Phylogenetic Analyses of Xanthomonads Causing Bacterial Leaf Spot of Tomato and Pepper: Xanthomonas euvesicatoria Revealed Homologous Populations Despite Distant Geographical Distribution


Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and DNA Extraction
2.2. Gene Selection and Primer Design
2.3. PCR, Sequencing, and Identity Confirmation
2.4. Phylogenetic Analysis of X. euvesicatoria and X. vesicatoria
2.5. Phylogenetic Analysis of X. euvesicatoria along with Other Xanthomonas Species
2.6. ELISA Analysis
3. Results
3.1. PCR Amplification, Sequencing, and Identity Confirmation
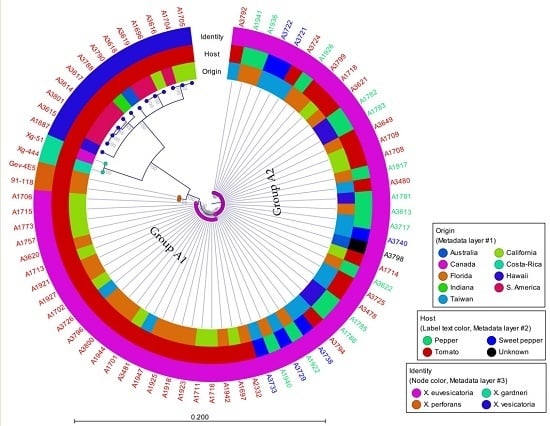
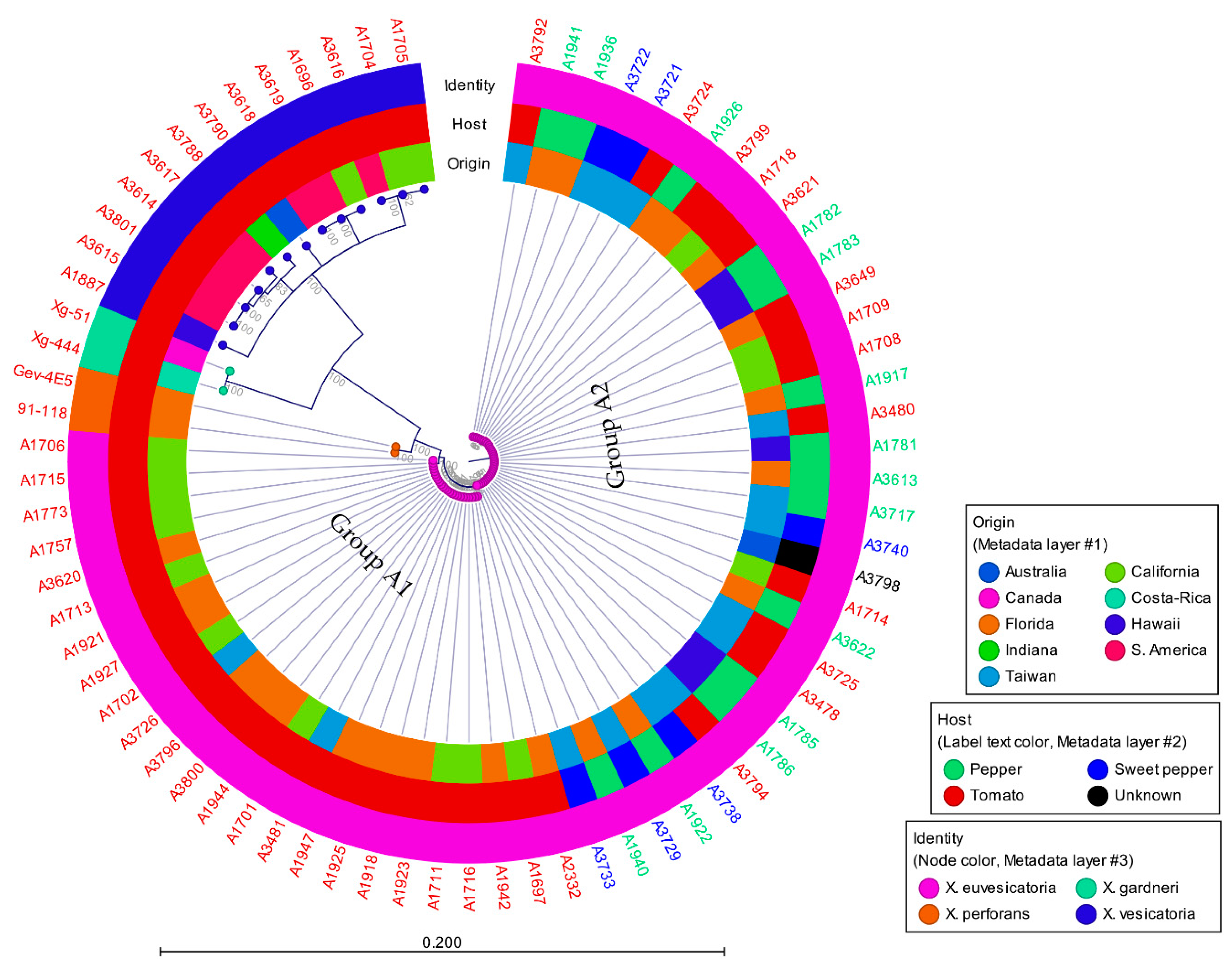
3.2. Phylogenetic Analysis
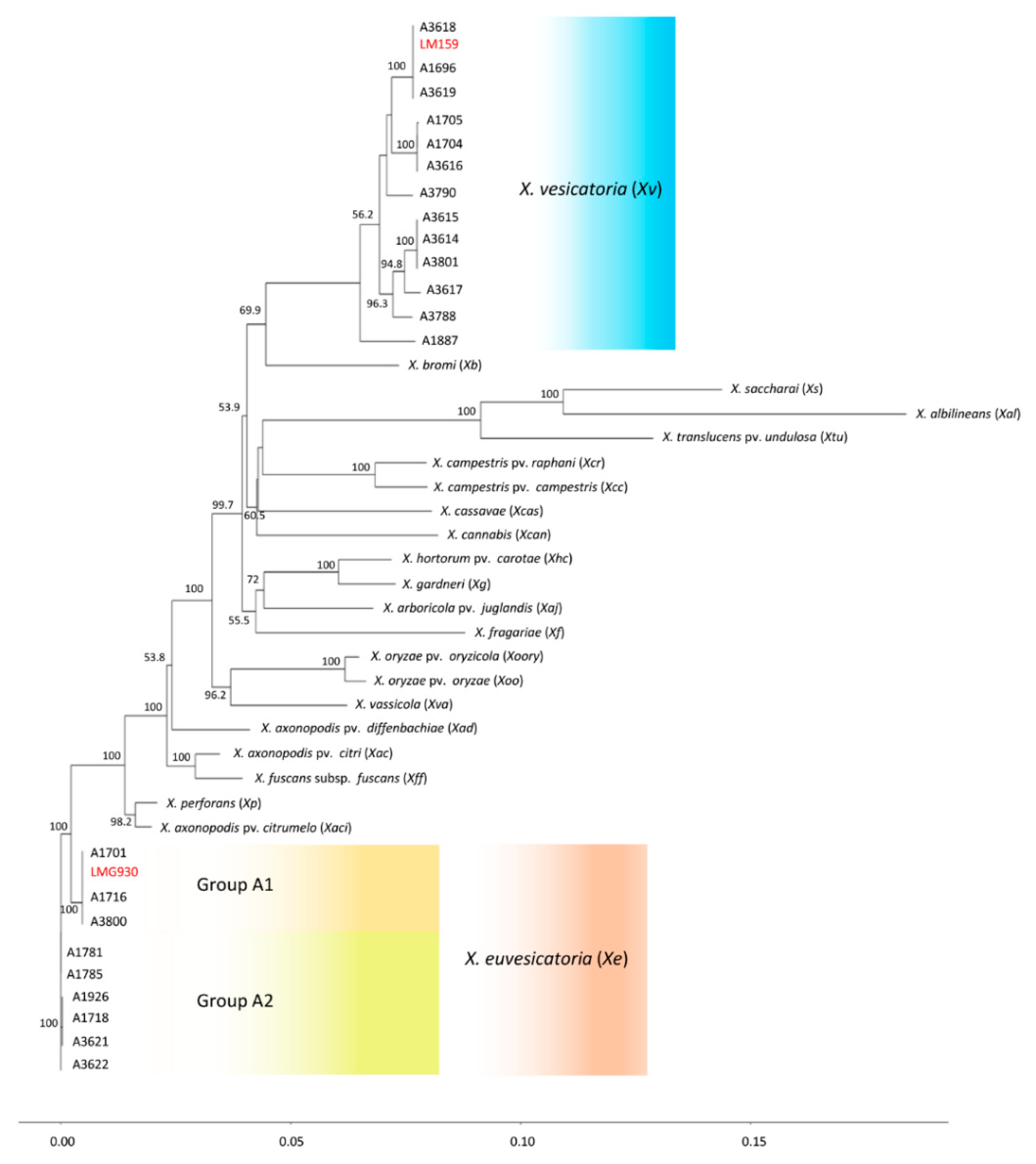
3.3. Phylogenetic Position of X. vesicatoria and X. euvesicatoria Relative to Other Xanthomonads
3.4. Phylogenetic Analysis of Antibody Data
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bradbury, J.F. Guide to Plant Pathogenic Bacteria; CAB International Mycological Institute: Farnham House, Slough, UK, 1986. [Google Scholar]
- Boch, J.; Bonas, U. Xanthomonas AvrBs3 Family-Type III effectors: Discovery and function. Annu. Rev. Phytopathol. 2010, 48, 419–436. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.B.; Lacy, G.H.; Bouzar, H.; Stall, R.E.; Schaad, N.W. Reclassification of the xanthomonads associated with bacterial spot disease of tomato and pepper. Syst. Appl. Microbiol. 2004, 27, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.W.; Somodi, G.C.; Jones, J.B. Resistance to bacterial spot fruit infection in tomato. HortScience 1989, 24, 825–827. [Google Scholar]
