Defining Kinetic Properties of HIV-Specific CD8+ T-Cell Responses in Acute Infection



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Experimental Data
2.2. Mathematical Model of CD8 T-Cell Response to a Viral Infection
2.3. Statistics
2.4. Ethics Statement
2.5. Competing Interests Statement
3. Results
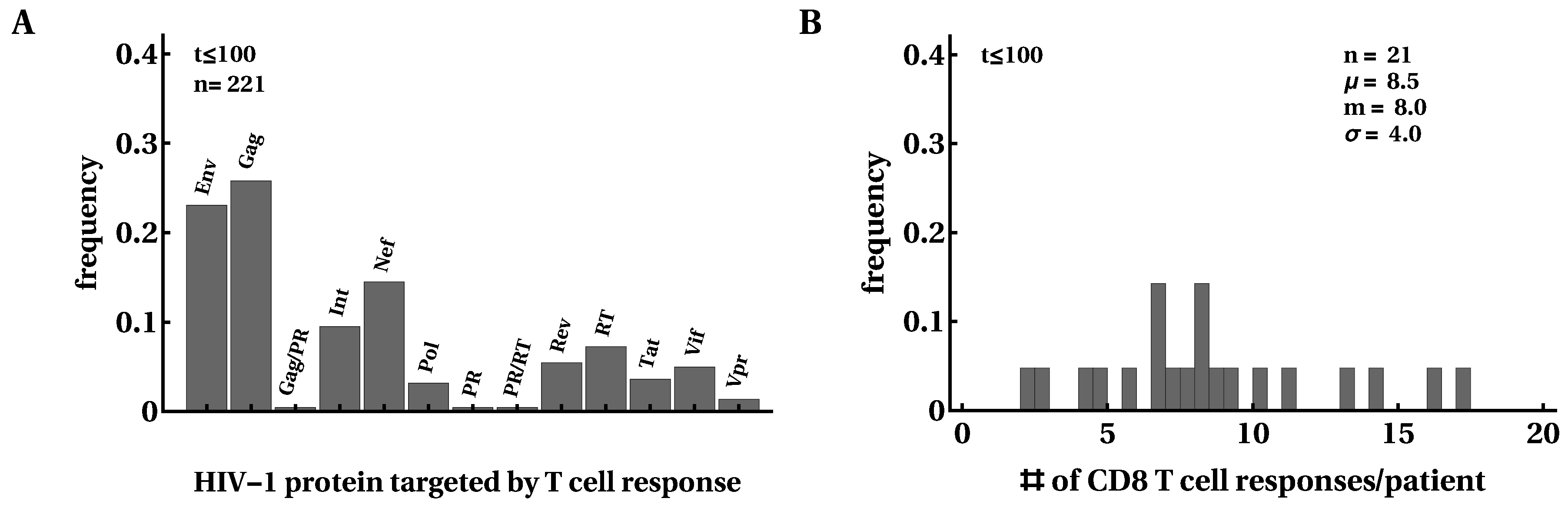
3.1. Moderate Changes in the Breadth of HIV-Specific CD8 T-Cell Response over the Course of Infection
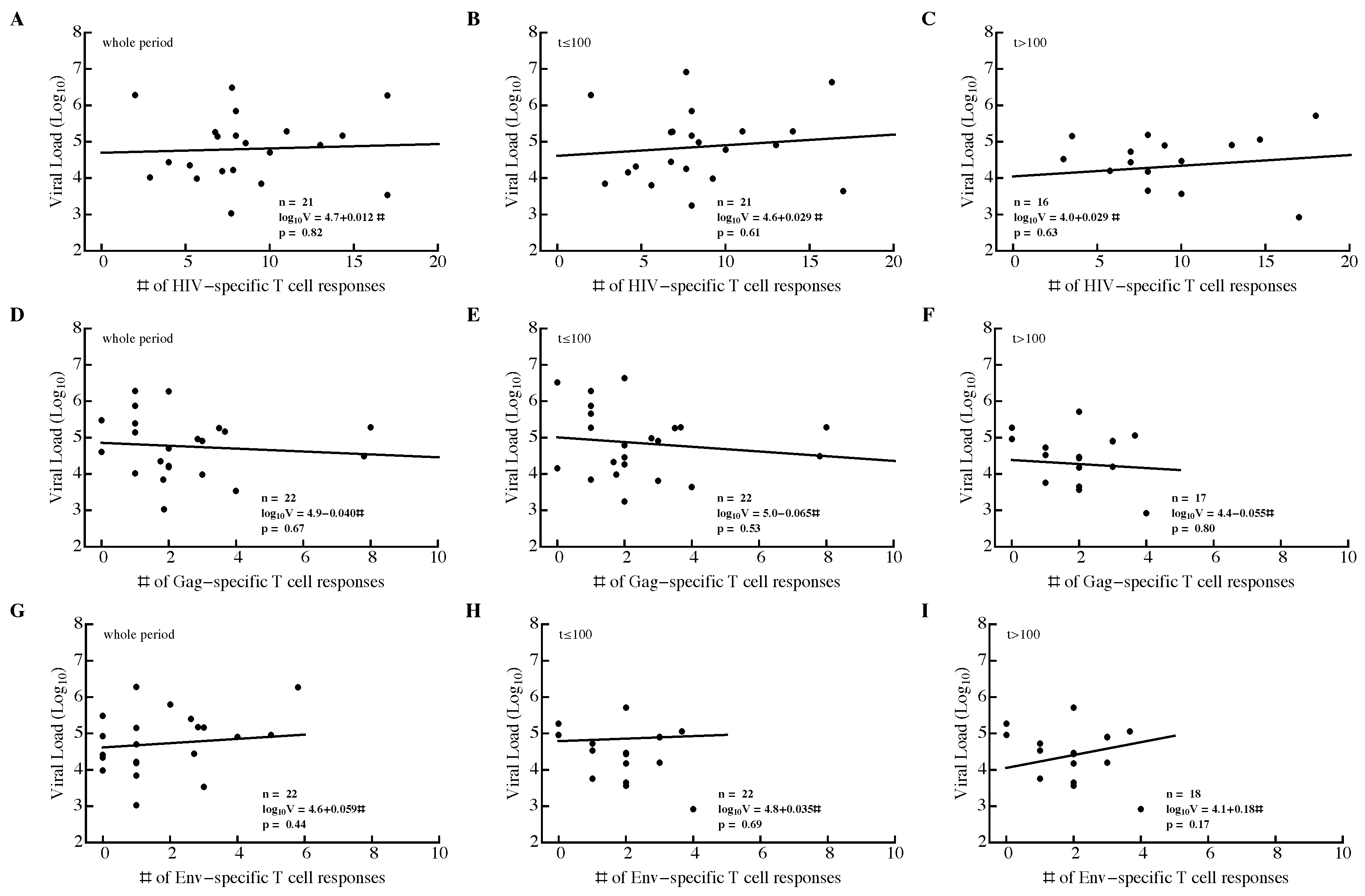
3.2. Variable Correlations between Immune Response Breadth and Viral Load
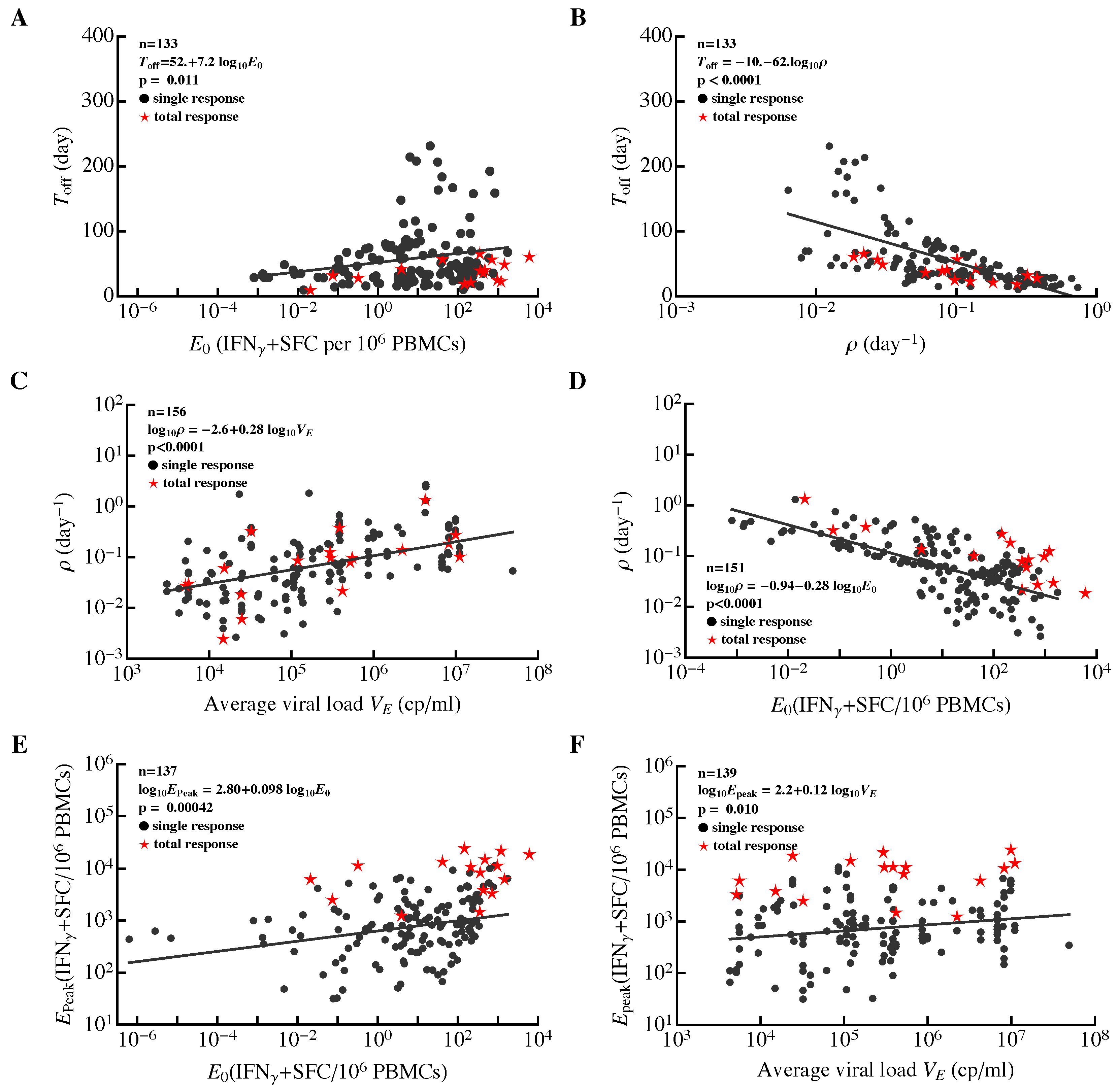
3.3. Most HIV-Specific CD8 T-Cell Responses Expand Slowly and Peak Early
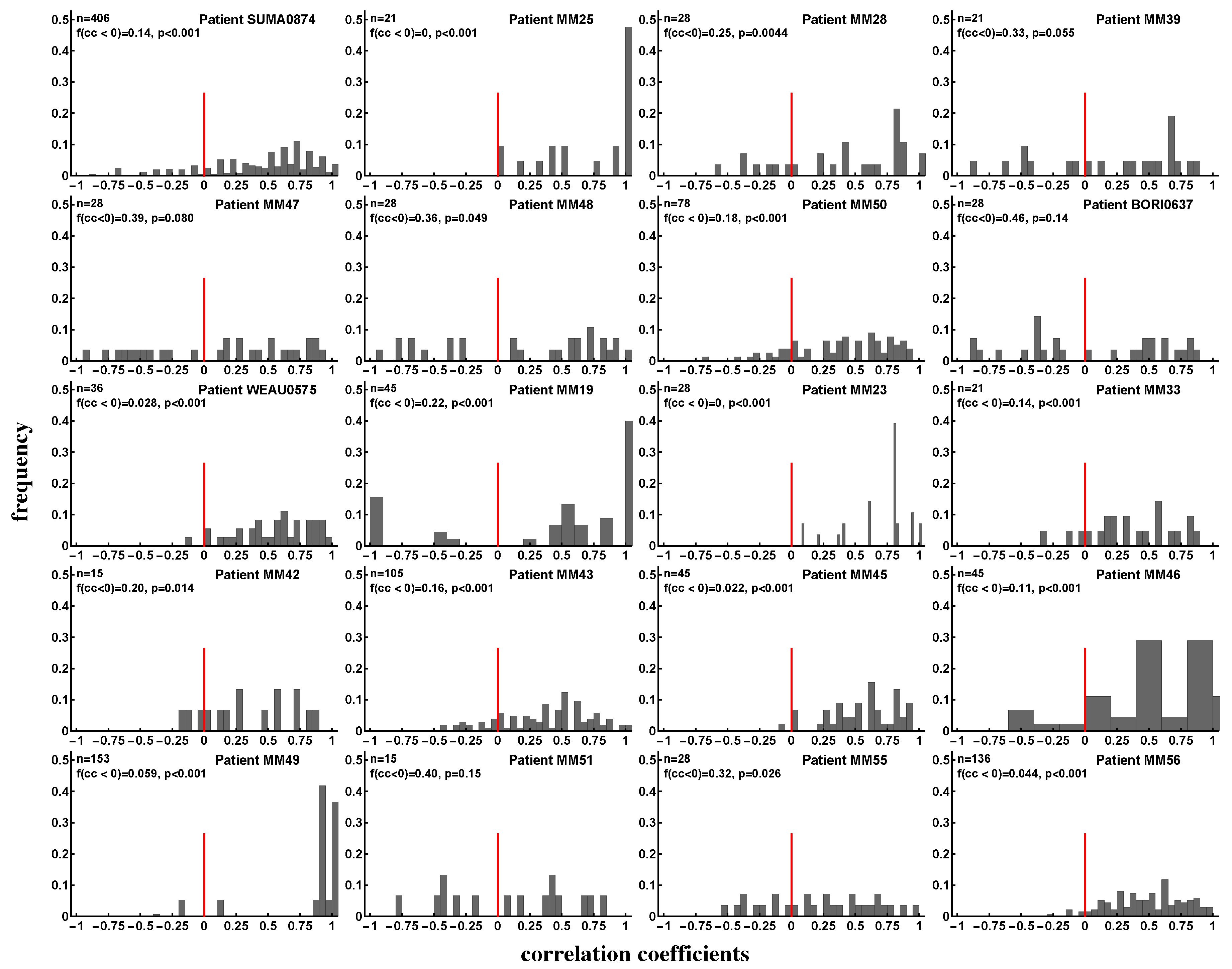
3.4. Evidence of Intraclonal Competition of CD8 T Cells
3.5. Evidence of Interclonal Competition of CD8 T Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CTL | cytotoxic T lymphocyte |
| HIV | human immunodeficiency virus |
| SE | Shannon entropy |
| EI | Evenness index |
| PBMC | peripheral blood mononuclear cells |
| SFC | spot-forming cells |
| IFN | interferon |
References
- Demberg, T.; Robert-Guroff, M. Controlling the HIV/AIDS epidemic: Current status and global challenges. Front. Immunol. 2012, 3, 250. [Google Scholar] [CrossRef] [PubMed]
- Maartens, G.; Celum, C.; Lewin, S.R. HIV infection: Epidemiology, pathogenesis, treatment, and prevention. Lancet 2014, 384, 258–271. [Google Scholar] [CrossRef]
- Uberla, K. HIV vaccine development in the aftermath of the STEP study: Re-focus on occult HIV infection? PLoS Pathog. 2008, 4, e1000114. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J. HIV/AIDS research. Beyond Thailand: Making sense of a qualified AIDS vaccine “success”. Science 2009, 326, 652–653. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, J.D.; Sobieszczyk, M.E.; Hammer, S.M.; Buchbinder, S.P. Lessons drawn from recent HIV vaccine efficacy trials. J. Acquir. Immune Defic. Syndr. 2010, 55 (Suppl. 2), S128–S131. [Google Scholar] [CrossRef] [PubMed]
- Barouch, D.; Santra, S.; Schmitz, J.; Kuroda, M.; Fu, T.; Wagner, W.; Bilska, M.; Craiu, A.; Zheng, X.; Krivulka, G.; et al. Control of viremia and prevention of clinical AIDS in rhesus monkeys by cytokine-augmented DNA vaccination. Science 2000, 290, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Shiver, J.W.; Fu, T.M.; Chen, L.; Casimiro, D.R.; Davies, M.E.; Evans, R.K.; Zhang, Z.Q.; Simon, A.J.; Trigona, W.L.; Dubey, S.A.; et al. Replication-incompetent adenoviral vaccine vector elicits effective anti-immunodeficiency-virus immunity. Nature 2002, 415, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Watkins, D.I.; Burton, D.R.; Kallas, E.G.; Moore, J.P.; Koff, W.C. Nonhuman primate models and the failure of the Merck HIV-1 vaccine in humans. Nat. Med. 2008, 14, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Watkins, D.I. The hope for an HIV vaccine based on induction of CD8+ T lymphocytes—A review. Memórias Inst. Oswaldo Cruz 2008, 103, 119–129. [Google Scholar] [CrossRef]
- Haynes, B.F.; Shaw, G.M.; Korber, B.; Kelsoe, G.; Sodroski, J.; Hahn, B.H.; Borrow, P.; McMichael, A.J. HIV-Host Interactions: Implications for Vaccine Design. Cell Host Microbe 2016, 19, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Mascola, J.R.; Haynes, B.F. HIV-1 neutralizing antibodies: Understanding nature’s pathways. Immunol. Rev. 2013, 254, 225–244. [Google Scholar] [CrossRef] [PubMed]
- Barouch, D.H.; Picker, L.J. Novel vaccine vectors for HIV-1. Nat. Rev. Microbiol. 2014, 12, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Haynes, B.F. New approaches to HIV vaccine development. Curr. Opin. Immunol. 2015, 35, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Sadanand, S.; Suscovich, T.J.; Alter, G. Broadly Neutralizing Antibodies Against HIV: New Insights to Inform Vaccine Design. Annu. Rev. Med. 2016, 67, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Makedonas, G.; Betts, M.R. Living in a house of cards: Re-evaluating CD8+ T-cell immune correlates against HIV. Immunol. Rev. 2011, 239, 109–124. [Google Scholar] [CrossRef] [PubMed]
