The Role of Pseudomonas in Heterotrophic Nitrification: A Case Study on Shrimp Ponds (Litopenaeus vannamei) in Soc Trang Province

 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples Collection
2.2. Sequence Processing and Analysis
2.3. Predictive Metagenome Functions Analysis
2.4. Statistical Analysis
2.5. Isolation and Identification of Heterotrophic Nitrifying Bacteria
2.6. Ammonia-Oxidizing Activity Test
3. Results
3.1. Alpha Diversity and Beta Diversity Analysis
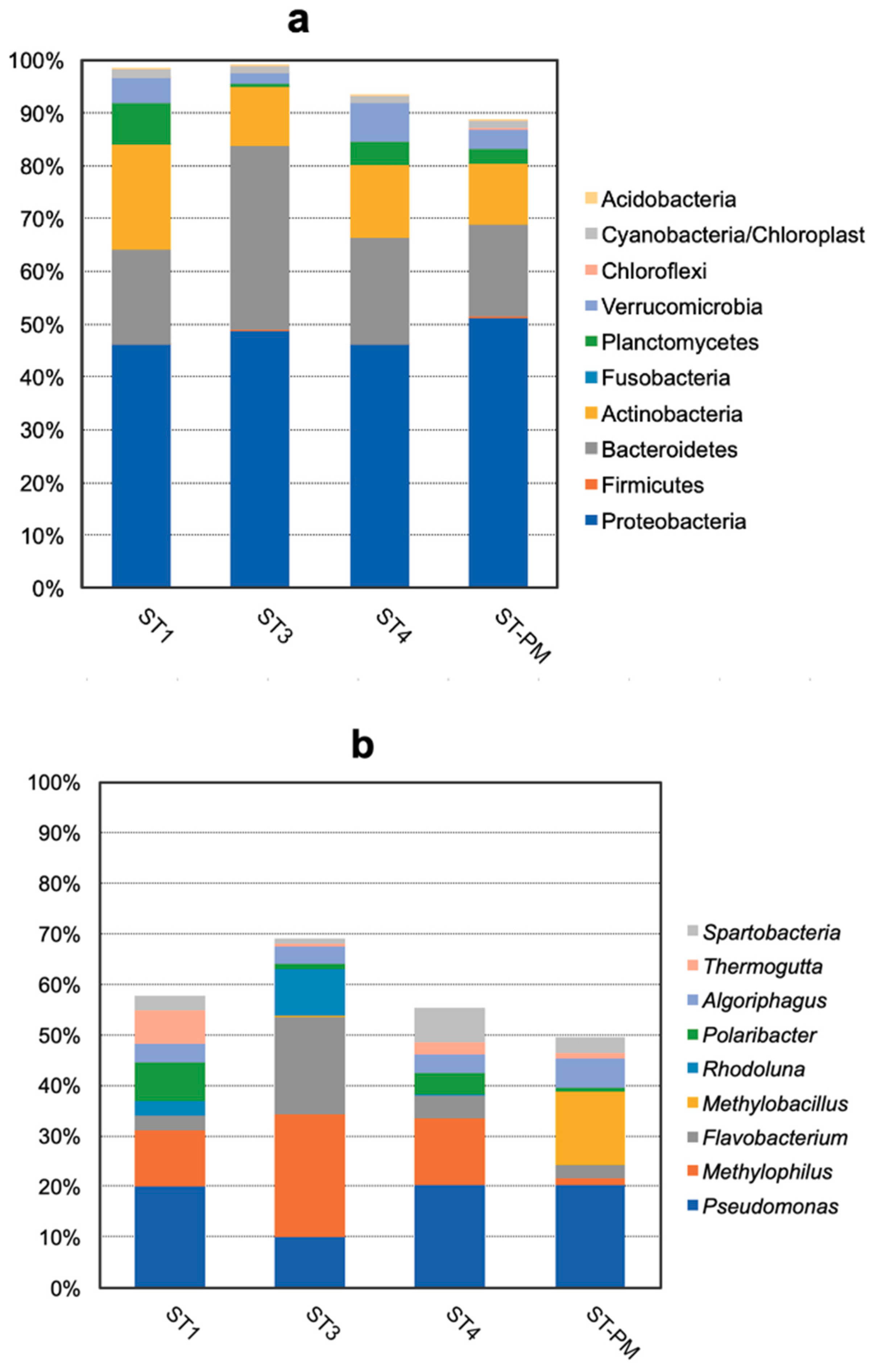
3.2. Compositions and Abundances of Bacterial Communities Analysis
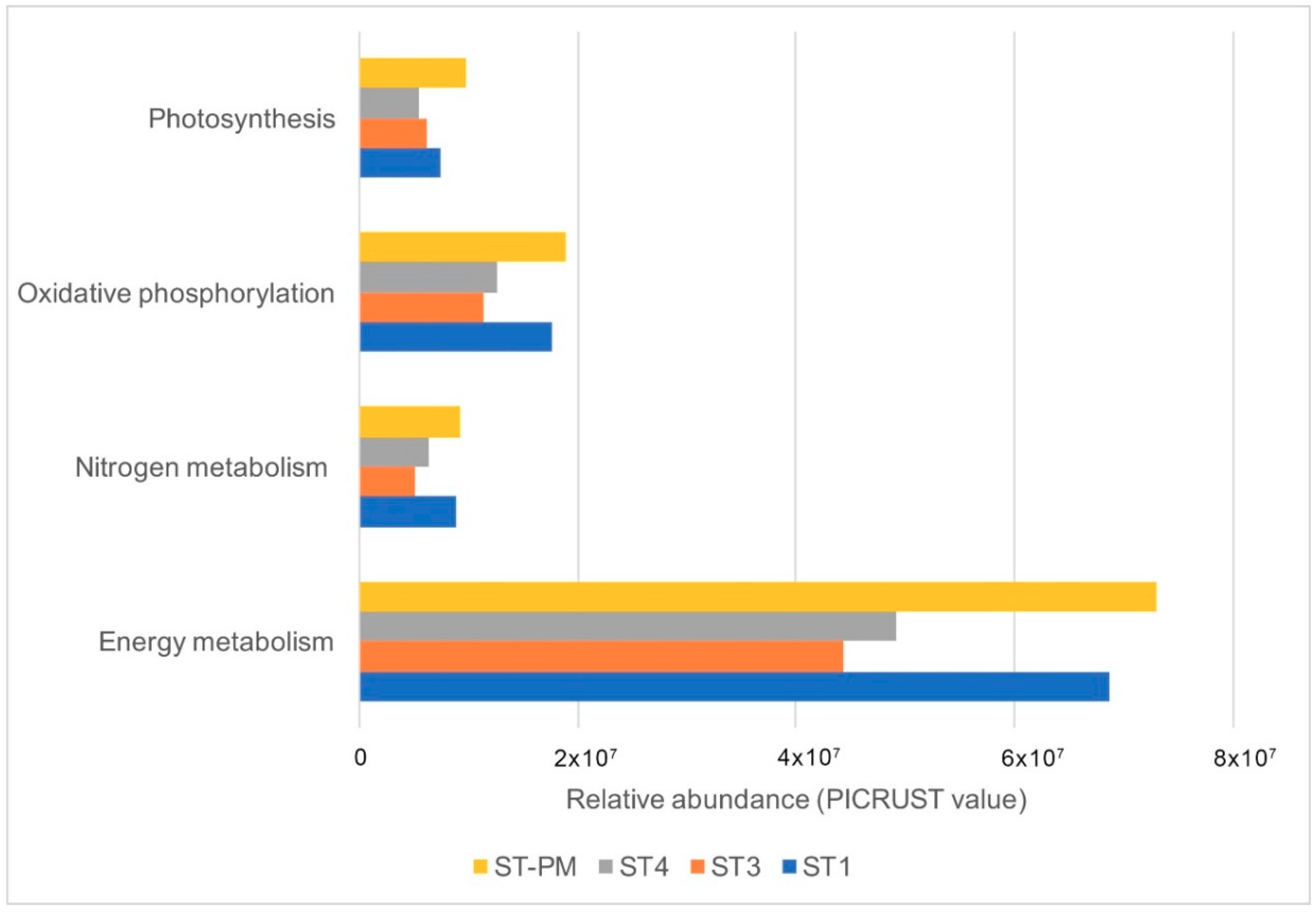
3.3. Predictive Functions of Microorganism Communities Analysis by PICRUST
3.4. Relationship Among Environmental Factors, Predictive Functions, and Bacterial Abundance
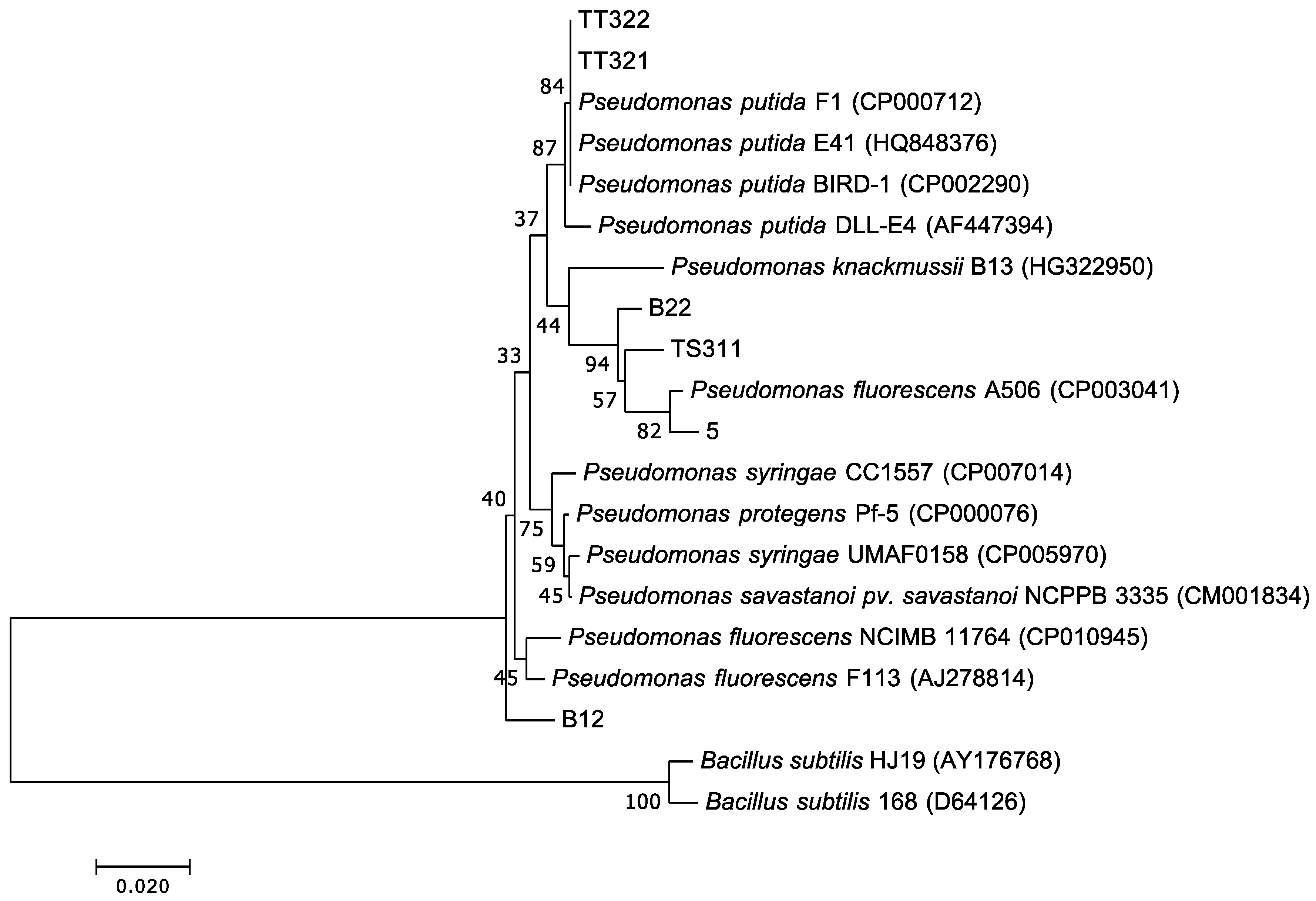
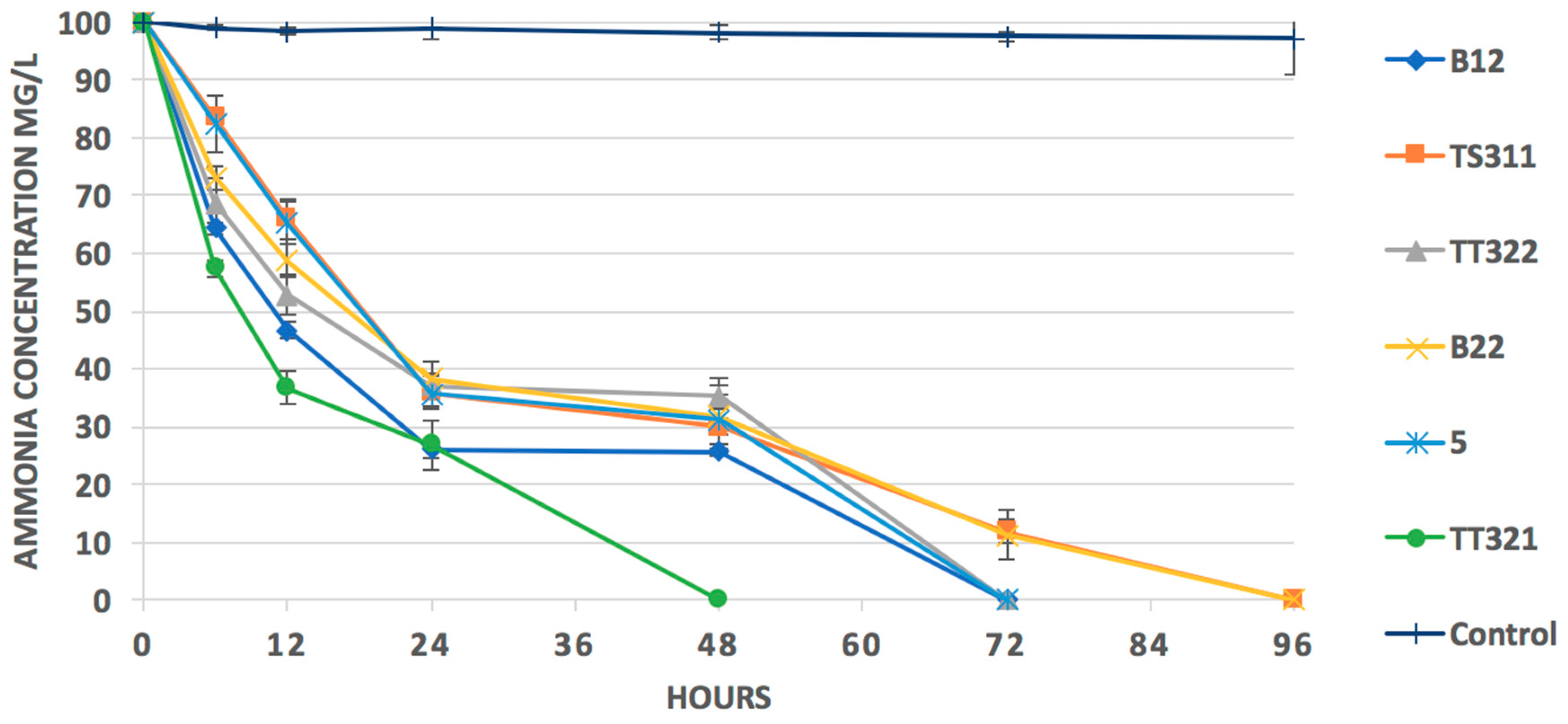
3.5. Isolation, Identification, and Heterotrophic Nitrifying Activity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lakshmi, B.; Viswanath, B.; Sai Gopal, D.V.R. Probiotics as Antiviral Agents in Shrimp Aquaculture. J. Pathog. 2013, 13. [Google Scholar] [CrossRef]
- FAO. The State of World Fisheries and Aquaculture; Food and Agriculture Organization: Rome, Italy, 2012; p. 209. [Google Scholar]
- Hopkins, J.S.; Sandifer, P.A.; DeVoe, M.R.; Holland, A.F.; Browdy, C.L.; Stokes, A.D. Environmental Impacts of Shrimp Farming with Special Reference to the Situation in the Continental United States. Estuaries 1995, 18, 25–42. [Google Scholar] [CrossRef]