- Stall, R.E.; Beaulieu, C.; Egel, D.; Hodge, N.C.; Leite, R.P.; Minsavage, G.V.; Bouzar, H.; Jones, J.B.; Alvarez, A.M.; Benedict, A.A. Two genetically diverse groups of strains are included in Xanthomonas campestris pv. vesicatoria. Int. J. Syst. Evol. Microbiol. 1994, 44, 47–53. [Google Scholar] [CrossRef]
- Bouzar, H.; Jones, J.B.; Stall, R.E.; Hodge, N.C.; Minsavage, G.V.; Benedict, A.A.; Alvarez, A.M. Physiological, chemical, serological, and pathogenic analyses of a worldwide collection of Xanthomonas campestris pv. vesicatoria strains. Phytopathology 1994, 84, 663–671. [Google Scholar] [CrossRef]
- Vauterin, L.; Hoste, B.; Kersters, K.; Swings, J. Reclassification of Xanthomonas. Int. J. Syst. Evol. Microbiol. 1995, 45, 472–489. [Google Scholar] [CrossRef]
- Roach, R.; Mann, R.; Gambley, C.G.; Shivas, R.G.; Rodoni, B. Identification of Xanthomonas species associated with bacterial leaf spot of tomato, capsicum and chili crops in eastern Australia. Eur. J. Plant Pathol. 2017, 150, 595–608. [Google Scholar] [CrossRef]
- Larrea-Sarmiento, A.; Dhakal, U.; Boluk, G.; Fatdal, L.; Alvarez, A.; Strayer-Scherer, A.; Paret, M.; Jones, J.; Jenkins, D.; Arif, M. Development of a genome-informed loop-mediated isothermal amplification assay for rapid and specific detection of Xanthomonas euvesicatoria. Sci. Rep. 2018, 8, 14298. [Google Scholar] [CrossRef]
- Potnis, N.; Timilsina, S.; Strayer, A.; Shantharaj, D.; Barak, J.D.; Paret, M.L.; Vallad, G.E.; Jones, J.B. Bacterial spot of tomato and pepper: Diverse Xanthomonas species with a wide variety of virulence factors posing a worldwide challenge. Mol. Plant Pathol. 2015, 16, 907–920. [Google Scholar] [CrossRef]