- McMichael, A.J.; Borrow, P.; Tomaras, G.D.; Goonetilleke, N.; Haynes, B.F. The immune response during acute HIV-1 infection: Clues for vaccine development. Nat. Rev. Immunol. 2010, 10, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Hersperger, A.R.; Migueles, S.A.; Betts, M.R.; Connors, M. Qualitative features of the HIV-specific CD8+ T-cell response associated with immunologic control. Curr. Opin. HIV AIDS 2011, 6, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Demers, K.R.; Reuter, M.A.; Betts, M.R. CD8(+) T-cell effector function and transcriptional regulation during HIV pathogenesis. Immunol. Rev. 2013, 254, 190–206. [Google Scholar] [CrossRef] [PubMed]
- Borrow, P.; Lewicki, H.; Hahn, B.H.; Shaw, G.M.; Oldstone, M.B. Virus-specific CD8+ cytotoxic T-lymphocyte activity associated with control of viremia in primary human immunodeficiency virus type 1 infection. J. Virol. 1994, 68, 6103–6110. [Google Scholar] [PubMed]
- Koup, R.A.; Safrit, J.T.; Cao, Y.; Andrews, C.A.; McLeod, G.; Borkowsky, W.; Farthing, C.; Ho, D.D. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J. Virol. 1994, 68, 4650–4655. [Google Scholar] [PubMed]
- Abdel-Motal, U.M.; Gillis, J.; Manson, K.; Wyand, M.; Montefiori, D.; Stefano-Cole, K.; Montelaro, R.C.; Altman, J.D.; Johnson, R.P. Kinetics of expansion of SIV Gag-specific CD8+ T lymphocytes following challenge of vaccinated macaques. Virology 2005, 333, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Newberg, M.H.; McEvers, K.J.; Gorgone, D.A.; Lifton, M.A.; Baumeister, S.H.; Veazey, R.S.; Schmitz, J.E.; Letvin, N.L. Immunodomination in the evolution of dominant epitope-specific CD8+ T lymphocyte responses in simian immunodeficiency virus-infected rhesus monkeys. J. Immunol. 2006, 176, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Carrington, M.; Nelson, G.W.; Martin, M.P.; Kissner, T.; Vlahov, D.; Goedert, J.J.; Kaslow, R.; Buchbinder, S.; Hoots, K.; O’Brien, S.J. HLA and HIV-1: Heterozygote advantage and B*35-Cw*04 disadvantage. Science 1999, 283, 1748–1752. [Google Scholar] [CrossRef] [PubMed]
- Carrington, M.; O’Brien, S. The influence of HLA genotype on AIDS. Annu. Rev. Med. 2003, 54, 535–551. [Google Scholar] [CrossRef] [PubMed]
- McLaren, P.J.; Carrington, M. The impact of host genetic variation on infection with HIV-1. Nat. Immunol. 2015, 16, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Goulder, P.; Watkins, D. HIV and SIV CTL escape: Implications for vaccine design. Nat. Rev. Immunol. 2004, 4, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Ogg, G.S.; Jin, X.; Bonhoeffer, S.; Dunbar, P.R.; Nowak, M.A.; Monard, S.; Segal, J.P.; Cao, Y.; Rowland-Jones, S.L.; Cerundolo, V.; et al. Quantitation of HIV-1-specific cytotoxic T lymphocytes and plasma load of viral RNA. Science 1998, 279, 2103–2106. [Google Scholar] [CrossRef] [PubMed]
- Kalams, S.A.; Buchbinder, S.P.; Rosenberg, E.S.; Billingsley, J.M.; Colbert, D.S.; Jones, N.G.; Shea, A.K.; Trocha, A.K.; Walker, B.D. Association between virus-specific cytotoxic T-lymphocyte and helper responses in human immunodeficiency virus type 1 infection. J. Virol. 1999, 73, 6715–6720. [Google Scholar] [PubMed]
- Betts, M.; Ambrozak, D.; Douek, D.; Bonhoeffer, S.; Brenchley, J.; Casazza, J.; Koup, R.; Picker, L. Analysis of total human immunodeficiency virus (HIV)-specific CD4(+) and CD8(+) T-cell responses: Relationship to viral load in untreated HIV infection. J. Virol. 2001, 75, 11983–11991. [Google Scholar] [CrossRef] [PubMed]
