Reanalysis of Lactobacillus paracasei Lbs2 Strain and Large-Scale Comparative Genomics Places Many Strains into Their Correct Taxonomic Position

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Brief Description of the Key Software Used in This Study
2.2. Processing of Reads, Re-Assembly, and Acquisition of Publicly Available Assemblies
2.3. Quality Assessment of Genomes
2.4. Average Nucleotide Identity (ANI) and Average Amino Acid Identity (AAI) Calculation
2.5. Gene Prediction and Annotation
2.6. Construction of Pan/Core-Genome Families and Unique Genes
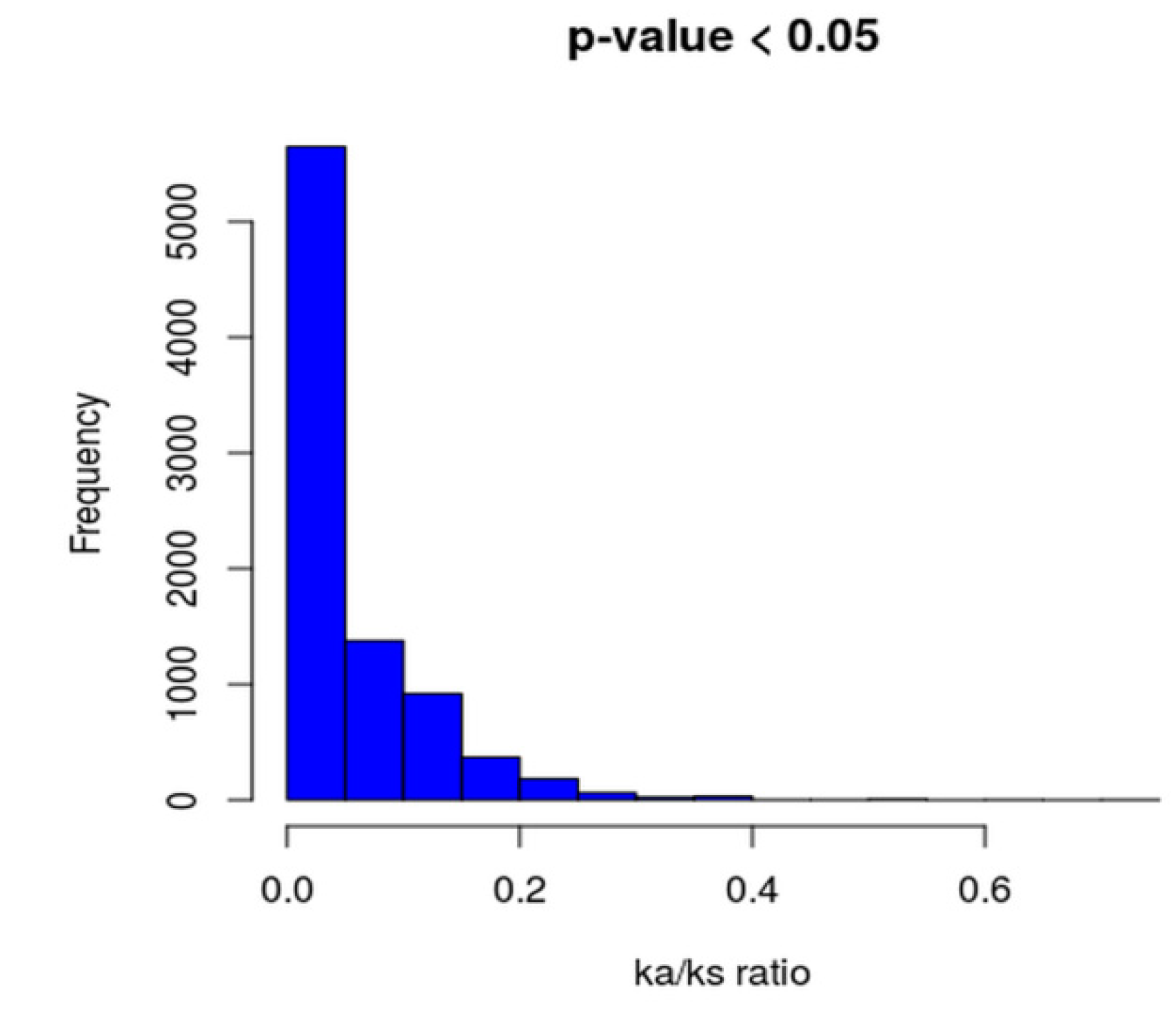
2.7. Phylogenomic Analysis and Ka/Ks Calculation of Core Genomes
2.8. Comparative Genomic Analysis
2.9. Data Availability
3. Results and Discussion
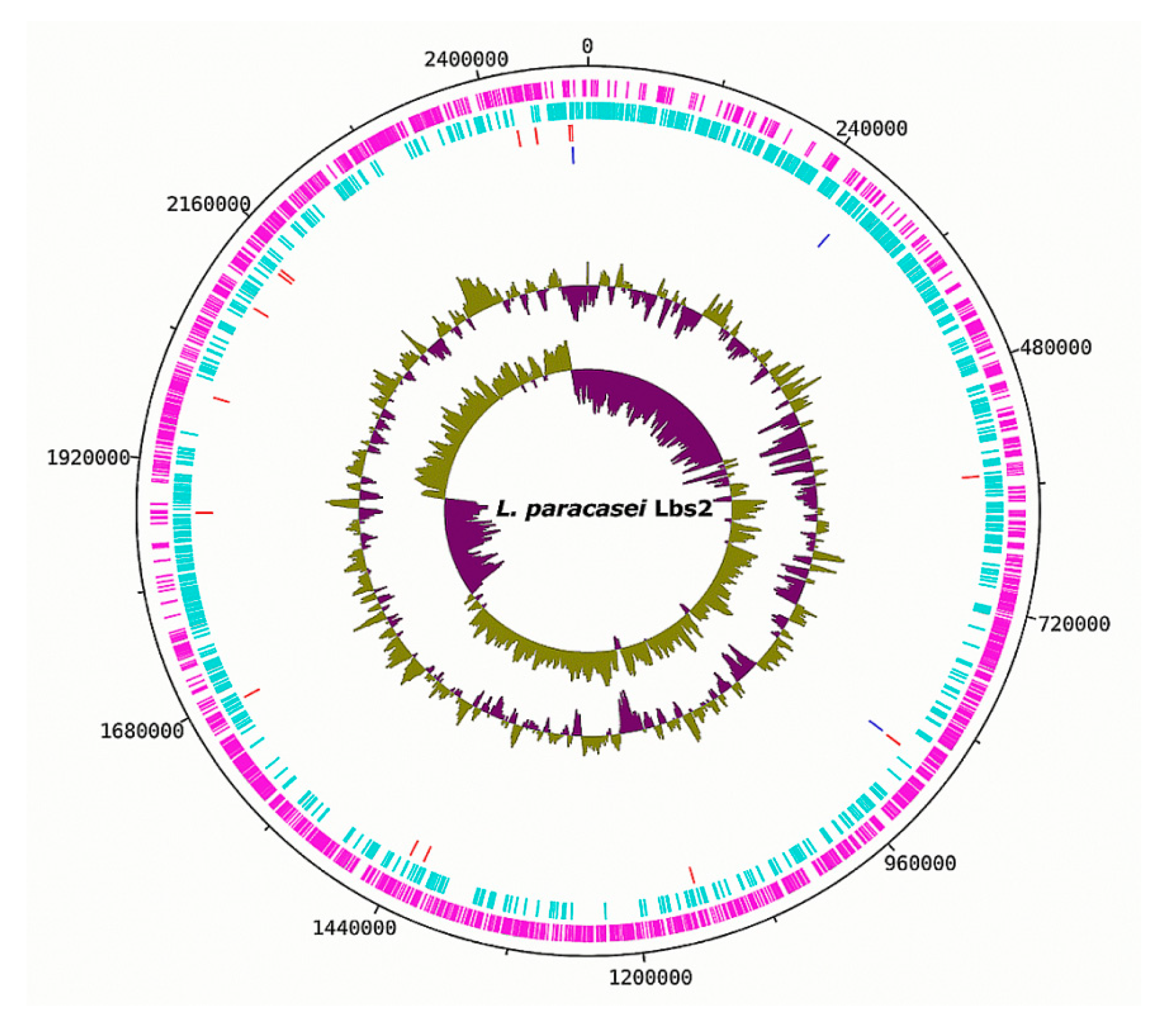
3.1. Genomic Properties of the L. paracasei Strains
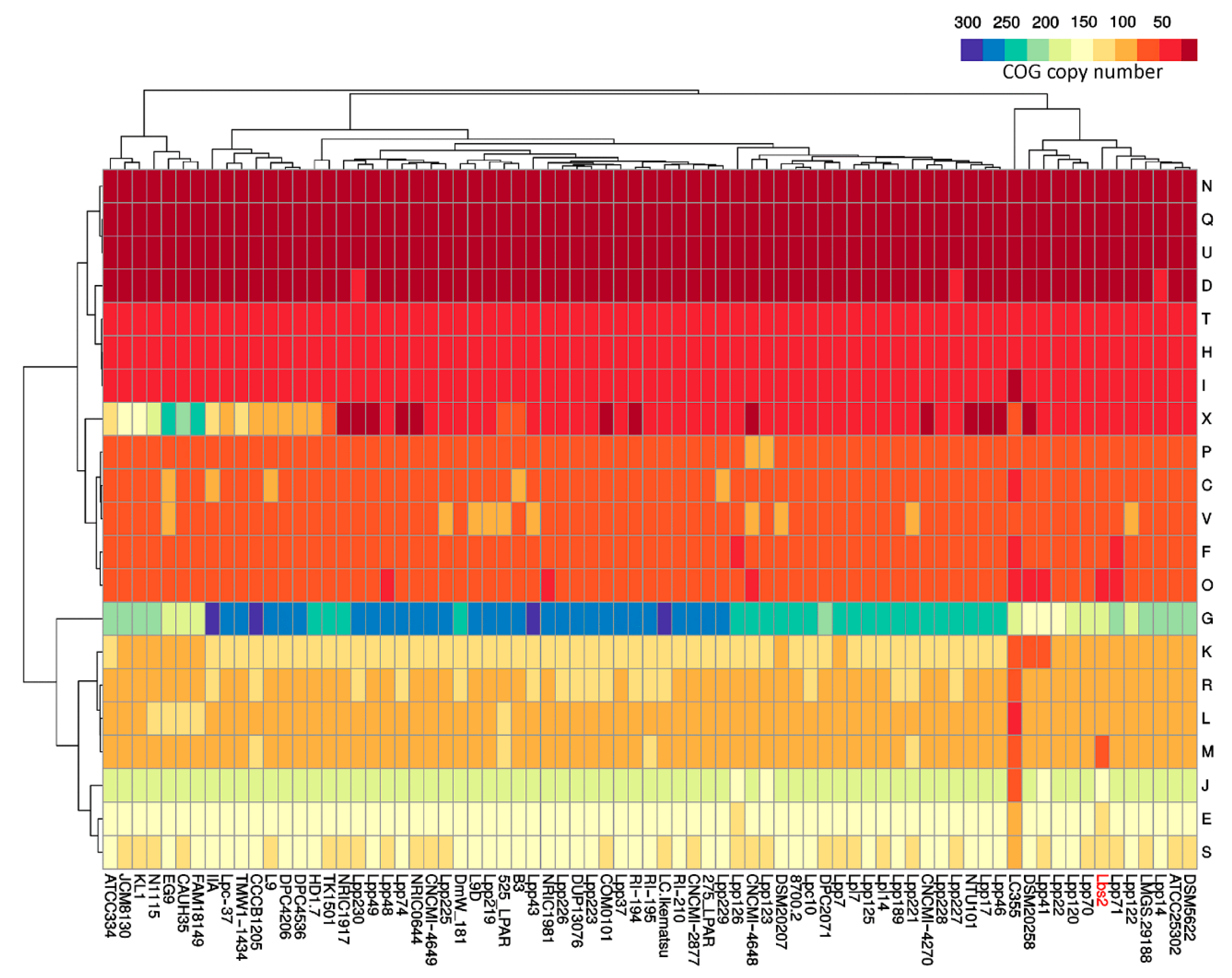
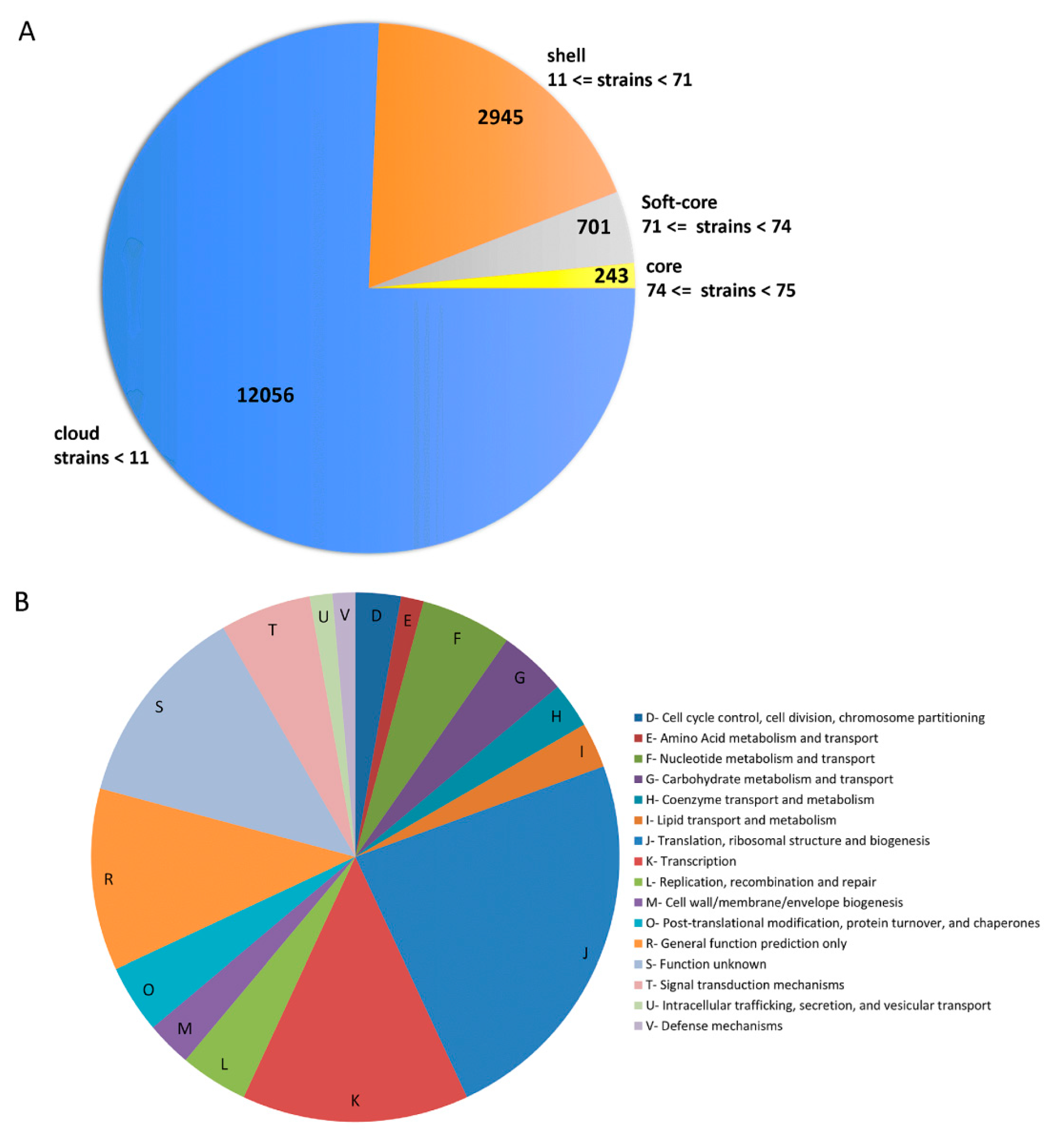
3.2. Pan/Core-Genome and Unique Genes Analysis
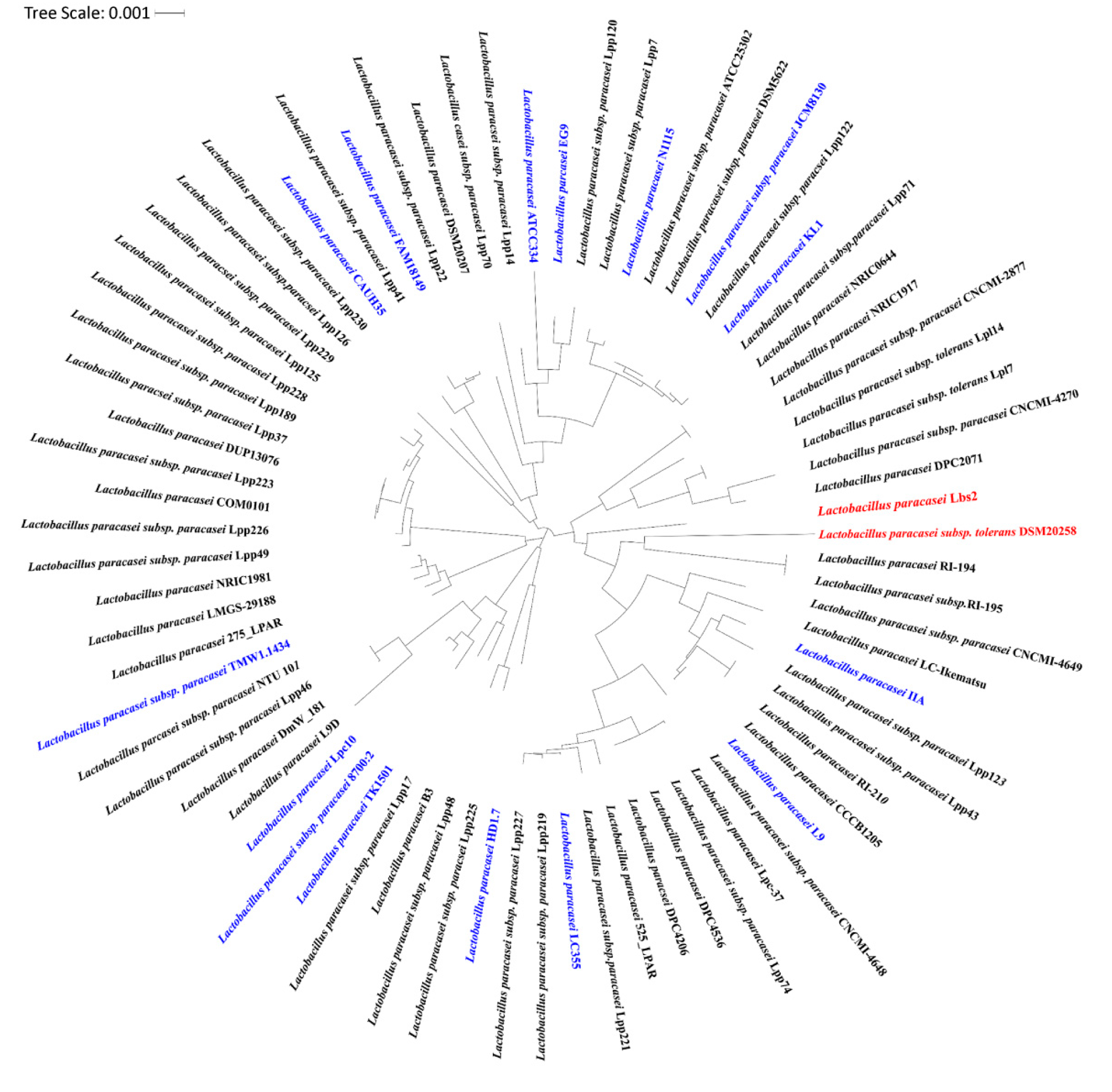
3.3. Whole Genome Phylogenetic Analysis Reveals Closeness between L. paracasei Lbs2 and L. paracasei subsp. Tolerans DSM20258 Strain
3.4. Horizontal Gene Transfer Analysis Indicates L. paracasei Lbs2 Strain Acquired Important Niche-Specific Genes
3.5. Extracellular Properties of the L. paracasei Strains
3.6. Various Stress Factors Characterized Across the L. paracasei Strains
3.7. Bacteriocins Identified Among the Lactobacillus paracasei Strains
3.8. A Wide Distribution of Mobile-Genetic Elements and CRISPR-Cas Systems
3.9. Broad Range Carbohydrate-Active Enzymes (CAZymes) and Carbohydrate Transporters Identified in the Pan-Genome of Lactobacillus paracasei
3.10. In-Depth Comparative Analysis of L. paracasei Lbs2 Against L. paracasei subsp. tolerans DSM20258 and L. paracasei Lpc-37
3.11. Evaluating the Technological and Probiotic Traits of L. paracasei Lbs2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sun, Z.; Harris, H.M.; McCann, A.; Guo, C.; Argimon, S.; Zhang, W.; Yang, X.; Jeffery, I.B.; Cooney, J.C.; Kagawa, T.F.; et al. Expanding the biotechnology potential of lactobacilli through comparative genomics of 213 strains and associated genera. Nat. Commun. 2015, 6, 8322. [Google Scholar] [CrossRef] [PubMed]