- Vancheva, T.; Stoyanova, M.; Tatyozova, M.; Bogatzevska, N.; Moncheva, P. Sub-species diversity of Xanthomonas euvesicatoria Bulgarian and Macedonian strains from pepper. Biotechnol. Biotec. Eq. 2014, 28, 592–601. [Google Scholar] [CrossRef]
- Parkinson, N.; Aritua, V.; Heeney, J.; Cowie, C.; Bew, J.; Stead, D. Phylogenetic analysis of Xanthomonas species by comparison of partial gyrase B gene sequences. Int. J. Syst. Evol. Microbiol. 2007, 57, 2881–2887. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, N.; Cowie, C.; Heeney, J.; Stead, D. Phylogenetic structure of Xanthomonas determined by comparison of gyrB sequences. Int. J. Syst. Evol. Microbiol. 2009, 59, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Arif, M.; Busot, G.Y.; Mann, R.; Rodoni, B.; Liu, S.; Stack, J.P. Emergence of a new population of Rathayibacter toxicus: An ecologically complex, geographically isolated bacterium. PLoS ONE 2016, 11, e0156182. [Google Scholar] [CrossRef]
- Young, J.M.; Wilkie, J.P.; Park, D.C.; Watson, D.R.W. New Zealand strains of plant pathogenic bacteria classified by multi-locus sequence analysis; proposal of Xanthomonas dyei sp. nov. Plant Pathol. 2010, 59, 270–281. [Google Scholar] [CrossRef]
- Kebede, M.; Timilsina, S.; Ayalew, A.; Admassu, B.; Potnis, N.; Minsavage, G.V.; Goss, E.M.; Hong, J.C.; Strayer, A.; Paret, M.; et al. Molecular characterization of Xanthomonas strains responsible for bacterial spot of tomato in Ethiopia. Eur. J. Plant. Pathol. 2014, 140, 677–688. [Google Scholar] [CrossRef]
