholar] [CrossRef]
- Abecasis, A.B.; Deforche, K.; Snoeck, J.; Bacheler, L.T.; McKenna, P.; Carvalho, A.P.; Gomes, P.; Camacho, R.J.; Vandamme, A.M. Protease mutation M89I/V is linked to therapy failure in patients infected with the HIV-1 non-B subtypes C, F or G. AIDS 2005, 19, 1799–1806. [Google Scholar] [CrossRef]
- Abecasis, A.B.; Deforche, K.; Bacheler, L.T.; McKenna, P.; Carvalho, A.P.; Gomes, P.; Vandamme, A.M.; Camacho, R.J. Investigation of baseline susceptibility to protease inhibitors in HIV-1 subtypes C, F, G and CRF02_AG. Antivir. Ther. 2006, 11, 581. [Google Scholar]
- Camacho, R.J.; Vandamme, A.M. Antiretroviral resistance in different HIV-1 subtypes impact on therapy outcomes and resistance testing interpretation. Curr. Opin. HIV AIDS 2007, 2, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Brenner, B.; Turner, D.; Oliveira, M.; Moisi, D.; Detorio, M.; Carobene, M.; Marlink, R.G.; Schapiro, J.; Roger, M.; Wainberg, M.A. A V106M mutation in HIV-1 clade C viruses exposed to efavirenz confers cross-resistance to non nucleoside reverse transcriptase inhibitors. AIDS 2003, 17, F1–F5. [Google Scholar] [CrossRef] [PubMed]
- Serwanga, J.; Nakiboneka, R.; Mugaba, S.; Magambo, B.; Ndembi, N.; Gotch, F.; Kaleebu, P. Frequencies of Gag-restricted T-cell escape ‘footprints’ differ across HIV-1 clades A1 and D chronically infected Ugandans irrespective of host HLA B alleles. Vaccine 2015, 33, 1664–1672. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bartolo, I.; Abecasis, A.B.; Borrego, P.; Barroso, H.; McCutchan, F.; Gomes, P.; Camacho, R.; Taveira, N. Origin and epidemiological history of HIV-1 CRF14 BG. PLoS ONE 2011, 6, e24130. [Google Scholar] [CrossRef] [PubMed]
- Renjifo, B.; Gilbert, P.; Chaplin, B.; Msamanga, G.; Mwakagile, D.; Fawzi, W.; Essex, M. Tanzanian Vitamin and HIV Study Group. Preferential in-utero transmission of HIV-1 subtype C as compared to HIV-1 subtype A or D. AIDS 2004, 18, 1629–1636. [Google Scholar] [CrossRef] [PubMed]
- John-Stewart, G.C.; Nduati, R.W.; Rousseau, C.M.; Mbori-Ngacha, D.A.; Richardson, B.A.; Rainwater, S.; Panteleff, D.D.; Overbaugh, J. Subtype C is associated with increase vaginal shedding of HIV-1. J. Infect. Dis. 2005, 192, 492–496. [Google Scholar] [CrossRef]
- Lai, A.; Simonetti, F.R.; Zehender, G.; De Luca, A.; Micheli, V.; Mervaiglia, P.; Corsi, P.; Bagnarelli, P.; Almi, P.; Zoncada, A.; et al. HIV-1 subtype F1 epidemiological networks among Italian heterosexual males are associated with introduction events from South America. PLoS ONE 2012, 7, e42223. [Google Scholar] [CrossRef][Green Version]
- Lai, A.; Bozzi, G.; Franzetti, M.; Binda, F.; Simonetti, F.R.; Micheli, V.; Meraviglia, P.; Corsi, P.; Bagnarelli, P.; De Luca, A.; et al. Phylogenetic analysis provides evidence of interactions between Italian heterosexual and South American homosexual males as the main source of National HIV-1 subtype C epidemics. J. Med. Virol. 2014, 86, 729–736. [Google Scholar] [CrossRef]
- Lai, A.; Bozzi, G.; Franzetti, M.; Binda, F.; Simonetti, F.R.; De Luca, A.; Micheli, V.; Meraviglia, P.; Bagnarelli, P.; Di Biagio, A.; et al. HIV-1 A1 subtype epidemic in Italy originated from Africa and Eastern Europe and shows a high frequency of transmission chains involving intravenous drug users. PLoS ONE 2016, 11, e0146097. [Google Scholar] [CrossRef][Green Version]
- Izzo, I.; Quiros-Roldan, E.; Casari, S.; Gargiulo, F.; Caruso, A.; Castelli, F. Characteristics and outcome of a cohort of HIV-1 non-B subtype-infected patients after a 10-year follow-up period. A single centre experience. J. AIDS 2017, 71, e23–e24. [Google Scholar] [CrossRef]
- Rossetti, B.; Di Gianbenedetto, S.; Torti, C.; Postorino, M.C.; Punzi, G.; Saladini, F.; Gennari, W.; Borghi, V.; Monno, L.; Pignataro, A.R.; et al. Evolution of transmitted HIV-1 drug resistance and viral subtypes circulation in Italy from 2006 to 2016. HIV Med. 2018, 19, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Sulle attività di prevenzione e promozione della salute dell’ATS di Brescia. In Rapporto 2017; ATS Brescia: Brescia, Italy, 2017.