- Smokvina, T.; Wels, M.; Polka, J.; Chervaux, C.; Brisse, S.; Boekhorst, J.; van HylckamaVlieg, J.E.; Siezen, R.J. Lactobacillus paracasei comparative genomics: Towards species pan-genome definition and exploitation of diversity. PLoS ONE 2013, 8, e68731. [Google Scholar] [CrossRef] [PubMed]
- Wuyts, S.; Wittouck, S.; De Boeck, I.; Allonsius, C.N.; Pasolli, E.; Segata, N.; Lebeer, S. Large-Scale Phylogenomics of the Lactobacillus casei Group Highlights Taxonomic Inconsistencies and Reveals Novel Clade-Associated Features. MSystems 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Salvetti, E.; Harris, H.M.B.; Felis, G.E.; O’Toole, P.W. Comparative Genomics of the Genus Lactobacillus Reveals Robust Phylogroups That Provide the Basis for Reclassification. Appl. Env. Microbiol. 2018, 84. [Google Scholar] [CrossRef]
- Huang, C.H.; Li, S.W.; Huang, L.; Watanabe, K. Identification and Classification for the Lactobacillus casei Group. Front. Microbiol. 2018, 9, 1974. [Google Scholar] [CrossRef] [PubMed]
- Call, E.K.; Goh, Y.J.; Selle, K.; Klaenhammer, T.R.; O’Flaherty, S. Sortase-deficient lactobacilli: Effect on immunomodulation and gut retention. Microbiology 2015, 161, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Boekhorst, J.; de Been, M.W.; Kleerebezem, M.; Siezen, R.J. Genome-wide detection and analysis of cell wall-bound proteins with LPxTG-like sorting motifs. J. Bacteriol. 2005, 187, 4928–4934. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Provencio, D.; Rodriguez-Diaz, J.; Collado, M.C.; Langella, P.; Bermudez-Humaran, L.G.; Monedero, V. Functional analysis of the Lactobacillus casei BL23 sortases. Appl. Env. Microbiol. 2012, 78, 8684–8693. [Google Scholar] [CrossRef]
- Dicks, L.M.T.; Dreyer, L.; Smith, C.; van Staden, A.D. A Review: The Fate of Bacteriocins in the Human Gastro-Intestinal Tract: Do They Cross the Gut-Blood Barrier? Front. Microbiol. 2018, 9, 2297. [Google Scholar] [CrossRef]
- Cotter, P.D.; Hill, C.; Ross, R.P. Bacteriocins: Developing innate immunity for food. Nat. Rev. Microbiol. 2005, 3, 777–788. [Google Scholar] [CrossRef]
- Corr, S.C.; Li, Y.; Riedel, C.U.; O’Toole, P.W.; Hill, C.; Gahan, C.G. Bacteriocin production as a mechanism for the antiinfective activity of Lactobacillus salivarius UCC118. Proc. Natl. Acad. Sci. USA 2007, 104, 7617–7621. [Google Scholar] [CrossRef] [PubMed]