- Miranda-Filho, K.C.; Pinho, G.L.; Wasielesky, W., Jr.; Bianchini, A. Long-term ammonia toxicity to the pink-shrimp Farfantepenaeus paulensis. Comp. Biochem. Physiol. Toxicol. Pharmacol. 2009, 150, 377–382. [Google Scholar] [CrossRef]
- Sahu, M.K.; Swarnakumar, N.S.; Sivakumar, K.; Thangaradjou, T.; Kannan, L. Probiotics in aquaculture: importance and future perspectives. Indian J. Microbiol. 2008, 48, 299–308. [Google Scholar] [CrossRef]
- Gong, Y.; Tang, X.; Shao, K.; Hu, Y.; Gao, G. Dynamics of bacterial abundance and the related environmental factors in large shallow eutrophic Lake Taihu. J. Freshw. Ecol. 2017, 32, 133–145. [Google Scholar] [CrossRef]
- Kingdom, T.; Hart, A.; Erondu, E. Effects of environmental factors on the abundance of shrimps, Macrobrachium species in the lower Taylor Creek, Niger Delta, Nigeria. Afr. J. Environ. Pollut. Health 2013, 10, 71–79. [Google Scholar]
- Tang, Y.; Tao, P.; Tan, J.; Mu, H.; Peng, L.; Yang, D.; Tong, S.; Chen, L. Identification of bacterial community composition in freshwater aquaculture system farming of Litopenaeus vannamei reveals distinct temperature-driven patterns. Int. J. Mol. Sci. 2014, 15, 13663–13680. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ke, X.; Hernández, M.; Wang, B.; Dumont, M.G.; Jia, Z.; Conrad, R. Autotrophic Growth of Bacterial and Archaeal Ammonia Oxidizers in Freshwater Sediment Microcosms Incubated at Different Temperatures. Appl. Environ. Microbiol. 2013, 79, 3076. [Google Scholar] [CrossRef]