- Suligoi, B.; Buttò, S.; Galli, C.; Bernasconi, D.; Salata, R.A.; Tavoschi, L.; Chiappi, M.; Mugyenyi, P.; Pimpinelli, F.; Kityo, C.; et al. Detection of recent HIV infections in African individuals infected by HIV-1 non-B subtypes using HIV antibody avidity. J. Clin. Virol 2008, 41, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Aitken, S.C.; Kliphuis, A.; Wallis, C.L.; Chu, M.L.; Fillekes, Q.; Barth, R.; Stevens, W.; Rinke de Wit, T.F.; Schuurman, R. Development and evaluation of an assay for HIV-1 protease and reverse drug resistance genotyping of all major group-M subtypes. J. Clin. Virol. 2012, 54, 21–25. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7, Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- De Oliveira, T.; Deforche, K.; Cassol, S.; Salminen, M.; Paraskevis, D.; Seebregts, C.; Snoeck, J.; van Rensburg, E.J.; Wensing, A.M.; Van de Vijver, D.A.; et al. An automated genotyping system for analysis of HIV-1 and other microbial sequences. Bioinformatics 2005, 21, 3798–3800. [Google Scholar] [CrossRef] [PubMed]
- Struck, D.; Lawyer, G.; Ternes, A.M.; Scmit, J.C.; Bercoff, D.P. COMET, Adaptive context-based modeling for ultrafast HIV-1 subtype identification. Nucleic Acids Res. 2015, 42, e144. [Google Scholar] [CrossRef]
- Kosakovski Pond, S.L.; Posada, D.; Stawiski, E.; Chappey, C.; Poon, A.F.; Hughes, G.; Fearnhill, E.; Gravenor, M.B.; Leigh Brown, A.J.; Frost, S.D. An evolutionary model based algorithm for accurate phylogenetic breakpoint mapping and subtype prediction in HIV-1. PLoS Comput. Biol. 2009, 5, e1000581. [Google Scholar] [CrossRef]
- Schultz, A.K.; Zhang, M.; Bulla, I.; Leitner, T.; Korber, B.; Morgenstern, B.; Stanke, M. jpHMM improving the reliability of recombination prediction in HIV-1. Nucleic Acids Res. 2009, 37, W647–W651. [Google Scholar] [CrossRef]
- Rozanov, M.; Plikat, U.; Chappey, C.; Kochergin, A.; Tatusova, T. A web-based genotyping resource for viral sequences. Nucleic Acids Res. 2004, 32, W654–W659. [Google Scholar] [CrossRef]
- Liu, T.F.; Shafer, R.W. Web Resources for HIV Type 1 genotypic resistance test interpretation. Clin. Infect. Dis. 2006, 42, 1608–1618. [Google Scholar] [CrossRef]
- Wensing, A.M.; Calvez, V.; Günthard, H.F.; Johnson, V.A.; Paredes, R.; Pillay, D.; Shafer, R.W.; Richman, D.D. 2017 update of the drug resistance mutations in HIV-1. Top Antivir. Med. 2017, 24, 132–133. [Google Scholar]
- Gifford, R.J.; Liu, T.F.; Rhee, S.Y.; Kiuchi, M.; Hue, S.; Pillay, D.; Shafer, R.W. The calibrated population resistance tool standardized genotypic estimation of transmitted HIV-1 drug resistance. Bioinformatics 2009, 25, 1197–1198. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.A.; Strimmer, K.; Vingron, M.; von Haeseler, A. TREE-PUZZLE: Maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 2002, 18, 502–504. [Google Scholar] [CrossRef] [PubMed]