- Timilsina, S.; Jibrin, M.O.; Potnis, N.; Minsavage, G.V.; Kebede, M.; Schwartz, A.; Bart, R.; Staskawicz, B.; Boyer, C.; Vallad, G.E.; et al. Multilocus sequence analysis of xanthomonads causing bacterial spot of tomato and pepper plants reveals strains generated by recombination among species and recent global spread of Xanthomonas gardneri. Appl. Environ. Microbiol. 2015, 81, 1520–1529. [Google Scholar] [CrossRef]
- Roach, R.; Mann, R.; Gambley, C.G.; Chapman, T.; Shivas, R.G.; Rodoni, B. Genomic sequence analysis reveals diversity of Australian Xanthomonas species associated with bacterial leaf spot of tomato, capsicum and chilli. BMC Genomics 2019, 20, 310. [Google Scholar] [CrossRef]
- Schneider, K.L.; Marrero, G.; Alvarez, A.M.; Presting, G.G. Classification of plant associated bacteria using RIF, a computationally derived DNA marker. PLoS ONE 2011, 6, e18496. [Google Scholar] [CrossRef]
- Darling, A.E.; Mau, B.; Perna, N.T. Progressive mauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef]
- Rozen, S.; Skaletsky, H. Primer3 on the WWW for General Users and for Biologist Programmers. In Bioinformatics Methods and Protocols: Methods in Molecular Biology; Krawetz, S., Misener, S., Eds.; Humana Press: Totowa, NJ, USA, 2000; pp. 365–386. [Google Scholar] [CrossRef]
- Ocenar, J.; Arizala, D.; Boluk, G.; Dhakal, U.; Gunarathne, S.; Paudel, S.; Dobhal, S.; Arif, M. Development of a robust, field-deployable loop-mediated isothermal amplification (LAMP) assay for specific detection of potato pathogen Dickeya dianthicola targeting a unique genomic region. PLoS ONE 2019, 14, e0218868. [Google Scholar] [CrossRef]
- RStudio, version 3.4.3; Integrated Development for R; RStudio, Inc.: Boston, MA, USA, 2015; Available online: http://www.rstudio.com/ (accessed on 23 August 2017).