- De Vos, W.M.; Vaughan, E.E. Genetics of lactose utilization in lactic acid bacteria. FEMS Microbiol. Rev. 1994, 15, 217–237. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Rodriguez, B.T.; Zhang, W.; Broadbent, J.R.; Steele, J.L. Genotypic and phenotypic characterization of Lactobacillus casei strains isolated from different ecological niches suggests frequent recombination and niche specificity. Microbiology 2007, 153, 2655–2665. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, J.G.; Milani, C.; de Giori, G.S.; Sesma, F.; van Sinderen, D.; Ventura, M. Bacteria as vitamin suppliers to their host: A gut microbiota perspective. Curr. Opin. Biotechnol. 2013, 24, 160–168. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, J.G.; Chain, F.; Martin, R.; Bermudez-Humaran, L.G.; Courau, S.; Langella, P. Beneficial effects on host energy metabolism of short-chain fatty acids and vitamins produced by commensal and probiotic bacteria. Microb. Cell Fact. 2017, 16, 79. [Google Scholar] [CrossRef]
- Bhowmick, S.; Malar, M.; Das, A.; Kumar Thakur, B.; Saha, P.; Das, S.; Rashmi, H.M.; Batish, V.K.; Grover, S.; Tripathy, S. Draft Genome Sequence of Lactobacillus casei Lbs2. Genome Announc. 2014, 2. [Google Scholar] [CrossRef]
- O’Connell, J.; Schulz-Trieglaff, O.; Carlson, E.; Hims, M.M.; Gormley, N.A.; Cox, A.J. NxTrim: Optimized trimming of Illumina mate pair reads. Bioinformatics 2015, 31, 2035–2037. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Enright, A.J.; Van Dongen, S.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef]
- Wang, Y.; Coleman-Derr, D.; Chen, G.; Gu, Y.Q. OrthoVenn: A web server for genome wide comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2015, 43, W78–W84. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A toolkit incorporating gamma-series methods and sliding window strategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef]
- Zhang, Z.; Xiao, J.; Wu, J.; Zhang, H.; Liu, G.; Wang, X.; Dai, L. ParaAT: A parallel tool for constructing multiple protein-coding DNA alignments. Biochem. Biophys. Res. Commun. 2012, 419, 779–781. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [PubMed]
- Carver, T.; Thomson, N.; Bleasby, A.; Berriman, M.; Parkhill, J. DNAPlotter: Circular and linear interactive genome visualization. Bioinformatics 2009, 25, 119–120. [Google Scholar] [CrossRef]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Mao, X.; Yang, J.; Chen, X.; Mao, F.; Xu, Y. dbCAN: A web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012, 40, W445–W451. [Google Scholar] [CrossRef]