- Kosakovski Pond, S.L.; Weaver, S.; Leigh Brown, A.J.; Wertheim, J.O. HIV-TRACE (TRAnsmission Cluster Engine): A tool for large scale molecular epidemiology of HIV-1 and other rapidly evolving pathogens. Mol. Biol. Evol. 2018, 35, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.S.; Choi, J.Y.; Yoo, M.S.; Seong, J.H.; Choi, B.S.; Kang, C. Phylogenetic transmission clusters among newly diagnosed antiretroviral drug-naive patients with human immunodeficiency virus-1 in Korea, A study from 1999 to 2012. PLoS ONE 2019, 14, e0217817. [Google Scholar] [CrossRef]
- Rolland, M.; Tovanabutra, S.; decamp, A.C.; Frahm, N.; Gilbert, P.B.; Sanders-Buell, E.; Heath, L.; Magaret, C.A.; Bose, M.; Bradfield, A.; et al. Genetic impact of vaccination on breakthrough HIV-1 sequences from the STEP trial. Nat. Med. 2011, 17, 366–372. [Google Scholar] [CrossRef]
- Patiño-Galindo, J.Á.; González-Candelas, F. The substitution rate of HIV-1 subtypes, A genomic approach. Virus Evol. 2017, 3, vex029. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Kouyos, R.D.; von Wil, V.; Yerly, S.; Böni, J.; Taffé, P.; Shah, C.; Börgisser, P.; Klimkait, T.; Weber, R.; Hirschel, B.; et al. Molecular epidemiology reveals long-term changes in HIV type 1 subtype B transmission in Switzerland. J. Infect. Dis. 2010, 201, 1488–1497. [Google Scholar] [CrossRef]
- Alizon, S.; von Wil, V.; Stadler, T.; Kouyos, R.D.; Yerly, S.; Hirschel, B.; Böni, J.; Shah, C.; Klimkait, T.; Furrer, H.; et al. Phylogenetic approach reveals that virus genotype largely determines HIV set point viral load. PLoS Pathog. 2010, 6, e1001123. [Google Scholar] [CrossRef]
- Esbjörnsson, J.; Mild, M.; Audelin, A.; Fonager, J.; Skar, H.; JØrgenssen, L.B.; Liitsola, K.; Biörkman, P.; Brat, G.; Gisslèn, M.; et al. HIV transmission between MSM and heterosexuals, and increasing proportions of circulating recombinant forms in the Nordic Countries. Virus Evol. 2016, 2, vew010. [Google Scholar] [CrossRef] [PubMed]
- Snoeck, J.; Van Laethem, K.; Hermans, P.; Van Wijngaerden, E.; Derdelinckx, I.; Scheooten, Y.; vand Vijver, D.A.; De Wit, S.; Clumeck, N.; Vandamme, A.M. Rising prevalence of HIV-1 non-B subtypes in Belgium, 1983–2001. J. Acquir. Immune Defic. Syndr. 2004, 35, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Holguin, A.; de Mulder, M.; Yebra, G.; Lopez, M.; Soriano, V. Increase of non-B subtypes and recombinants among newly diagnosed HIV-1 native Spaniards and immigrants in Spain. Curr. HIV Res. 2008, 6, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Descamps, D.; Chaix, M.L.; Montes, B.; Pakianather, S.; Charpentier, C.; Storto, A.; Barin, F.; Dos santos, G.; Krivine, A.; Delaugerre, C.; et al. Increasing prevalence of transmitted drug resistance mutations and non-B subtype circulation in antiretroviral-naive chronically HIV-infected patients from 2001 to 2006/2007 in France. J Antimicrob. Chemother. 2010, 65, 2620–2627. [Google Scholar] [CrossRef] [PubMed]
- Neogi, U.; Häggblom, A.; Santacatterina, M.; Bratt, G.; Gisslén, M.; Albert, J.; Sonnerborg, A. Temporal trends in the swedish HIV-1 epidemic: Increase in Non-B subtypes and recombinant forms over three decades. PLoS ONE 2014, 9, e99390. [Google Scholar] [CrossRef]