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.Y. ggtree: An r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Alvarez, A.; Benedict, A.; Mizumoto, C. Identification of xanthomonads and grouping of strains of Xanthomonas campestris pv. campestris with monoclonal antibodies. Phytopathology 1985, 75, 722–728. [Google Scholar] [CrossRef]
- Hamza, A.A.; Robene-Soustrade, I.; Jouen, E.; Lefeuvre, P.; Chiroleu, F.; Fisher-Le Saux, M.; Gagnevin, L.; Pruvost, O. MultiLocus Sequence Analysis- and Amplified Fragment Length Polymorphism-based characterization of xanthomonads associated with bacterial spot of tomato and pepper and their relatedness to Xanthomonas species. Syst. Appl. Microbiol. 2012, 35, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Constantin, E.C.; Cleenwerck, I.; Maes, M.; Baeyen, S.; Van Malderghem, C.; De Vos, P.; Cottyn, B. Genetic characterization of strains named as Xanthomonas axonopodis pv. dieffenbachiae leads to a taxonomic revision of the X. axonopodis species complex. Plant Pathol. 2016, 65, 729–806. [Google Scholar] [CrossRef]
- Potnis, N.; Krasileva, K.; Chow, V.; Almeida, N.F.; Patil, P.B.; Ryan, R.P.; Sharlach, M.; Behlau, F.; Dow, J.M.; Momol, M.T.; et al. Comparative genomics reveals diversity among xanthomonads infecting tomato and pepper. BMC Genomics 2011, 12, 146. [Google Scholar] [CrossRef]
- Young, J.M.; Park, D.C.; Shearman, H.M.; Fargier, E. A multilocus sequence analysis of the genus Xanthomonas. Syst. Appl. Microbiol. 2008, 31, 366–377. [Google Scholar] [CrossRef]
- Ngoc, L.B.T.; Vernière, C.; Jouen, E.; Ah-You, N.; Lefeuvre, P.; Chiroleu, F.; Gagnevin, L.; Pruvost, O. Amplified fragment length polymorphism and multilocus sequence analysis-based genotypic relatedness among pathogenic variants of Xanthomonas citri pv. citri and Xanthomonas campestris pv. bilvae. Int. J. Syst. Evol. Microbiol. 2010, 60, 515–525. [Google Scholar] [CrossRef]
- Horvath, D.M.; Stall, R.E.; Jones, J.B.; Pauly, M.H.; Vallad, G.E.; Dahlbeck, D.; Staskawicz, B.J.; Scott, J.W. Transgenic resistance confers effective field level control of bacterial spot disease in tomato. PLoS ONE 2012, 7, e42036. [Google Scholar] [CrossRef]
- Posada, D.; Crandall, K.A.; Holmes, E.C. Recombination in evolutionary genomics. Annu. Rev. Genet. 2002, 36, 75–97. [Google Scholar] [CrossRef]
- Comas, I.; Moya, A.; Azad, R.K.; Lawrence, J.G.; Gonzalez-Candelas, F. The evolutionary origin of xanthomonadales genomes and the nature of the horizontal gene transfer process. Mol. Biol. Evol. 2006, 23, 2049–2057. [Google Scholar] [CrossRef] [PubMed]
- Susko, E.; Leigh, J.; Doolittle, W.F.; Bapteste, E. Visualizing and assessing phylogenetic congruence of core gene sets: A case study of the γ-proteobacteria. Mol. Biol. Evol. 2006, 23, 1019–1030. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A Number | Other ID | Origin | Host | Identity | Acquired Date |
