The Impact of Bioinformatics Pipelines on Microbiota Studies: Does the Analytical “Microscope” Affect the Biological Interpretation?

 , ,
, ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Bioinformatics Analysis
2.3. Comparison of Bacterial Diversity, Richness and Composition and Statistical Analysis
3. Results and Discussion
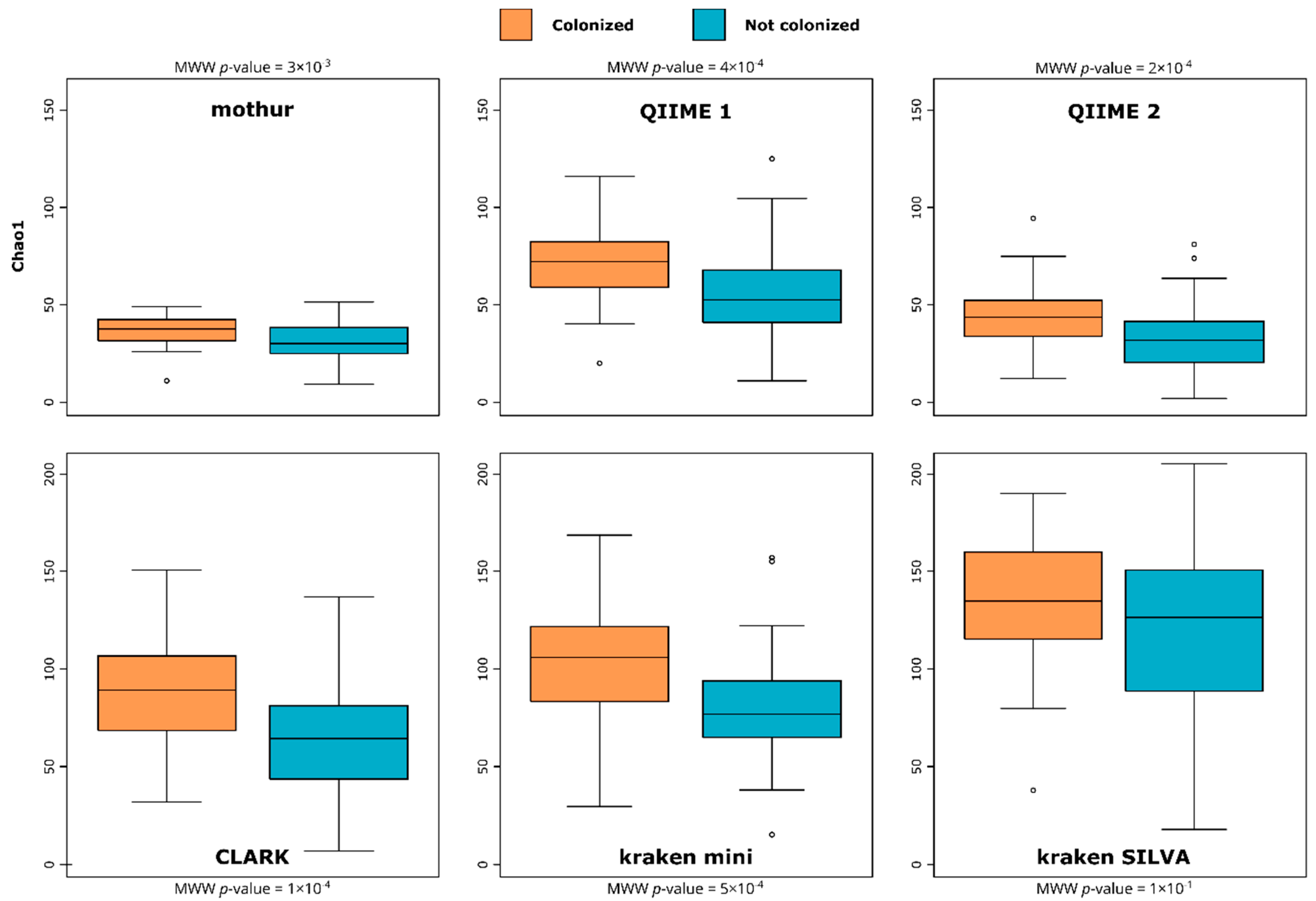
3.1. Richness/Diversity
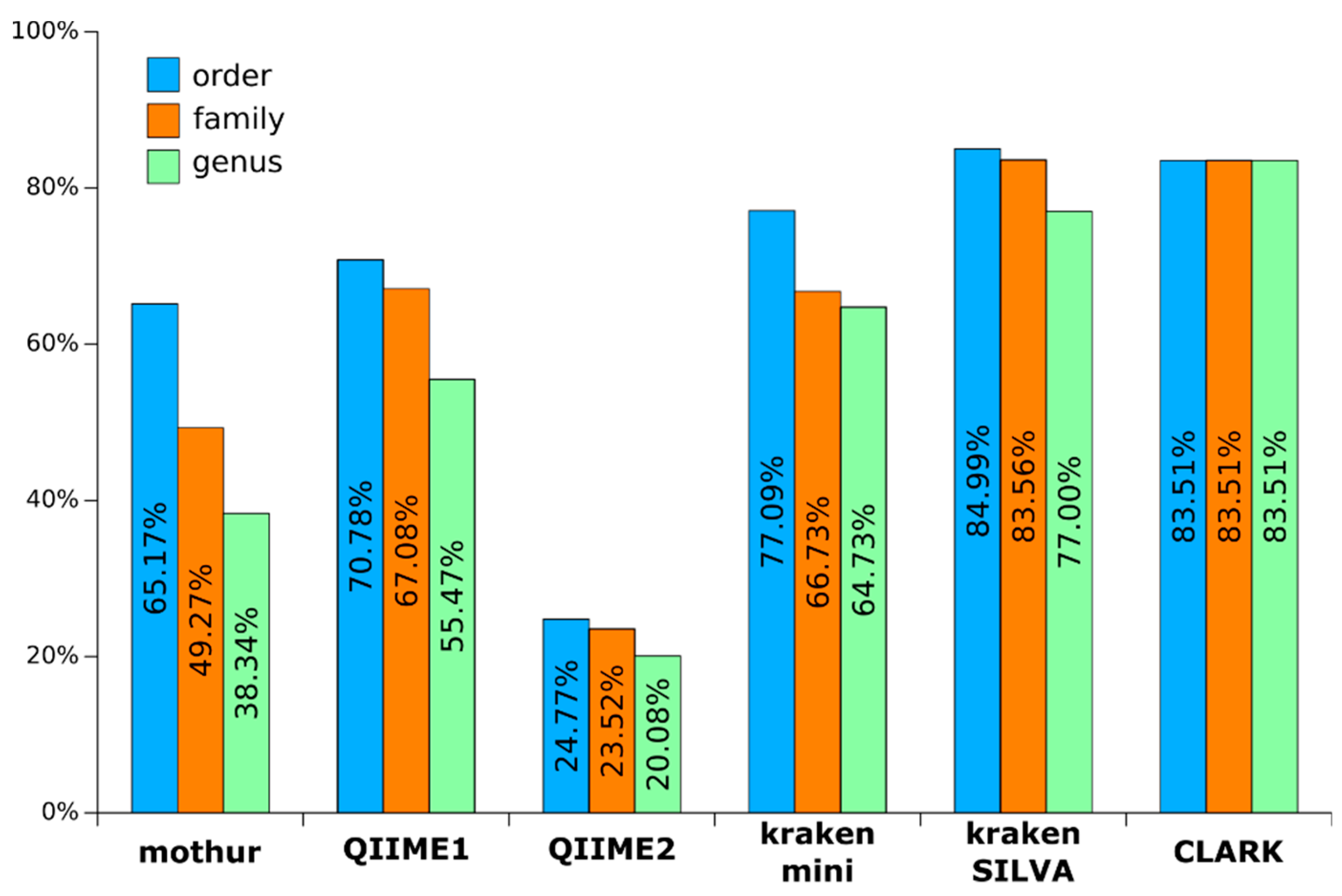
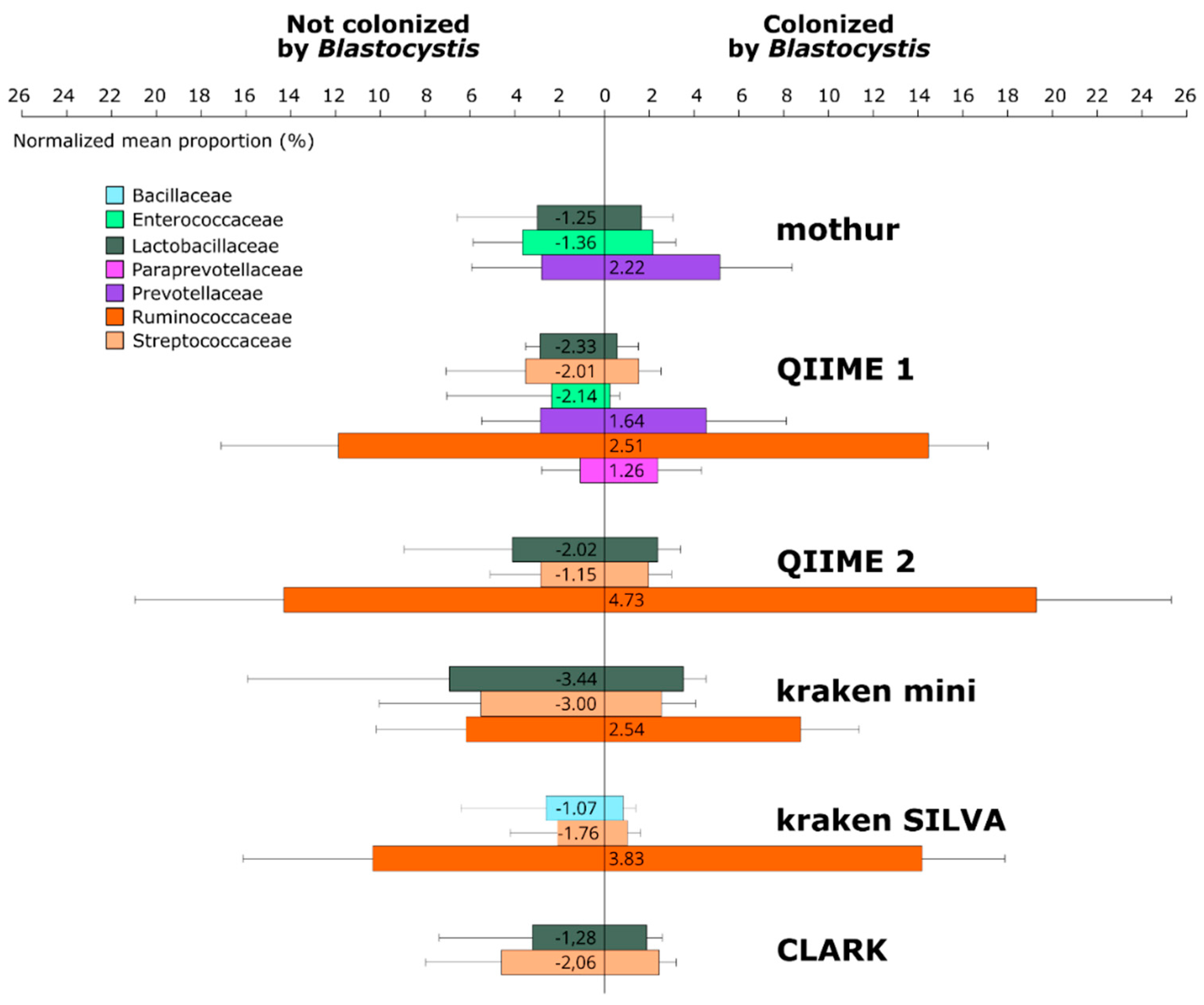
3.2. Composition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Pollock, J.; Glendinning, L.; Wisedchanwet, T.; Watson, M. The Madness of Microbiome: Attempting To Find Consensus “Best Practice” for 16S Microbiome Studies. Appl. Environ. Microbiol. 2018, 84. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.; Abu-Ali, G.; Vogtmann, E.; Fodor, A.A.; Ren, B.; Amir, A.; Schwager, E.; Crabtree, J.; Ma, S.; Abnet, C.C.; et al. Assessment of variation in microbial community amplicon sequencing by the Microbiome Quality Control (MBQC) project consortium. Nat. Biotechnol. 2017, 35, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Brooks, J.P.; Edwards, D.J.; Harwich, M.D.; Rivera, M.C.; Fettweis, J.M.; Serrano, M.G.; Reris, R.A.; Sheth, N.U.; Huang, B.; Girerd, P.; et al. The truth about metagenomics: Quantifying and counteracting bias in 16S rRNA studies. BMC Microbiol. 2015, 15, 66. [Google Scholar] [CrossRef] [PubMed]
- Wasserstein, R.L.; Lazar, N.A. The ASA’s Statement on p -Values: Context, Process, and Purpose. Am. Stat. 2016, 70, 129–133. [Google Scholar] [CrossRef]
- Nuzzo, R. Scientific method: Statistical errors. Nature 2014, 506, 150–152. [Google Scholar] [CrossRef] [PubMed]