- Struck, D.; Roman, F.; De Landtsheer, S.; Servais, J.Y.; Lambert, C.; Masquelier, C.; Venard, V.; Ruelle, J.; Nijhuis, M.; Schmit, J.C.; et al. Near Full-Length Characterization and Population Dynamics of the Human Immunodeficiency Virus Type I Circulating Recombinant Form 42 (CRF42_BF) in Luxembourg. AIDS Res. Hum. Retrovir. 2015, 31, 554–558. [Google Scholar] [CrossRef]
- Lai, A.; Riva, C.; Marconi, A.; Balestrieri, M.; Razzolini, F.; Meini, G.; Vicenti, I.; Rosi, A.; Saladini, F.; Caramma, I.; et al. Changing patterns in HIV-1 non-B clade prevalence and diversity in Italy over three decades. HIV Med. 2010, 11, 593–602. [Google Scholar] [CrossRef]
- Torti, C.; Lapadula, G.; Izzo, I.; Brindicci, G.; Labbate, G.; Quiros-Roldan, E.; Diallo, I.; Gargiulo, F.; Castelnbuovo, F.; Calabresi, A.; et al. Heterogeneity and penetration of HIV-1 non- subtype B viruses in an Italian province, Public health implications. Epidemiol. Infect. 2010, 138, 1298–1307. [Google Scholar] [CrossRef]
- Paraskevis, D.; Beloukas, A.; Stasinos, K.; Pantazis, N.; de Mendoza, C.; Bannert, N.; Meyer, L.; Zangerle, R.; Gill, J.; Prins, M.; et al. HIV-1 molecular transmission clusters in nine European countries and Canada: Association with demographic and clinical factors. BMC Med. 2019, 17, 4. [Google Scholar] [CrossRef]
- Abecasis, A.B.; Wensing, A.M.; Paraskevis, D.; Vercauteren, J.; Theys, K.; Van de Vijer, D.A.; Albert, J.; Asjö, B.; Balotta, C.; Beshkov, D.; et al. HIV-1 subtype distribution and its demographic determinants in newly diagnosed patients in Europe suggest highly compartmentalized epidemics. Retrovirology 2013, 10, 7. [Google Scholar] [CrossRef]
- Tebit, D.M.; Arts, E.J. Tracking a century of global expansion and evolution of HIV to drive understanding and to combat disease. Lancet Infect. Dis. 2011, 11, 45–56. [Google Scholar] [CrossRef]
- Ratmann, O.; van Sighem, A.; Bezemer, D.; Gavryushkina, A.; Jurriaans, S.; Wensing, A.; de Wolf, F.; Reiss, P.; Fraser, C. Sources of HIV infection among men having sex with men and implications for prevention. Sci. Transl. Med. 2016, 8, 320. [Google Scholar] [CrossRef] [PubMed]
- Chaillon, A.; Essat, A.; Frange, P.; Smith, D.M.; Delaugerre, C.; Barin, F.; Ghosn, J.; Piaoloux, G.; Robineaum, O.; Rouzioux, C.; et al. Spatiotemporal dynamics of HIV-1 transmission in France (1999–2014) and impact of targeted prevention strategies. Retrovirology 2017, 14, 15. [Google Scholar] [CrossRef] [PubMed]
- German, D.; Grabowski, M.K.; Beyrer, C. Enhanced use of phylogenetic data to inform public health approaches to HIV among men who have sex with men. Sex Health 2017, 14, 89–96. [Google Scholar] [CrossRef]
- Oster, A.M.; France, A.M.; Panneer, N.; Bañez Ocfemia, M.C.; Campbell, E.; Dasgupta, s.; Switzer, W.M.; Wertheim, J.O.; Hernandez, A.L. Identifying clusters of recent and rapid HIV transmission through analysis of molecular surveillance data. J. Acquir. Immune Defic. Syndr. 2018, 79, 543–550. [Google Scholar] [CrossRef]