|---|---|---|---|---|---|
| A1701 | B94 | California | Tomato | Xanthomonas euvesicatoria | 1986 |
| A1711 | K625/B63 | California | Tomato | X. euvesicatoria | 1986 |
| A3620 | Xv 153 | Florida | Tomato | X. euvesicatoria | 1990 |
| A1782 | K337 | Hawaii | Pepper | X. euvesicatoria | 1986 |
| A1781 | K336 | Hawaii | Pepper | X. euvesicatoria | 1986 |
| A1786 | K339 | Hawaii | Pepper | X. euvesicatoria | 1986 |
| A3480 | XVT20 | Taiwan | Tomato | X. euvesicatoria | 1990 |
| A3478 | K348/XVT8 | Taiwan | Tomato | X. euvesicatoria | 1990 |
| A1702 | K618/B111 | California | Tomato | X. euvesicatoria | 1986 |
| A1706 | K622/B62 | California | Tomato | X. euvesicatoria | 1986 |
| A1708 | K623/B93 | California | Tomato | X. euvesicatoria | 1986 |
| A1709 | K624/B108 | California | Tomato | X. euvesicatoria | 1986 |
| A1713 | K626/B78 | California | Tomato | X. euvesicatoria | 1986 |
| A1714 | K627/B81 | California | Tomato | X. euvesicatoria | 1986 |
| A1715 | K628/B92 | California | Tomato | X. euvesicatoria | 1986 |
| A1716 | K629/B95 | California | Tomato | X. euvesicatoria | 1986 |
| A1718 | K630/B106 | California | Tomato | X. euvesicatoria | 1986 |
| A1757 | K641/XCV1 | California | Tomato | X. euvesicatoria | 1986 |
| A1773 | K645/XCV2 | California | Tomato | X. euvesicatoria | 1986 |
| A1783 | MCG | Hawaii | Pepper | X. euvesicatoria | 1986 |
| A1785 | EWCII | Hawaii | Pepper | X. euvesicatoria | 1986 |
| A1917 | 62-8 | Florida | Pepper | X. euvesicatoria | 1986 |
| A1918 | 65-2a | Florida | Tomato | X. euvesicatoria | 1986 |
| A3799 | Xv158 | Florida | Tomato | X. euvesicatoria | 1991 |
| A1921 | 69-13 | Florida | Tomato | X. euvesicatoria | 1986 |
| A1922 | 71-21 | Florida | Pepper | X. euvesicatoria | 1986 |
| A1923 | 71-39 | Florida | Tomato | X. euvesicatoria | 1986 |
| A1925 | 75-4 | Florida | Tomato | X. euvesicatoria | 1986 |
| A1926 | 77-3 | Florida | Pepper | X. euvesicatoria | 1986 |
| A3794 | Xv150 | Taiwan | Tomato | X. euvesicatoria | 1991 |
| A3796 | Xv155 | Florida | Tomato | X. euvesicatoria | 1991 |
| A3792 | Xv148 | Taiwan | Tomato | X. euvesicatoria | 1991 |
| A3798 | Xv157 | Australia | NA | X. euvesicatoria | 1991 |
| A3800 | Xv159 | Florida | Tomato | X. euvesicatoria | 1991 |
| A1697 | B79 | California | Tomato | X. euvesicatoria | 1986 |
| A1936 | 82-12 | Florida | Pepper | X. euvesicatoria | 1986 |
| A1940 | 82-16 | Florida | Pepper | X. euvesicatoria | 1986 |
| A1941 | 82-17 | Florida | Pepper | X. euvesicatoria | 1986 |
| A3621 | Xv158 | Florida | Tomato | X. euvesicatoria | 1990 |
| A3481 | XVT14 | Taiwan | Tomato | X. euvesicatoria | 1990 |
| A3613 | Xv134 | Florida | Pepper | X. euvesicatoria | 1990 |
| A3622 | XV173 | Florida | Pepper | X. euvesicatoria | 1990 |
| A3649 | XV154 | Florida | Tomato | X. euvesicatoria | 1990 |
| A3717 | XVP28 | Taiwan | Pepper | X. euvesicatoria | 1991 |