- Novitsky, V.; Gilbert, P.; Peter, T.; McLane, M.F.; Gaolekwe, S.; Rybak, N.; Thior, I.; Ndungu, T.; Marlink, R.; Lee, T.H.; et al. Association between Virus-Specific T-Cell Responses and Plasma Viral Load in Human Immunodeficiency Virus Type 1 Subtype C Infection. J. Virol. 2003, 77, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Day, C.L.; Kiepiela, P.; Leslie, A.J.; van der Stok, M.; Nair, K.; Ismail, N.; Honeyborne, I.; Crawford, H.; Coovadia, H.M.; Goulder, P.J.; et al. Proliferative capacity of epitope-specific CD8 T-cell responses is inversely related to viral load in chronic human immunodeficiency virus type 1 infection. J. Virol. 2007, 81, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Gray, C.M.; Mlotshwa, M.; Riou, C.; Mathebula, T.; de Assis Rosa, D.; Mashishi, T.; Seoighe, C.; Ngandu, N.; van Loggerenberg, F.; Morris, L.; et al. Human immunodeficiency virus-specific gamma interferon enzyme-linked immunospot assay responses targeting specific regions of the proteome during primary subtype C infection are poor predictors of the course of viremia and set point. J. Virol. 2009, 83, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Kiepiela, P.; Ngumbela, K.; Thobakgale, C.; Ramduth, D.; Honeyborne, I.; Moodley, E.; Reddy, S.; de Pierres, C.; Mncube, Z.; Mkhwanazi, N.; et al. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat. Med. 2007, 13, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Geldmacher, C.; Currier, J.R.; Herrmann, E.; Haule, A.; Kuta, E.; McCutchan, F.; Njovu, L.; Geis, S.; Hoffmann, O.; Maboko, L.; et al. CD8 T-cell recognition of multiple epitopes within specific Gag regions is associated with maintenance of a low steady-state viremia in human immunodeficiency virus type 1-seropositive patients. J. Virol. 2007, 81, 2440–2448. [Google Scholar] [CrossRef] [PubMed]
- Rolland, M.; Heckerman, D.; Deng, W.; Rousseau, C.M.; Coovadia, H.; Bishop, K.; Goulder, P.J.R.; Walker, B.D.; Brander, C.; Mullins, J.I. Broad and Gag-biased HIV-1 epitope repertoires are associated with lower viral loads. PLoS ONE 2008, 3, e1424. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.A.; Ibarrondo, F.J.; Sugar, C.A.; Hausner, M.A.; Shih, R.; Ng, H.L.; Detels, R.; Margolick, J.B.; Rinaldo, C.R.; Phair, J.; et al. Early HLA-B*57-restricted CD8+ T lymphocyte responses predict HIV-1 disease progression. J. Virol. 2012, 86, 10505–10516. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Bauer, D.E.; Tuttleton, S.E.; Lewin, S.; Gettie, A.; Blanchard, J.; Irwin, C.E.; Safrit, J.T.; Mittler, J.; Weinberger, L.; et al. Dramatic rise in plasma viremia after CD8(+) T cell depletion in simian immunodeficiency virus-infected macaques. J. Exp. Med. 1999, 189, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.E.; Kuroda, M.J.; Santra, S.; Sasseville, V.G.; Simon, M.A.; Lifton, M.A.; Racz, P.; Tenner-Racz, K.; Dalesandro, M.; Scallon, B.J.; et al. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science 1999, 283, 857–860. [Google Scholar] [CrossRef] [PubMed]
- Klatt, N.R.; Shudo, E.; Ortiz, A.M.; Engram, J.C.; Paiardini, M.; Lawson, B.; Miller, M.D.; Else, J.; Pandrea, I.; Estes, J.D.; et al. CD8+ lymphocytes control viral replication in SIVmac239-infected rhesus macaques without decreasing the lifespan of productively infected cells. PLoS Pathog. 2010, 6, e1000747. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.K.; Strain, M.C.; Porrata, R.; Reay, E.; Sankaran-Walters, S.; Ignacio, C.C.; Russell, T.; Pillai, S.K.; Looney, D.J.; Dandekar, S. In vivo CD8+ T-cell suppression of SIV viremia is not mediated by CTL clearance of productively infected cells. PLoS Pathog. 2010, 6, e1000748. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.G.; Ford, J.C.; Lewis, M.S.; Ventura, A.B.; Hughes, C.M.; Coyne-Johnson, L.; Whizin, N.; Oswald, K.; Shoemaker, R.; Swanson, T.; et al. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 2011, 473, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, K.E.; Li, H.; Walker, B.D.; Michael, N.L.; Barouch, D.H. Gag-specific cellular immunity determines in vitro viral inhibition and in vivo virologic control following simian immunodeficiency virus challenges of vaccinated rhesus monkeys. J. Virol. 2012, 86, 9583–9589. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.G.; Piatak, M.; Ventura, A.B.; Hughes, C.M.; Gilbride, R.M.; Ford, J.C.; Oswald, K.; Shoemaker, R.; Li, Y.; Lewis, M.S.; et al. Immune clearance of highly pathogenic SIV infection. Nature 2013, 502, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, N.; Takahashi, N.; Seki, S.; Nomura, T.; Yamamoto, H.; Inoue, M.; Shu, T.; Naruse, T.K.; Kimura, A.; Matano, T. Control of simian immunodeficiency virus replication by vaccine-induced Gag- and Vif-specific CD8+ T cells. J. Virol. 2014, 88, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.S.; Shaw, G.M.; McMichael, A.J.; Haynes, B.F. Acute HIV-1 Infection. N. Engl. J. Med. 2011, 364, 1943–1954. [Google Scholar] [CrossRef] [PubMed]