- Wertheim, J.O.; Murrell, B.; Mehta, S.R.; Forgione, L.A.; Kosakovsky Pond, S.L.; Smith, D.M.; Torian, L.V. Growth of HIV-1 molecular transmission clusters in NewYork City. J. Infect. Dis. 2018, 218, 1943–1953. [Google Scholar] [CrossRef]
- Magiorkinis, G.; Karamitros, T.; Vasylyeva, T.I.; Williams, L.D.; Mbisa, J.L.; Hatzakis, A.; Paraskevis, D.; Friedman, S.R. An innovative study design to assess the community effect of interventions to mitigate HIV epidemics using transmission-chain phylodynamics. Am. J. Epidemiol. 2018, 187, 2615–2622. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | HIV-1 Subtypes | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| F1 n = 50 | C n = 27 | G n = 18 | A1 n = 9 | CRF_02AG n = 150 | CRF06_CPX n = 12 | CRF01_AE n = 17 | CRF12_BF n = 5 | CRF09_CPX n = 5 | Total | |
| Gender | ||||||||||
| Male | 37 (74%) | 17 (63%) | 9 (50%) | 3 (33%) | 104 (69%) | 6 (50%) | 9 (53%) | 4 (80%) | 1 (20%) | 190 (65%) |
| Female | 13 (26%) | 10 (37%) | 9 (50%) | 6 (67%) | 46 (31%) | 6 (50%) | 8 (47%) | 1 (20%) | 4 (80%) | 103 (35%) |
| Age (years), Median (IQR) | 46 (17) | 42 (16) | 40 (15) | 43 (18) | 41 (18) | 41 (15) | 37 (18) | 35 (19.5) | 32 (17) | |
| Risk factors, n (%) | ||||||||||
| MSM | 9 (18%) | 6 (22%) | 1 (6%) | 1 (11%) | 29 (19%) | 2 (17%) | 5 (29%) | 0 | 0 | 53 (18%) |
| Heterosexual | 32 (64%) | 15 (56%) | 17 (94%) | 6 (67%) | 108 (72%) | 10 (83%) | 10 (59%) | 3 (60%) | 4 (80%) | 205 (70%) |
| IDU | 3 (6%) | 1 (4%) | 0 | 1 (11%) | 3 (2%) | 0 | 0 | 1 (20%) | 0 | 9 (3%) |
| Others | 6 (12%) | 5 (18%) | 0 | 1 (11%) | 10 (7%) | 0 | 2 (12%) | 1 (20%) | 1 (20%) | 26 (9%) |
| CD4 count, mean (SD) cells/mL | 253 (257) | 396 (310) | 325 (300) | 453 (311) | 330 (233) | 403 (280) | 260 (147) | 228 (261) | 227 (193) | |
| CD4 count < 200 cell/mL, n (%) | 27 (54%) | 7 (26%) | 7 (39%) | 2 (22%) | 48 (32%) | 3 (25%) | 6 (35%) | 3 (60%) | 2 (40%) | |
| PVL, mean copies/mL | 733,602 | 380,374 | 153,839 | 366,040 | 304,435 | 38,707 | 151,028 | 628,462 | 43,099 | |
| Geographic origin, n (%) | ||||||||||
| Italy | 43 (86%) | 17 (63%) | 2 (11%) | 4 (44%) | 80 (53.3%) | 4 (34%) | 4 (23.5%) | 3 (60%) | 0 | 157 (54%) |
| Europe | 1 (2%) | 0 | 0 | 2 (22%) | 5 (3%) | 0 | 6 (35%) | 2 (40%) | 0 | 16 (5%) |
| America | 2 (4%) | 0 | 0 | 0 | 2 (1%) | 1 (8%) | 0 | 0 | 0 | 5 (2%) |
| Africa | 2 (4%) | 2 (7%) | 16 (89%) | 2 (22%) | 62 (41%) | 7 (58%) | 3 (18%) | 0 | 5 (100%) | 99 (34%) |
| Asia | 2 (4%) | 8 (30%) | 0 | 1 (11%) | 1 (0.7%) | 0 | 4 (57.1%) | 0 | 0 | 16 (5%) |
| Coinfection with HCV | 7 (14%) | 1 (4%) | 0 | 1 (11%) | 9 (6%) | 0 | 1 (6%) | 1 (20%) | 0 | 20 (7%) |
| Coinfection with HBV | 2 (4%) | 1 (4%) | 4 (22%) | 1 (11%) | 12 (8%) | 2 (17%) | 1 (6%) | 0 | 1 (20%) | 24 (8%) |
| HIV Subtypes | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| F1 n = 50 | C n = 27 | G n = 18 | A1 n = 9 | CRF_02AG n = 150 | CRF06_CPX n = 12 | CRF01_AE n = 17 | CRF12_BF n = 5 | CRF09_CPX n = 5 | |
| No cluster | 17 (34%) | 17 (63%) | 18 (100%) | 9 (100%) | 58 (38%) | 8 (67%) | 12 (70.5%) | 2 (40%) | 5 (100%) |
| Small Cluster (2–3) | 8 (16%) | 10 (37%) | 0 | 0 | 17 (11.3%) | 4 (33%) | 4 (23.5%) | 3 (60%) | 0 |