| A3721 | XVP41 | Taiwan | Sweet pepper | X. euvesicatoria | 1991 |
| A3722 | XVP42 | Taiwan | Sweet pepper | X. euvesicatoria | 1991 |
| A3724 | XVT7 | Taiwan | Tomato | X. euvesicatoria | 1991 |
| A3725 | XVT8 | Taiwan | Tomato | X. euvesicatoria | 1991 |
| A3726 | XVT14 | Taiwan | Tomato | X. euvesicatoria | 1991 |
| A3729 | XVP1 | Taiwan | Sweet pepper | X. euvesicatoria | 1991 |
| A3733 | XVP5 | Taiwan | Sweet pepper | X. euvesicatoria | 1991 |
| A3738 | XVP10 | Taiwan | Sweet pepper | X. euvesicatoria | 1991 |
| A3740 | XVP12 | Taiwan | Sweet pepper | X. euvesicatoria | 1991 |
| A1927 | 80-1 | Florida | Tomato | X. euvesicatoria | 1986 |
| A1942 | 83-4 | Florida | Tomato | X. euvesicatoria | 1986 |
| A1944 | 83-13 | Florida | Tomato | X. euvesicatoria | 1986 |
| A1947 | E3 | Florida | Tomato | X. euvesicatoria | 1986 |
| A2332 | X298 | Florida | Tomato | X. euvesicatoria | 1990 |
| A3617 | XV145 | S. America | Tomato | X. vesicatoria | 1990 |
| A3616 | XV144 | S. America | Tomato | X. vesicatoria | 1990 |
| A3618 | XV146 | S. America | Tomato | X. vesicatoria | 1990 |
| A3788 | CC12, Xv138 | Indiana | Tomato | X. vesicatoria | 1991 |
| A1696 | K613/B71 | California | Tomato | X. vesicatoria | 1986 |
| A1703 | K619/B118 | California | Tomato | X. vesicatoria | 1986 |
| A1704 | K620/B122 | California | Tomato | X. vesicatoria | 1986 |
| A1705 | K621/XV-1 | California | Tomato | X. vesicatoria | 1986 |
| A1887 | K663/A135-1 | Hawaii | Tomato | X. vesicatoria | 1986 |
| A3801 | Xv142a | S. America | Tomato | X. vesicatoria | 1991 |
| A3790 | Xv140 | Australia | Tomato | X. vesicatoria | 1991 |
| A3614 | XV142b | S. America | Tomato | X. vesicatoria | 1990 |
| A3615 | XV143 | S. America | Tomato | X. vesicatoria | 1990 |
| A3619 | XV147 | S. America | Tomato | X. vesicatoria | 1990 |
| Xg-51 | Canada | Tomato | X. gardneri | * | |
| Xg-444 | Costa-Rica | Tomato | X. gardneri | * | |
| Gev 4E5 | Florida | Tomato | X. perforans | * | |
| 91-118 | Florida | Tomato | X. perforans | * |
| Target Gene | Primers Name | Primer Sequences (5′-3′) | Product Size |
|---|---|---|---|
| hrcN | X-hrcN-F | TCGGCACCATGCTCAAGGT | 846 |
| X-hrcN-F | GTGTAGAACGCGGTGATCGA | ||
| dnaA | dnaA-F | CAGCACGGTGGTGTGGTC | 928 |
| dnaA-R | CCTGGATTCGCATTACACC | ||
| gyrB | GyrB-F2 | GAGGTGATCCTCACCGTGCT | 841 |
| GyrB-R2 | TGATGGCCTTGGCTTCGTTC | ||
| gapA | gapA-F1 | TGGCCATCAATGACCTGCTC | 865 |
| gapA-R1 | TAGCCCCACTCGTTGTCGTA | ||
| pdg | pdg-F | CCACCCACCAGACCAAGAA | 990 |
| pdg-R | CAGGTACATGCCCTTGATGA | ||
| hmbs | Hmbs-F | GTATCGCCACCCGCAAAA | 873 |
| Hmbs-R | CCTTGTCGAACAGCCCTTG | ||
| Hmbs-F2 | TTGCATCGCCACCCGCAAGA | 837 | |
| Hmbs-R2 | TCCTTGTCGAACAGGCCTTG | ||
| Hmbs-F10 | AGGGCCTGTTTTTGAAGGAA | 595 | |
| Hmbs-R10 | AACCCCTCGCCTTCCCAGGT |
| Species | Accession Numbers | Host | Geographic Location |
|---|---|---|---|
| X. perforans | NZ_CP019725 | Tomato | Mauritius |