- Migueles, S.A.; Connors, M. Success and failure of the cellular immune response against HIV-1. Nat. Immunol. 2015, 16, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Migueles, S.; Laborico, A.; Shupert, W.; Sabbaghian, M.; Rabin, R.; Hallahan, C.; Van Baarle, D.; Kostense, S.; Miedema, F.; McLaughlin, M.; et al. HIV-specific CD8+ T cell proliferation is coupled to perforin expression and is maintained in nonprogressors. Nat. Immunol. 2002, 3, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.D. Elite control of HIV Infection: Implications for vaccines and treatment. Top. HIV Med. 2007, 15, 134–136. [Google Scholar] [PubMed]
- Lobritz, M.A.; Lassen, K.G.; Arts, E.J. HIV-1 replicative fitness in elite controllers. Curr. Opin. HIV AIDS 2011, 6, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Zaunders, J.; Dyer, W.B.; Churchill, M. The Sydney Blood Bank Cohort: Implications for viral fitness as a cause of elite control. Curr. Opin. HIV AIDS 2011, 6, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Poropatich, K.; Sullivan, D.J. Human immunodeficiency virus type 1 long-term non-progressors: The viral, genetic and immunological basis for disease non-progression. J. Gen. Virol. 2011, 92, 247–268. [Google Scholar] [CrossRef] [PubMed]
- Goulder, P.J.; Walker, B.D. HIV and HLA class I: An evolving relationship. Immunity 2012, 37, 426–440. [Google Scholar] [CrossRef] [PubMed]
- Kløverpris, H.N.; Leslie, A.; Goulder, P. Role of HLA adaptation in HIV evolution. Front. Immunol. 2015, 6, 665. [Google Scholar] [CrossRef] [PubMed]
- Goonetilleke, N.; Liu, M.K.; Salazar-Gonzalez, J.F.; Ferrari, G.; Giorgi, E.; Ganusov, V.V.; Keele, B.F.; Learn, G.H.; Turnbull, E.L.; Salazar, M.G.; et al. The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. J. Exp. Med. 2009, 206, 1253–1272. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, E.L.; Wong, M.; Wang, S.; Wei, X.; Jones, N.A.; Conrod, K.E.; Aldam, D.; Turner, J.; Pellegrino, P.; Keele, B.F.; et al. Kinetics of expansion of epitope-specific T cell responses during primary HIV-1 infection. J. Immunol. 2009, 182, 7131–7145. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.K.P.; Hawkins, N.; Ritchie, A.J.; Ganusov, V.V.; Whale, V.; Brackenridge, S.; Li, H.; Pavlicek, J.W.; Cai, F.; Rose-Abrahams, M.; et al. Vertical T cell immunodominance and epitope entropy determine HIV-1 escape. J. Clin. Investig. 2013, 123, 380–393. [Google Scholar] [CrossRef] [PubMed]
- Riou, C.; Ganusov, V.V.; Campion, S.; Mlotshwa, M.; Liu, M.K.P.; Whale, V.E.; Goonetilleke, N.; Borrow, P.; Ferrari, G.; Betts, M.R.; et al. Distinct kinetics of Gag-specific CD4(+) and CD8(+) T cell responses during acute HIV-1 infection. J. Immunol. 2012, 188, 2198–2206. [Google Scholar] [CrossRef] [PubMed]
- Yue, L.; Pfafferott, K.J.; Baalwa, J.; Conrod, K.; Dong, C.C.; Chui, C.; Rong, R.; Claiborne, D.T.; Prince, J.L.; Tang, J.; et al. Transmitted virus fitness and host T cell responses collectively define divergent infection outcomes in two HIV-1 recipients. PLoS Pathog. 2015, 11, e1004565. [Google Scholar] [CrossRef] [PubMed]
- Ndhlovu, Z.M.; Kamya, P.; Mewalal, N.; Klaverpris, H.N.; Nkosi, T.; Pretorius, K.; Laher, F.; Ogunshola, F.; Chopera, D.; Shekhar, K.; et al. Magnitude and Kinetics of CD8(+) T Cell Activation during Hyperacute HIV Infection Impact Viral Set Point. Immunity 2015, 43, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.; van der Most, R.; Akondy, R.; Glidewell, J.; Albott, S.; Masopust, D.; Murali-Krishna, K.; Mahar, P.; Edupuganti, S.; Lalor, S.; et al. Human effector and memory CD8+ T cell responses to smallpox and yellow fever vaccines. Immunity 2008, 28, 710–722. [Google Scholar] [CrossRef] [PubMed]