| Medium Cluster (4–9) | 15 (30%) | 0 | 0 | 0 | 1 (0.6%) | 0 | 1 (5.8%) | 0 | 0 |
| Large cluster (≥10) | 10 (20%) | 0 | 0 | 0 | 74 (50%) | 0 | 0 | 0 | 0 |
| Univariate Analysis | Multivariate Analysis | ||||
|---|---|---|---|---|---|
| Not in Cluster n = 146 | In Cluster n = 147 | p-Value | OR (95% CI) | p-Value | |
| Gender | |||||
| Male | 82 (56%) | 108 (73.4%) | 0.0022 | 0.73 (0.34–1.58) | 0.43 |
| Female | 64 (44%) | 39 (26.5%) | |||
| Age (years), Median (IQR) | 39 (17) | 40 (18) | 0.45 | ||
| Risk factors, n (%) | |||||
| MSM | 24 (16%) | 29 (19.7%) | 0.54 | ||
| Heterosexual | 106 (73%) | 99 (67.3%) | 0.37 | ||
| IDU | 4 (3%) | 5 (3.4%) | 1 | ||
| Others | 12 (8%) | 14 (9.5%) | 0.83 | ||
| Geographic origin, n (%) | |||||
| Italy | 40 (27%) | 117 (79.5%) | 0.0001 | 8.73 (1.33–57) | 0.02 |
| Europe | 7 (5%) | 9 (6.1%) | 0.79 | ||
| America | 3 (2%) | 2 (1.3%) | 0.68 | ||
| Africa | 84 (58%) | 15 (10.2%) | 0.0001 | ||
| Asia | 12 (8%) | 4 (2.7%) | 0.04 | ||
| Subtypes | |||||
| F1 | 17 (11.6%) | 33 (22.4%) | 0.019 | 6.17 (1.24–30.73) | 0.026 |
| G | 18 (100%) | 0 | 0.0001 | ||
| C | 17 (11.6%) | 10 (6.8%) | 0.16 | ||
| A1 | 9 (100%) | 0 | 0.001 | ||
| CRF02_AG | 58 (40%) | 92 (60%) | 0.0001 | 0.093 (0.02–0.43) | 0.02 |
| CRF06_CPX | 8 (5.4%) | 4 (2.7%) | 0.25 | ||
| CRF01_AE | 12 (8.2%) | 5 (3.4%) | 0.08 | ||
| CRF12_BF | 2 (1.3%) | 3 (2%) | 1 | ||
| CRF09_CPX | 5 (100%) | 0 | 0.03 | ||
| CD4 count, mean (SD) cells/mL | 332 (244) | 354 (252) | 0.87 | ||
| PVL, mean copies/mL | 184,300 | 519,463 | 0.01 | 1 (Ref) | 0.2 |
| Transmission drug resistance | 24 (16.4%) | 9 (6.1%) | 0.005 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lorenzin, G.; Gargiulo, F.; Caruso, A.; Caccuri, F.; Focà, E.; Celotti, A.; Quiros-Roldan, E.; Izzo, I.; Castelli, F.; De Francesco, M.A. Prevalence of Non-B HIV-1 Subtypes in North Italy and Analysis of Transmission Clusters Based on Sequence Data Analysis. Microorganisms 2020, 8, 36. https://doi.org/10.3390/microorganisms8010036
Lorenzin G, Gargiulo F, Caruso A, Caccuri F, Focà E, Celotti A, Quiros-Roldan E, Izzo I, Castelli F, De Francesco MA. Prevalence of Non-B HIV-1 Subtypes in North Italy and Analysis of Transmission Clusters Based on Sequence Data Analysis. Microorganisms. 2020; 8(1):36. https://doi.org/10.3390/microorganisms8010036
Chicago/Turabian StyleLorenzin, Giovanni, Franco Gargiulo, Arnaldo Caruso, Francesca Caccuri, Emanuele Focà, Anna Celotti, Eugenia Quiros-Roldan, Ilaria Izzo, Francesco Castelli, and Maria A. De Francesco. 2020. "Prevalence of Non-B HIV-1 Subtypes in North Italy and Analysis of Transmission Clusters Based on Sequence Data Analysis" Microorganisms 8, no. 1: 36. https://doi.org/10.3390/microorganisms8010036
APA StyleLorenzin, G., Gargiulo, F., Caruso, A., Caccuri, F., Focà, E., Celotti, A., Quiros-Roldan, E., Izzo, I., Castelli, F., & De Francesco, M. A. (2020). Prevalence of Non-B HIV-1 Subtypes in North Italy and Analysis of Transmission Clusters Based on Sequence Data Analysis. Microorganisms, 8(1), 36. https://doi.org/10.3390/microorganisms8010036