- Norris, R.D. Handbook of Bioremediation; Taylor & Francis: Abingdon, UK, 1993; pp. 13–239. [Google Scholar]
- Sombatjinda, S.; Boonapatcharoen, N.; Ruengjitchatchawalya, M.; Wantawin, C.; Withyachumnarnkul, B.; Techkarnjanaruk, S. Dynamics of Microbial Communities in an Earthen Shrimp Pond during the Shrimp Growing Period. Environ. Nat. Resour. Res. 2011, 1, 171–180. [Google Scholar] [CrossRef]
- Sandifer, P.A.; Hopkins, J.S. Conceptual design of a sustainable pond-based shrimp culture system. Aquac. Eng. 1996, 15, 41–52. [Google Scholar] [CrossRef]
- Cardona, E.; Gueguen, Y.; Magré, K.; Lorgeoux, B.; Piquemal, D.; Pierrat, F.; Noguier, F.; Saulnier, D. Bacterial community characterization of water and intestine of the shrimp Litopenaeus stylirostris in a biofloc system. BMC Microbiol. 2016, 16, 157. [Google Scholar] [CrossRef]
- Garcia, Y.T.; Aragon, C.T.; Dator, M.A.L. Milkfish Bibliography A Compilation of Abstracts on Milkfish Studies; WorldFish: Penang, Malaysia, 2007; pp. 1–283. [Google Scholar]
- Gerardi, M.H. Nitrification and Denitrification in the Activated Sludge Process; John Wiley & Sons: Hoboken, NJ, USA, 2003; pp. 1–167. [Google Scholar]
- Cottrell, M.T.; Kirchman, D.L. Community composition of marine bacterioplankton determined by 16S rRNA gene clone libraries and fluorescence in situ hybridization. Appl. Environ. Microbiol. 2000, 66, 5116–5122. [Google Scholar] [CrossRef]
- Revetta, R.P.; Matlib, R.S.; Santo Domingo, J.W. 16S rRNA gene sequence analysis of drinking water using RNA and DNA extracts as targets for clone library development. Curr. Microbiol. 2011, 63, 50–59. [Google Scholar] [CrossRef]