- Siegwald, L.; Touzet, H.; Lemoine, Y.; Hot, D.; Audebert, C.; Caboche, S. Assessment of Common and Emerging Bioinformatics Pipelines for Targeted Metagenomics. PLoS ONE 2017, 12, e0169563. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, M.; Ijaz, U.Z.; D’Amore, R.; Hall, N.; Sloan, W.T.; Quince, C. Insight into biases and sequencing errors for amplicon sequencing with the Illumina MiSeq platform. Nucleic Acids Res. 2015, 43, e37. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Amir, A.; McDonald, D.; Navas-Molina, J.A.; Kopylova, E.; Morton, J.T.; Zech Xu, Z.; Kightley, E.P.; Thompson, L.R.; Hyde, E.R.; Gonzalez, A.; et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. MSystems 2017, 2, e00191-16. [Google Scholar] [CrossRef]
- Audebert, C.; Even, G.; Cian, A.; Blastocystis Investigation Group; Loywick, A.; Merlin, S.; Viscogliosi, E.; Chabé, M.; El Safadi, D.; Certad, G.; et al. Colonization with the enteric protozoa Blastocystis is associated with increased diversity of human gut bacterial microbiota. Sci. Rep. 2016, 6, 25255. [Google Scholar] [CrossRef]
- Hollister, E.B.; Gao, C.; Versalovic, J. Compositional and functional features of the gastrointestinal microbiome and their effects on human health. Gastroenterology 2014, 146, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Siegwald, L.; Audebert, C.; Even, G.; Viscogliosi, E.; Caboche, S.; Chabé, M. Targeted metagenomic sequencing data of human gut microbiota associated with Blastocystis colonization. Sci. Data 2017, 4, 170081. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [PubMed]