- Plyusnin, I.; Holm, L.; Kankainen, M. LOCP—locating pilus operons in gram-positive bacteria. Bioinformatics 2009, 25, 1187–1188. [Google Scholar] [CrossRef][Green Version]
- Van Heel, A.J.; de Jong, A.; Song, C.; Viel, J.H.; Kok, J.; Kuipers, O.P. BAGEL4: A user-friendly web server to thoroughly mine RiPPs and bacteriocins. Nucleic Acids Res. 2018, 46, W278–W281. [Google Scholar] [CrossRef]
- Grissa, I.; Vergnaud, G.; Pourcel, C. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinform. 2007, 8, 172. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Varani, A.M.; Siguier, P.; Gourbeyre, E.; Charneau, V.; Chandler, M. ISsaga is an ensemble of web-based methods for high throughput identification and semi-automatic annotation of insertion sequences in prokaryotic genomes. Genome Biol. 2011, 12, R30. [Google Scholar] [CrossRef] [PubMed]
- Ekstrom, A.; Yin, Y. ORFanFinder: Automated identification of taxonomically restricted orphan genes. Bioinformatics 2016, 32, 2053–2055. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Simon Fraser University Research Computing Group; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Konstantinidis, K.T.; Tiedje, J.M. Towards a genome-based taxonomy for prokaryotes. J. Bacteriol. 2005, 187, 6258–6264. [Google Scholar] [CrossRef]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef]
- Barrangou, R.; Azcarate-Peril, M.A.; Duong, T.; Conners, S.B.; Kelly, R.M.; Klaenhammer, T.R. Global analysis of carbohydrate utilization by Lactobacillus acidophilus using cDNA microarrays. Proc. Natl. Acad. Sci. USA 2006, 103, 3816–3821. [Google Scholar] [CrossRef]
- Watson, D.; O’Connell Motherway, M.; Schoterman, M.H.; van Neerven, R.J.; Nauta, A.; van Sinderen, D. Selective carbohydrate utilization by lactobacilli and bifidobacteria. J. Appl. Microbiol. 2013, 114, 1132–1146. [Google Scholar] [CrossRef]
- Salvetti, E.; Torriani, S.; Felis, G.E. The Genus Lactobacillus: A Taxonomic Update. Probiotics Antimicrob. Proteins 2012, 4, 217–226. [Google Scholar] [CrossRef]
- Claesson, M.J.; van Sinderen, D.; O’Toole, P.W. Lactobacillus phylogenomics—Towards a reclassification of the genus. Int. J. Syst. Evol. Microbiol. 2008, 58, 2945–2954. [Google Scholar] [CrossRef]