- Le, D.; Miller, J.D.; Ganusov, V.V. Mathematical modeling provides kinetic details of the human immune response to vaccination. Front. Cell. Infect. Microbiol. 2015, 4, 177. [Google Scholar] [CrossRef] [PubMed]
- Davenport, M.P.; Ribeiro, R.M.; Chao, D.L.; Perelson, A.S. Predicting the impact of a nonsterilizing vaccine against human immunodeficiency virus. J. Virol. 2004, 78, 11340–11351. [Google Scholar] [CrossRef] [PubMed]
- Davenport, M.P.; Ribeiro, R.M.; Perelson, A.S. Kinetics of virus-specific CD8+ T cells and the control of human immunodeficiency virus infection. J. Virol. 2004, 78, 10096–10103. [Google Scholar] [CrossRef] [PubMed]
- Althaus, C.L.; De Boer, R.J. Dynamics of immune escape during HIV/SIV infection. PLoS Comput. Biol. 2008, 4, e1000103. [Google Scholar] [CrossRef] [PubMed]
- Asquith, B.; Edwards, C.; Lipsitch, M.; McLean, A. Inefficient cytotoxic T lymphocyte-mediated killing of HIV-1-infected cells in vivo. PLoS Biol. 2006, 4, e90. [Google Scholar] [CrossRef] [PubMed]
- De Boer, R.J. Understanding the failure of CD8+ T-cell vaccination against simian/human immunodeficiency virus. J. Virol. 2007, 81, 2838–2848. [Google Scholar] [CrossRef] [PubMed]
- Ganusov, V.V.; De Boer, R.J. Estimating Costs and Benefits of CTL Escape Mutations in SIV/HIV Infection. PLoS Comput. Biol. 2006, 2, e24. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.A.; Bangham, C.R.M. Population dynamics of immune responses to persistent viruses. Science 1996, 272, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.A.; Maya, R.M.; Sigmund, K. Immune responses against multiple epitopes. J. Theor. Biol. 1995, 175, 325–353. [Google Scholar] [CrossRef] [PubMed]
- Martyushev, A.P.; Petravic, J.; Grimm, A.J.; Alinejad-Rokny, H.; Gooneratne, S.L.; Reece, J.C.; Cromer, D.; Kent, S.J.; Davenport, M.P. Epitope-specific CD8+ T cell kinetics rather than viral variability determine the timing of immune escape in simian immunodeficiency virus infection. J. Immunol. 2015, 194, 4112–4121. [Google Scholar] [CrossRef] [PubMed]
- van Deutekom, H.W.; Wijnker, G.; de Boer, R.J. The rate of immune escape vanishes when multiple immune responses control an HIV infection. J. Immunol. 2013, 191, 3277–3286. [Google Scholar] [CrossRef] [PubMed]
- van der Most, R.G.; Concepcion, R.J.; Oseroff, C.; Alexander, J.; Southwood, S.; Sidney, J.; Chesnut, R.W.; Ahmed, R.; Sette, A. Uncovering subdominant cytotoxic T-lymphocyte responses in lymphocytic choriomeningitis virus-infected BALB/c mice. J. Virol. 1997, 71, 5110–5114. [Google Scholar] [PubMed]
- Vijh, S.; Pilip, I.; Pamer, E. Noncompetitive Expansion of Cytotoxic T Lymphocytes Specific for Different Antigens during Bacterial Infection. Infect. Immun. 1999, 67, 1303–1309. [Google Scholar] [PubMed]
- Kedl, R.M.; Rees, W.A.; Hildeman, D.A.; Schaefer, B.; Mitchell, T.; Kappler, J.; Marrack, P. T cells compete for access to antigen-bearing antigen-presenting cells. J. Exp. Med. 2000, 192, 1105–1113. [Google Scholar] [CrossRef] [PubMed]
- Grayson, J.M.; Harrington, L.E.; Lanier, J.G.; Wherry, E.J.; Ahmed, R. Differential sensitivity of naive and memory CD8(+) T cells to apoptosis in vivo. J. Immunol. 2002, 169, 3760–3770. [Google Scholar] [CrossRef] [PubMed]
- Brehm, M.; Pinto, A.; Daniels, K.; Schneck, J.; Welsh, R.; Selin, L. T cell immunodominance and maintenance of memory regulated by unexpectedly cross-reactive pathogens. Nat. Immunol. 2002, 3, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Kedl, R.M.; Kappler, J.W.; Marrack, P. Epitope dominance, competition and T cell affinity maturation. Curr. Opin. Immunol. 2003, 15, 120–127. [Google Scholar] [CrossRef]