- Ounit, R.; Wanamaker, S.; Close, T.J.; Lonardi, S. CLARK: Fast and accurate classification of metagenomic and genomic sequences using discriminative k-mers. BMC Genom. 2015, 16, 236. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Wilcoxon, F. Individual Comparisons by Ranking Methods. Biom. Bull. 1945, 1, 80. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef]
- White, J.R.; Nagarajan, N.; Pop, M. Statistical Methods for Detecting Differentially Abundant Features in Clinical Metagenomic Samples. PLoS Comput. Biol. 2009, 5, e1000352. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.-M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Mosca, A.; Leclerc, M.; Hugot, J.P. Gut microbiota diversity and human diseases: Should we reintroduce key predators in our ecosystem? Front. Microbiol. 2016, 7, 455. [Google Scholar] [CrossRef] [PubMed]
- Mann, H.B.; Whitney, D.R. On a Test of Whether one of Two Random Variables is Stochastically Larger than the Other. Ann. Math. Stat. 1947, 18, 50–60. [Google Scholar] [CrossRef]
- Biddle, A.; Stewart, L.; Blanchard, J.; Leschine, S. Untangling the genetic basis of fibrolytic specialization by lachnospiraceae and ruminococcaceae in diverse gut communities. Diversity 2013, 5, 627–640. [Google Scholar] [CrossRef]
- Manichanh, C.; Borruel, N.; Casellas, F.; Guarner, F. The gut microbiota in IBD. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 599–608. [Google Scholar] [CrossRef]
- Lyra, A.; Lahtine, S. Dysbiosis of the Intestinal Microbiota in IBS. Curr. Concepts Colon. Disord. 2012, 261–276. [Google Scholar]
- Pham, T.A.N.; Lawley, T.D. Emerging insights on intestinal dysbiosis during bacterial infections. Curr. Opin. Microbiol. 2014, 17, 67–74. [Google Scholar] [CrossRef]