- Zhang, R.; Wu, Q.; Piceno, Y.M.; Desantis, T.Z.; Saunders, F.M.; Andersen, G.L.; Liu, W.T. Diversity of bacterioplankton in contrasting Tibetan lakes revealed by high-density microarray and clone library analysis. FEMS Microbiol. Ecol. 2013, 86, 277–287. [Google Scholar] [CrossRef][Green Version]
- Bekaert, K.; Devriese, L.; Maes, S.; Robbens, J. Characterization of the dominant bacterial communities during storage of Norway lobster and Norway lobster tails (Nephrops norvegicus) based on 16S rDNA analysis by PCR-DGGE. Food Microbiol. 2015, 46, 132–138. [Google Scholar] [CrossRef]
- Zhong, F.; Wu, J.; Dai, Y.; Yang, L.; Zhang, Z.; Cheng, S.; Zhang, Q. Bacterial community analysis by PCR-DGGE and 454-pyrosequencing of horizontal subsurface flow constructed wetlands with front aeration. Appl. Microbiol. Biotechnol. 2015, 99, 1499–1512. [Google Scholar] [CrossRef]
- Qin, Y.; Hou, J.; Deng, M.; Liu, Q.; Wu, C.; Ji, Y.; He, X. Bacterial abundance and diversity in pond water supplied with different feeds. Sci. Rep. 2016, 6, 35232. [Google Scholar] [CrossRef]
- Liu, H.; Guo, X.; Gooneratne, R.; Lai, R.; Zeng, C.; Zhan, F.; Wang, W. The gut microbiome and degradation enzyme activity of wild freshwater fishes influenced by their trophic levels. Sci. Rep. 2016, 6, 24340. [Google Scholar] [CrossRef]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814. [Google Scholar] [CrossRef]