- Lakin, S.M.; Dean, C.; Noyes, N.R.; Dettenwanger, A.; Ross, A.S.; Doster, E.; Rovira, P.; Abdo, Z.; Jones, K.L.; Ruiz, J.; et al. MEGARes: An antimicrobial resistance database for high throughput sequencing. Nucleic Acids Res. 2017, 45, D574–D580. [Google Scholar] [CrossRef] [PubMed]
- Jose, N.M.; Bunt, C.R.; Hussain, M.A. Implications of Antibiotic Resistance in Probiotics. Food Rev. Int. 2015, 31, 52–62. [Google Scholar] [CrossRef]
- Tynkkynen, S.; Singh, K.V.; Varmanen, P. Vancomycin resistance factor of Lactobacillus rhamnosus GG in relation to enterococcal vancomycin resistance (van) genes. Int. J. Food Microbiol. 1998, 41, 195–204. [Google Scholar] [CrossRef]
- Gueimonde, M.; Sanchez, B.; de los Reyes-Gavilán, C.G.; Margolles, A. Antibiotic resistance in probiotic bacteria. Front. Microbiol. 2013, 4, 202. [Google Scholar] [CrossRef]
- Sengupta, R.; Altermann, E.; Anderson, R.C.; McNabb, W.C.; Moughan, P.J.; Roy, N.C. The role of cell surface architecture of lactobacilli in host-microbe interactions in the gastrointestinal tract. Mediat. Inflamm. 2013, 2013, 237921. [Google Scholar] [CrossRef]
- Call, E.K.; Klaenhammer, T.R. Relevance and application of sortase and sortase-dependent proteins in lactic acid bacteria. Front. Microbiol. 2013, 4, 73. [Google Scholar] [CrossRef]
- Su, X.; Sun, F.; Wang, Y.; Hashmi, M.Z.; Guo, L.; Ding, L.; Shen, C. Identification, characterization and molecular analysis of the viable but nonculturableRhodococcusbiphenylivorans. Sci. Rep. 2015, 5, 18590. [Google Scholar] [CrossRef]
- Gul, N.; Poolman, B. Functional reconstitution and osmoregulatory properties of the ProU ABC transporter from Escherichia coli. Mol. Membr. Biol. 2013, 30, 138–148. [Google Scholar] [CrossRef]
- Liu, X.; Sun, M.; Cheng, Y.; Yang, R.; Wen, Y.; Chen, Z.; Li, J. OxyR Is a Key Regulator in Response to Oxidative Stress in Streptomyces avermitilis. Microbiology 2016, 162, 707–716. [Google Scholar] [CrossRef]
- Nilsen, T.; Nes, I.F.; Holo, H. Enterolysin A, a cell wall-degrading bacteriocin from Enterococcus faecalis LMG 2333. Appl. Env. Microbiol. 2003, 69, 2975–2984. [Google Scholar] [CrossRef]