- Andreansky, S.S.; Stambas, J.; Thomas, P.G.; Xie, W.; Webby, R.J.; Doherty, P.C. Consequences of immunodominant epitope deletion for minor influenza virus-specific CD8+-T-cell responses. J. Virol. 2005, 79, 4329–4339. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, W.; Hedrick, S. Cutting edge: Latecomer CD8 T cells are imprinted with a unique differentiation program. J. Immunol. 2006, 177, 777–781. [Google Scholar] [CrossRef] [PubMed]
- Badovinac, V.; Haring, J.; Harty, J. Initial T cell receptor transgenic cell precursor frequency dictates critical aspects of the CD8(+) T cell response to infection. Immunity 2007, 26, 827–841. [Google Scholar] [CrossRef] [PubMed]
- Gruta, N.L.L.; Rothwell, W.T.; Cukalac, T.; Swan, N.G.; Valkenburg, S.A.; Kedzierska, K.; Thomas, P.G.; Doherty, P.C.; Turner, S.J. Primary CTL response magnitude in mice is determined by the extent of naive T cell recruitment and subsequent clonal expansion. J. Clin. Investig. 2010, 120, 1885–1894. [Google Scholar] [CrossRef] [PubMed]
- Farrington, L.A.; Smith, T.A.; Grey, F.; Hill, A.B.; Snyder, C.M. Competition for antigen at the level of the APC is a major determinant of immunodominance during memory inflation in murine cytomegalovirus infection. J. Immunol. 2013, 190, 3410–3416. [Google Scholar] [CrossRef] [PubMed]
- Fryer, H.R.; Scherer, A.; Oxenius, A.; Phillips, R.; McLean, A.R. No evidence for competition between cytotoxic T-lymphocyte responses in HIV-1 infection. Proc. Biol. Sci. 2009, 276, 4389–4397. [Google Scholar] [CrossRef] [PubMed]
- Robb, M.L.; Eller, L.A.; Kibuuka, H.; Rono, K.; Maganga, L.; Nitayaphan, S.; Kroon, E.; Sawe, F.K.; Sinei, S.; Sriplienchan, S.; et al. Prospective Study of Acute HIV-1 Infection in Adults in East Africa and Thailand. N. Engl. J. Med. 2016, 374, 2120–2130. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.A.; Wei, X.; Flower, D.R.; Wong, M.; Michor, F.; Saag, M.S.; Hahn, B.H.; Nowak, M.A.; Shaw, G.M.; Borrow, P. Determinants of human immunodeficiency virus type 1 escape from the primary CD8+ cytotoxic T lymphocyte response. J. Exp. Med. 2004, 200, 1243–1256. [Google Scholar] [CrossRef] [PubMed]
- De Boer, R.J.; Oprea, M.; Antia, R.; Murali-Krishna, K.; Ahmed, R.; Perelson, A.S. Recruitment times, proliferation, and apoptosis rates during the CD8(+) T-cell response to lymphocytic choriomeningitis virus. J. Virol. 2001, 75, 10663–10669. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, M.K.; Moon, J.J. The role of naive T cell precursor frequency and recruitment in dictating immune response magnitude. J. Immunol. 2012, 188, 4135–4140. [Google Scholar] [CrossRef] [PubMed]
- De Boer, R.J.; Homann, D.; Perelson, A.S. Different dynamics of CD4+ and CD8+ T cell responses during and after acute lymphocytic choriomeningitis virus infection. J. Immunol. 2003, 171, 3928–3935. [Google Scholar] [CrossRef] [PubMed]
- Barton, J.P.; Goonetilleke, N.; Butler, T.C.; Walker, B.D.; McMichael, A.J.; Chakraborty, A.K. Relative rate and location of intra-host HIV evolution to evade cellular immunity are predictable. Nat. Commun. 2016, 7, 11660. [Google Scholar] [CrossRef] [PubMed]
- Bodine, E.; Lenhart, S.; Gross, L. Mathematics for the Life Sciences; Princeton University Press: Princeton, NJ, USA, 2014. [Google Scholar]
- Haas, G.; Samri, A.; Gomard, E.; Hosmalin, A.; Duntze, J.; Bouley, J.M.; Ihlenfeldt, H.G.; Katlama, C.; Autran, B. Cytotoxic T-cell responses to HIV-1 reverse transcriptase, integrase and protease. AIDS 1998, 12, 1427–1436. [Google Scholar] [CrossRef] [PubMed]
- Radebe, M.; Gounder, K.; Mokgoro, M.; Ndhlovu, Z.M.; Mncube, Z.; Mkhize, L.; van der Stok, M.; Jaggernath, M.; Walker, B.D.; Ndung’u, T. Broad and persistent Gag-specific CD8+ T-cell responses are associated with viral control but rarely drive viral escape during primary HIV-1 infection. AIDS 2015, 29, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Radebe, M.; Nair, K.; Chonco, F.; Bishop, K.; Wright, J.K.; van der Stok, M.; Bassett, I.V.; Mncube, Z.; Altfeld, M.; Walker, B.D.; et al. Limited immunogenicity of HIV CD8+ T-cell epitopes in acute Clade C virus infection. J. Infect. Dis. 2011, 204, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Ganusov, V.V.; Goonetilleke, N.; Liu, M.K.; Ferrari, G.; Shaw, G.M.; McMichael, A.J.; Borrow, P.; Korber, B.T.; Perelson, A.S. Fitness costs and diversity of the cytotoxic T lymphocyte (CTL) response determine the rate of CTL escape during acute and chronic phases of HIV infection. J. Virol. 2011, 85, 10518–10528. [Google Scholar] [CrossRef] [PubMed]