- Pal, R.R.; Khardenavis, A.A.; Purohit, H.J. Identification and monitoring of nitrification and denitrification genes in Klebsiella pneumoniae EGD-HP19-C for its ability to perform heterotrophic nitrification and aerobic denitrification. Funct. Integr. Genom. 2015, 15, 63–76. [Google Scholar] [CrossRef]
- Koeppel, A.F.; Wu, M. Surprisingly extensive mixed phylogenetic and ecological signals among bacterial Operational Taxonomic Units. Nucleic Acids Res. 2013, 41, 5175–5188. [Google Scholar] [CrossRef]
- Chao, A. Nonparametric Estimation of the Number of Classes in a Population. Scand. J. Stat. 1984, 11, 265–270. [Google Scholar]
- Magurran, A.E. Measuring Biological Diversity; Wiley: Hoboken, NJ, USA, 2013. [Google Scholar]
- Bryanskaya, A.V.; Malup, T.K.; Lazareva, E.V.; Taran, O.P.; Rozanov, A.S.; Efimov, V.M.; Peltek, S.E. The role of environmental factors for the composition of microbial communities of saline lakes in the Novosibirsk region (Russia). BMC Microbiol. 2016, 16. [Google Scholar] [CrossRef]
- Kaevska, M.; Videnska, P.; Sedlar, K.; Slana, I. Seasonal changes in microbial community composition in river water studied using 454-pyrosequencing. SpringerPlus 2016, 5, 409. [Google Scholar] [CrossRef]
- Zhang, Y.; Ruan, X.; Wan, Y.; Li, X. Effects of Environmental Factors on Anammox Bacterial Community Structure in Sediments of a Freshwater Aquaculture Farm, Yangcheng Lake. Geomicrobiol. J. 2016, 33, 479–487. [Google Scholar] [CrossRef]