- Perez, R.H.; Zendo, T.; Sonomoto, K. Novel bacteriocins from lactic acid bacteria (LAB): various structures and applications. Microb. Cell. Fact. 2014, 13, S3. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.C.; Liu, C.F.; Lin, J.F.; Li, A.C.; Lo, T.C.; Lin, T.H. Characterization of putative class II bacteriocins identified from a non-bacteriocin-producing strain Lactobacillus casei ATCC 334. Appl. Microbiol. Biotechnol. 2013, 97, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Riley, M.A.; Wertz, J.E. Bacteriocin diversity: Ecological and evolutionary perspectives. Biochimie 2002, 84, 357–364. [Google Scholar] [CrossRef]
- Majeed, H.; Gillor, O.; Kerr, B.; Riley, M.A. Competitive interactions in Escherichia coli populations: The role of bacteriocins. ISME J. 2011, 5, 71–81. [Google Scholar] [CrossRef]
- Meijerink, M.; van Hemert, S.; Taverne, N.; Wels, M.; de Vos, P.; Bron, P.A.; Savelkoul, H.F.; van Bilsen, J.; Kleerebezem, M.; Wells, J.M. Identification of genetic loci in Lactobacillus plantarum that modulate the immune response of dendritic cells using comparative genome hybridization. PLoS ONE 2010, 5, e10632. [Google Scholar] [CrossRef]
- Kaur, S. Bacteriocins as Potential Anticancer Agents. Front. Pharmacol. 2015, 6, 272. [Google Scholar] [CrossRef]
- Bondy-Denomy, J.; Qian, J.; Westra, E.R.; Buckling, A.; Guttman, D.S.; Davidson, A.R.; Maxwell, K.L. Prophages mediate defense against phage infection through diverse mechanisms. ISME J. 2016, 10, 2854–2866. [Google Scholar] [CrossRef]
- Douillard, F.P.; Ribbera, A.; Kant, R.; Pietila, T.E.; Jarvinen, H.M.; Messing, M.; Randazzo, C.L.; Paulin, L.; Laine, P.; Ritari, J.; et al. Comparative genomic and functional analysis of 100 Lactobacillus rhamnosus strains and their comparison with strain GG. PLoS Genet. 2013, 9, e1003683. [Google Scholar] [CrossRef]
- Ross, P.; Mayer, R.; Benziman, M. Cellulose biosynthesis and function in bacteria. Microbiol. Rev. 1991, 55, 35–58. [Google Scholar]
- Lorca, G.L.; Font de Valdez, G.; Ljungh, A. Characterization of the protein-synthesis dependent adaptive acid tolerance response in Lactobacillus acidophilus. J. Mol. Microbiol. Biotechnol. 2002, 4, 525–532. [Google Scholar]
- Jiao, Y.; Cody, G.D.; Harding, A.K.; Wilmes, P.; Schrenk, M.; Wheeler, K.E.; Banfield, J.F.; Thelen, M.P. Characterization of extracellular polymeric substances from acidophilic microbial biofilms. Appl. Environ. Microbiol. 2010, 76, 2916–2922. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wise, M.J. Glycogen with short average chain length enhances bacterial durability. Naturwissenschaften 2011, 98, 719–729. [Google Scholar] [CrossRef] [PubMed]