| X. gardneri | NZ_CP018731 | Tomato | New Zealand |
| X. vesicatoria | NZ_CP018470 | Tomato | New Zealand |
| X. euvesicatoria | NZ_CP018467 | Pepper | USA |
| X. fragariae | NZ_CP016830 | Strawberry | California, USA |
| X. axonopodis pv. diffenbachiae | NZ_CP014347 | Anthurium | Brazil |
| X. sacchari | NZ_CP010409 | Rice | China |
| X. translucens pv. undulosa | NZ_CP008714 | Wheat | Kansas, USA |
| X. hortum pv. carotae | NZ_CM002307 | Carrot | Oregon. USA |
| X. fuscans subsp. fuscans | NC_022541 | Bean | France |
| X. campestris pv. raphani | NC_017271 | Cabbage | East Asia |
| X. oryzae pv. oryzicola | NC_017267 | Rice | Philippines |
| X. axonopodis pv. citrumelo | NC_016010 | Citrus | Florida, USA |
| X. albilineans | NC_013722 | Sugarcane | France |
| X. oryzae pv. oryzae | NC_007705 | Rice | Japan |
| X. axonopodis pv. citri | NC_003919 | Mexican lime | Florida, USA |
| X. campestris pv. campestris | NC_003902 | Cabbage | UK |
| X. arboricola pv. juglandis | CP012251 | Walnut | USA |
| X. bromi | GCF_900092025.1 | Brome grass | France |
| X. cannabis | GCF_000802405.1 | Cannabis sativa | Japan |
| X. vassicola | GCF_000772705.2 | Sorghum | New Zealand |
| X. cassavae | GCF_000454545.1 | Cassava | Malawi |
| Mab Name | Clone Number | Subclass | Immunogen (s) |
|---|---|---|---|
| Xv1 | 106-41-1-1 | IgG2b | A1074 |
| Xv3 | 131-39-14-2 | IgG2b | 82-17 |
| Xv5 * | 131-10-9-1 | IgG3 | 82-17 |
| Xv7 * | 130-10-2-1 | NA | 65-2a |
| Xv8 | 4H5-3B1 | IgG1 | BA29-1, BV20-3A, X525-85 |
| Xcv15 | 209-C15-4-4 | IgM | B61, B80 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dhakal, U.; Dobhal, S.; Alvarez, A.M.; Arif, M. Phylogenetic Analyses of Xanthomonads Causing Bacterial Leaf Spot of Tomato and Pepper: Xanthomonas euvesicatoria Revealed Homologous Populations Despite Distant Geographical Distribution. Microorganisms 2019, 7, 462. https://doi.org/10.3390/microorganisms7100462
Dhakal U, Dobhal S, Alvarez AM, Arif M. Phylogenetic Analyses of Xanthomonads Causing Bacterial Leaf Spot of Tomato and Pepper: Xanthomonas euvesicatoria Revealed Homologous Populations Despite Distant Geographical Distribution. Microorganisms. 2019; 7(10):462. https://doi.org/10.3390/microorganisms7100462
Chicago/Turabian StyleDhakal, Upasana, Shefali Dobhal, Anne M. Alvarez, and Mohammad Arif. 2019. "Phylogenetic Analyses of Xanthomonads Causing Bacterial Leaf Spot of Tomato and Pepper: Xanthomonas euvesicatoria Revealed Homologous Populations Despite Distant Geographical Distribution" Microorganisms 7, no. 10: 462. https://doi.org/10.3390/microorganisms7100462
APA StyleDhakal, U., Dobhal, S., Alvarez, A. M., & Arif, M. (2019). Phylogenetic Analyses of Xanthomonads Causing Bacterial Leaf Spot of Tomato and Pepper: Xanthomonas euvesicatoria Revealed Homologous Populations Despite Distant Geographical Distribution. Microorganisms, 7(10), 462. https://doi.org/10.3390/microorganisms7100462