- Xu, Y.; He, T.; Li, Z.; Ye, Q.; Chen, Y.; Xie, E.; Zhang, X. Nitrogen Removal Characteristics of Pseudomonas putida Y-9 Capable of Heterotrophic Nitrification and Aerobic Denitrification at Low Temperature. BioMed Res. Int. 2017, 2017, 1429018. [Google Scholar] [CrossRef]
- Wang, J.; Gong, B.; Wang, Y.; Wen, Y.; Zhou, J.; He, Q. The potential multiple mechanisms and microbial communities in simultaneous nitrification and denitrification process treating high carbon and nitrogen concentration saline wastewater. Bioresour. Technol. 2017, 243, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Liu, T.; Liu, G.; Zhou, J.; Huang, J.; Wang, A. Simultaneous heterotrophic nitrification and aerobic denitrification by the marine origin bacterium Pseudomonas sp. ADN-42. Appl. Biochem. Biotechnol. 2015, 175, 2000–2011. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Ren, Y.X.; Yang, L.; Zhao, S.Q.; Xia, Z.H. [Characteristics of Nitrogen Removal by a Heterotrophic Nitrification-Aerobic Denitrification Bacterium YL]. Huanjing kexue 2015, 36, 1749–1756. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Ace | Chao1 | Observed OTUs | Shannon | Simpson |
|---|---|---|---|---|---|
| ST3 | 1343.40 | 1358.76 | 1049.00 | 4.47 | 0.86 |
| ST1 | 1644.29 | 1664.06 | 1341.00 | 5.54 | 0.95 |
| ST4 | 1778.10 | 1772.69 | 1363.00 | 5.05 | 0.92 |
| ST-PM | 1861.68 | 1848.19 | 1426.00 | 5.04 | 0.93 |
| Parameters | Pearson Correlation Formula | |
|---|---|---|
| r | p | |
| pH | −0.88 | 0.12 |
| Temperature | 0.99 | 0.015 |
| BOD | −0.17 | 0.83 |
| COD | −0.69 | 0.31 |
| Chlorophyll-a | 0.19 | 0.81 |
| TOM | −0.67 | 0.33 |
| Total N | −0.6 | 0.4 |