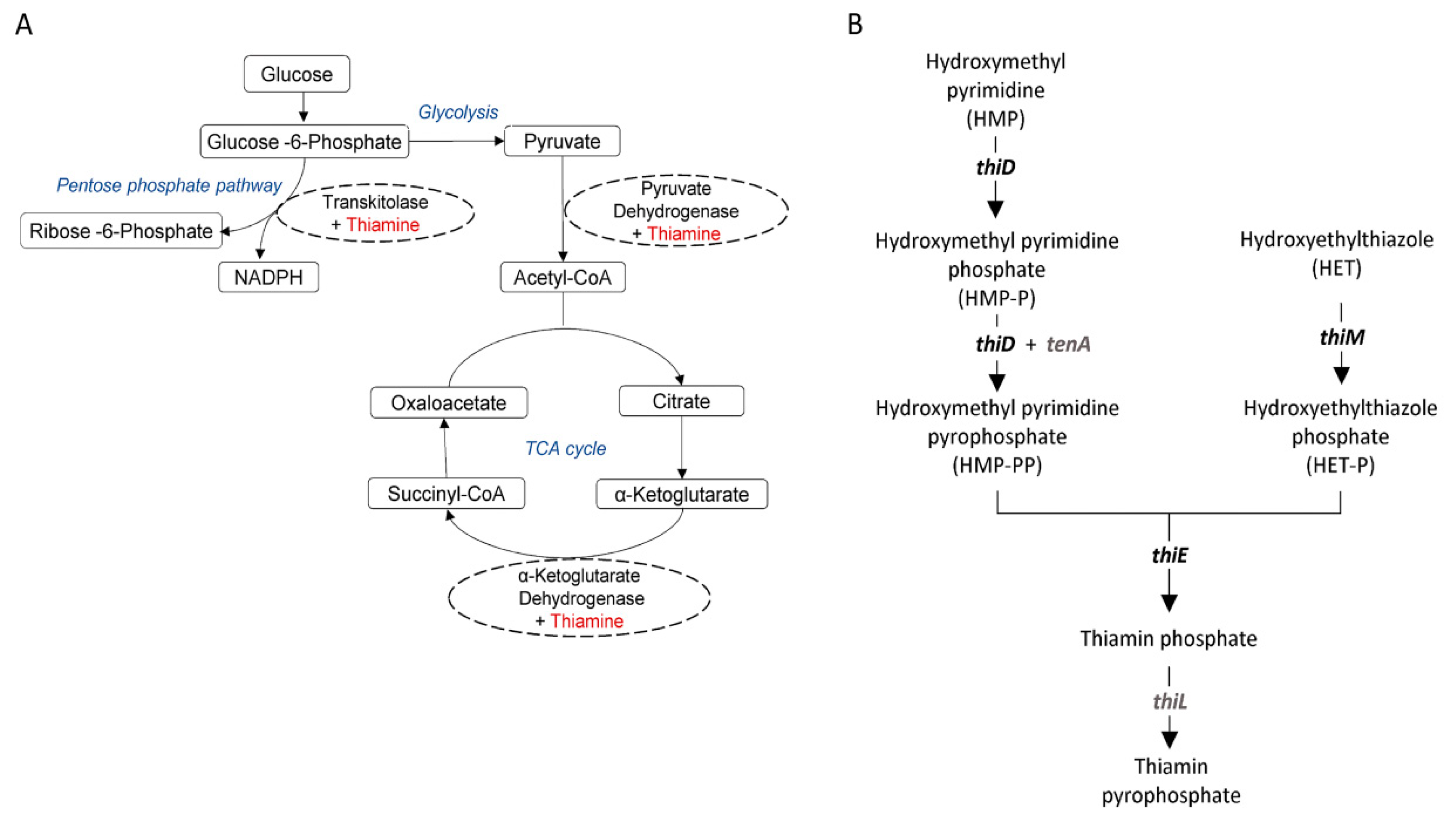
- Kerns, J.C.; Arundel, C.; Chawla, L.S. Thiamin deficiency in people with obesity. Adv. Nutr. 2015, 6, 147–153. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Existing Name in NCBI Database (GenBank) | Proposed Name |
|---|---|
| Lactobacillus casei 12A (Complete genome) | Lactobacillus paracasei 12A |
| Lactobacillus casei 12A (Draft) | Lactobacillus paracasei 12A |
| Lactobacillus casei 1316.rep1_LPAR (Scaf no.170) | Lactobacillus paracasei 1316.rep1_LPAR |
| Lactobacillus casei 1316.rep2_LPAR (Scaf no.264) | Lactobacillus paracasei 1316.rep2_LPAR |
| Lactobacillus casei 21/1 | Lactobacillus paracasei 21/1 |
| Lactobacillus casei 32G | Lactobacillus paracasei 32G |
| Lactobacillus casei 5b | Lactobacillus paracasei 5b |
| Lactobacillus casei 844_LCAS | Lactobacillus paracasei 844_LCAS |
| Lactobacillus casei A2-362 (Scaffold no. 162) | Lactobacillus paracasei A2-362 |
| Lactobacillus casei A2-362 (Contig no. 167) | Lactobacillus paracasei A2-362 |
| Lactobacillus casei BD II | Lactobacillus paracasei BD II |
| Lactobacillus casei BL23 | Lactobacillus paracasei BL23 |
| Lactobacillus casei BM-LC14617 | Lactobacillus paracasei BM-LC14617 |
| Lactobacillus casei CRF28 | Lactobacillus paracasei CRF28 |
| Lactobacillus casei DPC6800 | Lactobacillus paracasei DPC6800 |
| Lactobacillus casei DSM 20011 | Lactobacillus paracasei DSM 20011 |
| Lactobacillus casei GCRL163 | Lactobacillus paracasei GCRL163 |
| Lactobacillus casei HDS-01 | Lactobacillus paracasei HDS-01 |
| Lactobacillus casei HZ-1 | Lactobacillus paracasei HZ-1 |
| Lactobacillus casei KL1-Liu | Lactobacillus paracasei KL1-Liu |
| Lactobacillus casei LC2W | Lactobacillus paracasei LC2W |
| Lactobacillus casei LOCK919 | Lactobacillus paracasei LOCK919 |
| Lactobacillus casei Lc-10 | Lactobacillus paracasei Lc-10 |
| Lactobacillus casei Lc1542 | Lactobacillus paracasei Lc1542 |
| Lactobacillus casei LcY | Lactobacillus paracasei LcY |
| Lactobacillus casei Lpc-37 (Contig no.150) | Lactobacillus paracasei Lpc-37 |
| Lactobacillus casei M36 | Lactobacillus paracasei M36 |
| Lactobacillus casei MJA12 | Lactobacillus paracasei MJA12 |
| Lactobacillus casei T71499 | Lactobacillus paracasei T71499 |
| Lactobacillus casei UCD174 | Lactobacillus paracasei UCD174 |