- Ganusov, V.V.; Barber, D.L.; De Boer, R.J. Killing of targets by CD8 T cells in the mouse spleen follows the law of mass action. PLoS ONE 2011, 6, e15959. [Google Scholar] [CrossRef] [PubMed]
- Mellors, J.W.; Munoz, A.; Giorgi, J.V.; Margolick, J.B.; Tassoni, C.J.; Gupta, P.; Kingsley, L.A.; Todd, J.A.; Saah, A.J.; Detels, R.; et al. Plasma viral load and CD4+ lymphocytes as prognostic markers of HIV-1 infection. Ann. Intern. Med. 1997, 126, 946–954. [Google Scholar] [CrossRef] [PubMed]
- Mothe, B.; Llano, A.; Ibarrondo, J.; Daniels, M.; Miranda, C.; Zamarreño, J.; Bach, V.; Zuniga, R.; Pérez-Álvarez, S.; Berger, C.T.; et al. Definition of the viral targets of protective HIV-1-specific T cell responses. J. Transl. Med. 2011, 9, 208. [Google Scholar] [CrossRef] [PubMed]
- Geldmacher, C.; Gray, C.; Nason, M.; Currier, J.R.; Haule, A.; Njovu, L.; Geis, S.; Hoffmann, O.; Maboko, L.; Meyerhans, A.; et al. A high viral burden predicts the loss of CD8 T-cell responses specific for subdominant gag epitopes during chronic human immunodeficiency virus infection. J. Virol. 2007, 81, 13809–13815. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, R.M.; Qin, L.; Chavez, L.L.; Li, D.; Self, S.G.; Perelson, A.S. Estimation of the initial viral growth rate and basic reproductive number during acute HIV-1 infection. J. Virol. 2010, 84, 6096–6102. [Google Scholar] [CrossRef] [PubMed]
- Alanio, C.; Lemaitre, F.; Law, H.K.; Hasan, M.; Albert, M.L. Enumeration of human antigen-specific naive CD8+ T cells reveals conserved precursor frequencies. Blood 2010, 115, 3718–3725. [Google Scholar] [CrossRef] [PubMed]
- Murali-Krishna, K.; Altman, J.; Suresh, M.; Sourdive, D.; Zajac, A.; Miller, J.; Slansky, J.; Ahmed, R. Counting antigen-specific CD8+ T cells: A re-evaluation of bystander actiation during viral infection. Immunity 1998, 8, 177–187. [Google Scholar] [CrossRef]
- Homann, D.; Teyton, L.; Oldstone, M. Differential regulation of antiviral T-cell immunity results in stable CD8+ but declining CD4+ T-cell memory. Nat. Med. 2001, 7, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Obar, J.J.; Khanna, K.M.; Lefrancois, L. Endogenous naive CD8+ T cell precursor frequency regulates primary and memory responses to infection. Immunity 2008, 28, 859–869. [Google Scholar] [CrossRef] [PubMed]
- Akondy, R.S.; Johnson, P.L.F.; Nakaya, H.I.; Edupuganti, S.; Mulligan, M.J.; Lawson, B.; Miller, J.D.; Pulendran, B.; Antia, R.; Ahmed, R. Initial viral load determines the magnitude of the human CD8 T cell response to yellow fever vaccination. Proc. Natl. Acad. Sci. USA 2015, 112, 3050–3055. [Google Scholar] [CrossRef] [PubMed]
- Sergeev, R.A.; Batorsky, R.E.; Rouzine, I.M. Model with two types of CTL regulation and experiments on CTL dynamics. J. Theor. Biol. 2010, 263, 369–384. [Google Scholar] [CrossRef] [PubMed]
- Kemp, R.A.; Powell, T.J.; Dwyer, D.W.; Dutton, R.W. Cutting edge: Regulation of CD8+ T cell effector population size. J. Immunol. 2004, 173, 2923–2927. [Google Scholar] [CrossRef] [PubMed]
- Badovinac, V.P.; Harty, J.T. Manipulating the rate of memory CD8+ T cell generation after acute infection. J. Immunol. 2007, 179, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Bocharov, G.; Ludewig, B.; Bertoletti, A.; Klenerman, P.; Junt, T.; Krebs, P.; Luzyanina, T.; Fraser, C.; Anderson, R.M. Underwhelming the immune response: Effect of slow virus growth on CD8+-T-lymphocyte responses. J. Virol. 2004, 78, 2247–2254. [Google Scholar] [CrossRef] [PubMed]