| Total P | −0.53 | 0.47 |
| NO3-N | 0.097 | 0.9 |
| PO4-P | −0.58 | 0.42 |
| TAN | −0.97 | 0.025 |
| TDS | 0.59 | 0.41 |
| TSS | −0.86 | 0.14 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trung Tran, T.; Bott, N.J.; Dai Lam, N.; Trung Nguyen, N.; Hoang Thi Dang, O.; Hoang Le, D.; Tung Le, L.; Hoang Chu, H. The Role of Pseudomonas in Heterotrophic Nitrification: A Case Study on Shrimp Ponds (Litopenaeus vannamei) in Soc Trang Province. Microorganisms 2019, 7, 155. https://doi.org/10.3390/microorganisms7060155
Trung Tran T, Bott NJ, Dai Lam N, Trung Nguyen N, Hoang Thi Dang O, Hoang Le D, Tung Le L, Hoang Chu H. The Role of Pseudomonas in Heterotrophic Nitrification: A Case Study on Shrimp Ponds (Litopenaeus vannamei) in Soc Trang Province. Microorganisms. 2019; 7(6):155. https://doi.org/10.3390/microorganisms7060155
Chicago/Turabian StyleTrung Tran, Thanh, Nathan J. Bott, Nhan Dai Lam, Nam Trung Nguyen, Oanh Hoang Thi Dang, Duc Hoang Le, Lam Tung Le, and Ha Hoang Chu. 2019. "The Role of Pseudomonas in Heterotrophic Nitrification: A Case Study on Shrimp Ponds (Litopenaeus vannamei) in Soc Trang Province" Microorganisms 7, no. 6: 155. https://doi.org/10.3390/microorganisms7060155
APA StyleTrung Tran, T., Bott, N. J., Dai Lam, N., Trung Nguyen, N., Hoang Thi Dang, O., Hoang Le, D., Tung Le, L., & Hoang Chu, H. (2019). The Role of Pseudomonas in Heterotrophic Nitrification: A Case Study on Shrimp Ponds (Litopenaeus vannamei) in Soc Trang Province. Microorganisms, 7(6), 155. https://doi.org/10.3390/microorganisms7060155