- Cai, Y.; Sun, Y. ESPRIT-Tree: Hierarchical clustering analysis of millions of 16S rRNA pyrosequences in quasilinear computational time. Nucleic Acids Res. 2011, 39, e95. [Google Scholar] [CrossRef]
- Winter, S.E.; Bäumler, A.J. Why related bacterial species bloom simultaneously in the gut: Principles underlying the “like will to like” concept. Cell. Microbiol. 2014, 16, 179–184. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siegwald, L.; Caboche, S.; Even, G.; Viscogliosi, E.; Audebert, C.; Chabé, M. The Impact of Bioinformatics Pipelines on Microbiota Studies: Does the Analytical “Microscope” Affect the Biological Interpretation? Microorganisms 2019, 7, 393. https://doi.org/10.3390/microorganisms7100393
Siegwald L, Caboche S, Even G, Viscogliosi E, Audebert C, Chabé M. The Impact of Bioinformatics Pipelines on Microbiota Studies: Does the Analytical “Microscope” Affect the Biological Interpretation? Microorganisms. 2019; 7(10):393. https://doi.org/10.3390/microorganisms7100393
Chicago/Turabian StyleSiegwald, Léa, Ségolène Caboche, Gaël Even, Eric Viscogliosi, Christophe Audebert, and Magali Chabé. 2019. "The Impact of Bioinformatics Pipelines on Microbiota Studies: Does the Analytical “Microscope” Affect the Biological Interpretation?" Microorganisms 7, no. 10: 393. https://doi.org/10.3390/microorganisms7100393
APA StyleSiegwald, L., Caboche, S., Even, G., Viscogliosi, E., Audebert, C., & Chabé, M. (2019). The Impact of Bioinformatics Pipelines on Microbiota Studies: Does the Analytical “Microscope” Affect the Biological Interpretation? Microorganisms, 7(10), 393. https://doi.org/10.3390/microorganisms7100393