- Davenport, M.P.; Belz, G.T.; Ribeiro, R.M. The race between infection and immunity: How do pathogens set the pace? Trends Immunol. 2009, 30, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Kastenmuller, W.; Gasteiger, G.; Gronau, J.H.; Baier, R.; Ljapoci, R.; Busch, D.H.; Drexler, I. Cross-competition of CD8+ T cells shapes the immunodominance hierarchy during boost vaccination. J. Exp. Med. 2007, 204, 2187–2198. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.L.; Wikstrom, M.E.; Fazekas de St Groth, B. Visualizing T cell competition for peptide/MHC complexes: A specific mechanism to minimize the effect of precursor frequency. Immunity 2000, 13, 783–794. [Google Scholar] [CrossRef]
- Probst, H.C.; Dumrese, T.; van den Broek, M.F. Cutting edge: Competition for APC by CTLs of different specificities is not functionally important during induction of antiviral responses. J. Immunol. 2002, 168, 5387–5391. [Google Scholar] [CrossRef] [PubMed]
- Owen, R.E.; Heitman, J.W.; Hirschkorn, D.F.; Lanteri, M.C.; Biswas, H.H.; Martin, J.N.; Krone, M.R.; Deeks, S.G.; Norris, P.J.; NIAID Center for HIV/AIDS Vaccine Immunology. HIV+ elite controllers have low HIV-specific T-cell activation yet maintain strong, polyfunctional T-cell responses. AIDS 2010, 24, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Halsey, L.G.; Curran-Everett, D.; Vowler, S.L.; Drummond, G.B. The fickle P value generates irreproducible results. Nat. Methods 2015, 12, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.E.; Gossez, M.; Williams, J.P.; Stöhr, W.; Meyerowitz, J.; Leitman, E.M.; Goulder, P.; Porter, K.; Fidler, S.; Frater, J.; et al. Post-treatment control or treated controllers? Viral remission in treated and untreated primary HIV infection. AIDS 2017, 31, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Lanzavecchia, A. Lack of fair play in the T cell response. Nat. Immunol. 2002, 3, 9–10. [Google Scholar] [CrossRef] [PubMed]
- Tilman, D. Resource Competition and Community Structure; Princeton University Press: Princeton, NJ, USA, 1982. [Google Scholar]
- Amanna, I.J.; Carlson, N.E.; Slifka, M.K. Duration of humoral immunity to common viral and vaccine antigens. N. Engl. J. Med. 2007, 357, 1903–1915. [Google Scholar] [CrossRef] [PubMed]
- Althaus, C.L.; Ganusov, V.V.; De Boer, R.J. Dynamics of CD8+ T cell responses during acute and chronic lymphocytic choriomeningitis virus infection. J. Immunol. 2007, 179, 2944–2951. [Google Scholar] [CrossRef] [PubMed]
- Fiebig, E.W.; Wright, D.J.; Rawal, B.D.; Garrett, P.E.; Schumacher, R.T.; Peddada, L.; Heldebrant, C.; Smith, R.; Conrad, A.; Kleinman, S.H.; et al. Dynamics of HIV viremia and antibody seroconversion in plasma donors: Implications for diagnosis and staging of primary HIV infection. AIDS 2003, 17, 1871–1879. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, E.E.; Funkhouser, B.; Athreya, G.; Perelson, A.S.; Korber, B.T.; Bhattacharya, T. Estimating time since infection in early homogeneous HIV-1 samples using a poisson model. BMC Bioinform. 2010, 11, 532. [Google Scholar] [CrossRef] [PubMed]
- McMichael, A.J.; Haynes, B.F. Lessons learned from HIV-1 vaccine trials: New priorities and directions. Nat. Immunol. 2012, 13, 423–427. [Google Scholar] [CrossRef] [PubMed]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Ganusov, V.V. Defining Kinetic Properties of HIV-Specific CD8+ T-Cell Responses in Acute Infection. Microorganisms 2019, 7, 69. https://doi.org/10.3390/microorganisms7030069
Yang Y, Ganusov VV. Defining Kinetic Properties of HIV-Specific CD8+ T-Cell Responses in Acute Infection. Microorganisms. 2019; 7(3):69. https://doi.org/10.3390/microorganisms7030069
Chicago/Turabian StyleYang, Yiding, and Vitaly V. Ganusov. 2019. "Defining Kinetic Properties of HIV-Specific CD8+ T-Cell Responses in Acute Infection" Microorganisms 7, no. 3: 69. https://doi.org/10.3390/microorganisms7030069
APA StyleYang, Y., & Ganusov, V. V. (2019). Defining Kinetic Properties of HIV-Specific CD8+ T-Cell Responses in Acute Infection. Microorganisms, 7(3), 69. https://doi.org/10.3390/microorganisms7030069