| Lactobacillus casei UW1 | Lactobacillus paracasei UW1 |
| Lactobacillus casei UW4 (Contig no. 122) | Lactobacillus paracasei UW4 |
| Lactobacillus casei UW4 (Contig no. 144) | Lactobacillus paracasei UW4 |
| Lactobacillus casei W14 | Lactobacillus paracasei W14 |
| Lactobacillus casei W16 | Lactobacillus paracasei W16 |
| Lactobacillus casei W56 | Lactobacillus paracasei W56 |
| Lactobacillus casei Z11 | Lactobacillus paracasei Z11 |
| Lactobacillus casei Zhang | Lactobacillus paracasei Zhang |
| Strain | |||
|---|---|---|---|
| Features | Lpc-37 | Lbs2 | DSM20258 |
| Source | Microbial food product | Human Gut | Not available |
| Genome Status | Draft | Draft | Draft |
| Accession Number | NOKL00000000.1 | JPKN00000000.3 | AYYJ00000000.1 |
| N50 (bp) | 3,112,081 | 10,992 | 14,516 |
| L50 | 1 | 68 | 49 |
| Completeness (%) | 100 | 91.28 | 97.19 |
| Contamination (%) | 0 | 0.87 | 0 |
| Size (Mb) | 3.16 | 2.50 | 2.36 |
| GC% | 46.33 | 46.97 | 46.44 |
| Genes | 3125 | 2380 | 2424 |
| Proteins | 3010 | 2308 | 2339 |
| t-RNA | 59 | 20 | 37 |
| r-RNA | 15 | 3 | 2 |
| Other-RNA | 41 | 49 | 46 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghosh, S.; Sarangi, A.N.; Mukherjee, M.; Bhowmick, S.; Tripathy, S. Reanalysis of Lactobacillus paracasei Lbs2 Strain and Large-Scale Comparative Genomics Places Many Strains into Their Correct Taxonomic Position. Microorganisms 2019, 7, 487. https://doi.org/10.3390/microorganisms7110487
Ghosh S, Sarangi AN, Mukherjee M, Bhowmick S, Tripathy S. Reanalysis of Lactobacillus paracasei Lbs2 Strain and Large-Scale Comparative Genomics Places Many Strains into Their Correct Taxonomic Position. Microorganisms. 2019; 7(11):487. https://doi.org/10.3390/microorganisms7110487
Chicago/Turabian StyleGhosh, Samrat, Aditya Narayan Sarangi, Mayuri Mukherjee, Swati Bhowmick, and Sucheta Tripathy. 2019. "Reanalysis of Lactobacillus paracasei Lbs2 Strain and Large-Scale Comparative Genomics Places Many Strains into Their Correct Taxonomic Position" Microorganisms 7, no. 11: 487. https://doi.org/10.3390/microorganisms7110487
APA StyleGhosh, S., Sarangi, A. N., Mukherjee, M., Bhowmick, S., & Tripathy, S. (2019). Reanalysis of Lactobacillus paracasei Lbs2 Strain and Large-Scale Comparative Genomics Places Many Strains into Their Correct Taxonomic Position. Microorganisms, 7(11), 487. https://doi.org/10.3390/microorganisms7110487