Characterised Flavin-Dependent Two-Component Monooxygenases from the CAM Plasmid of Pseudomonas putida ATCC 17453 (NCIMB 10007): ketolactonases by Another Name


Abstract
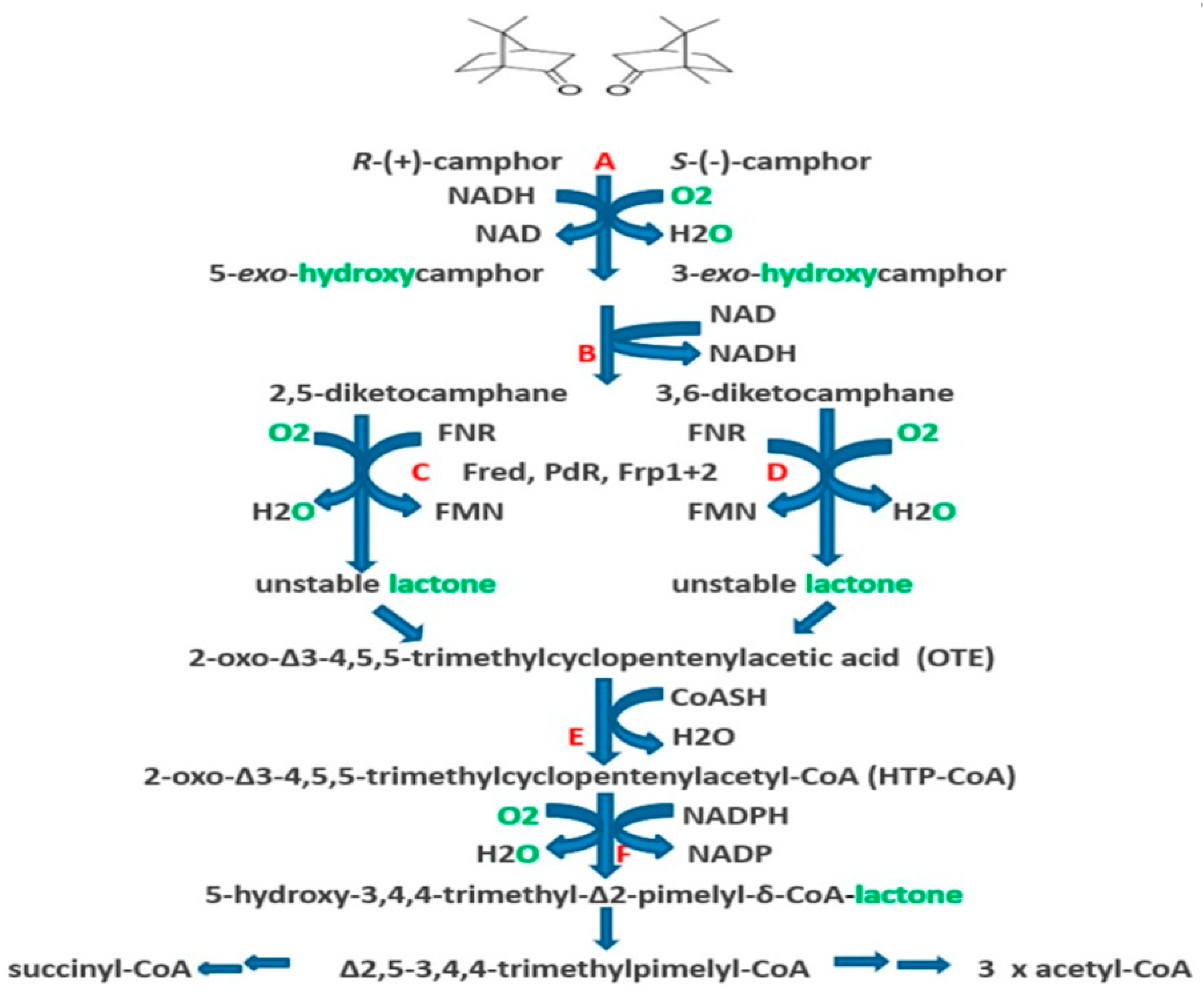
:1. Introduction
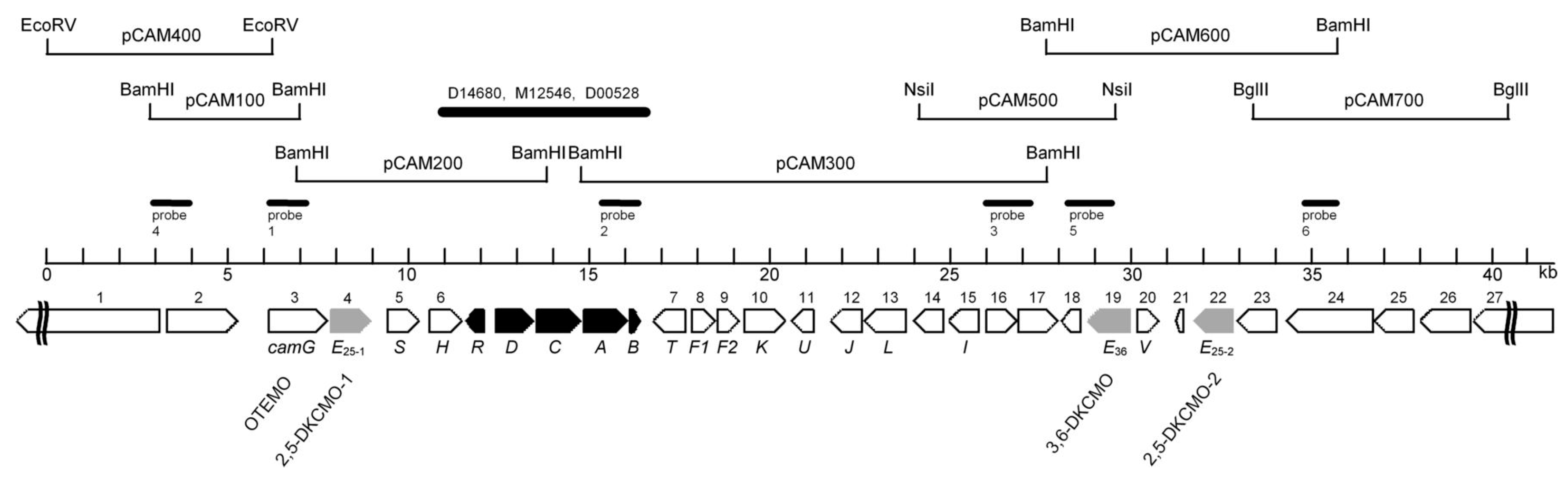
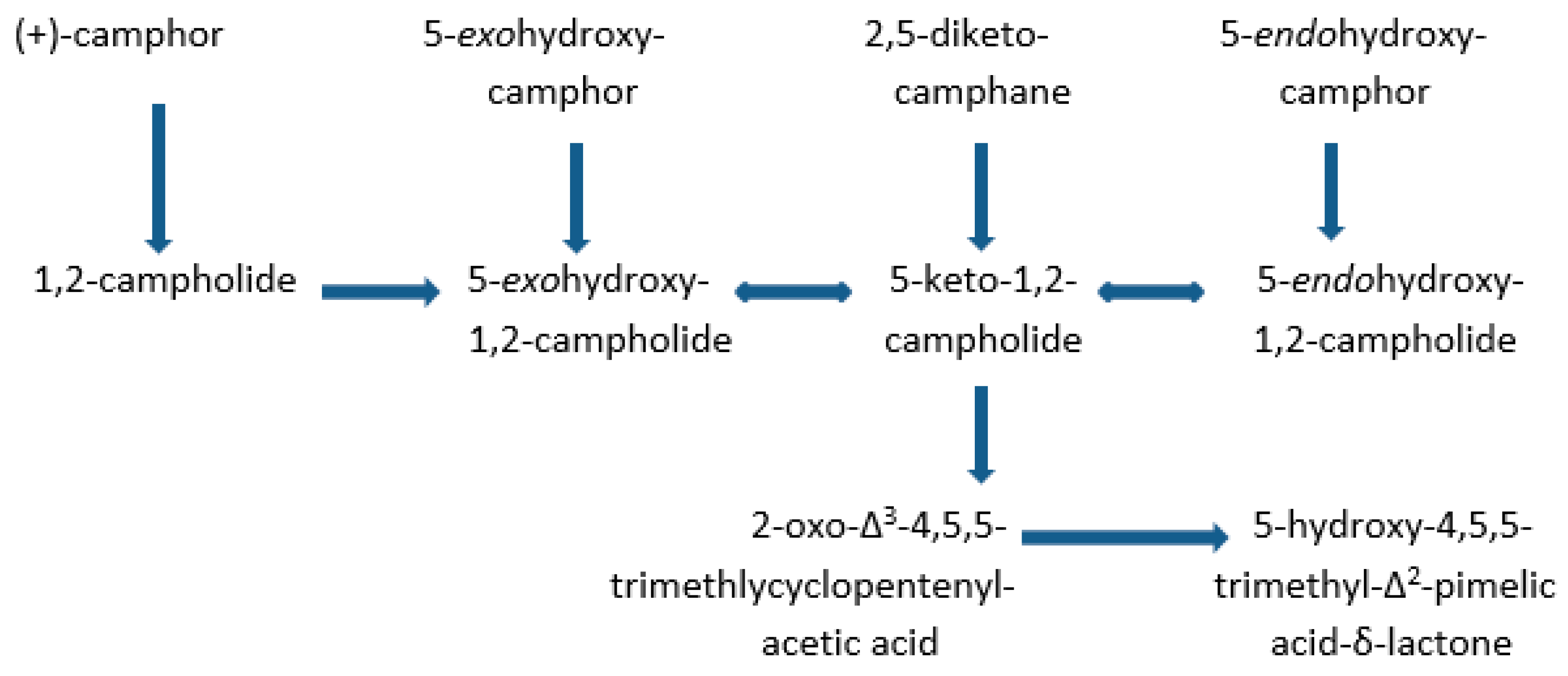
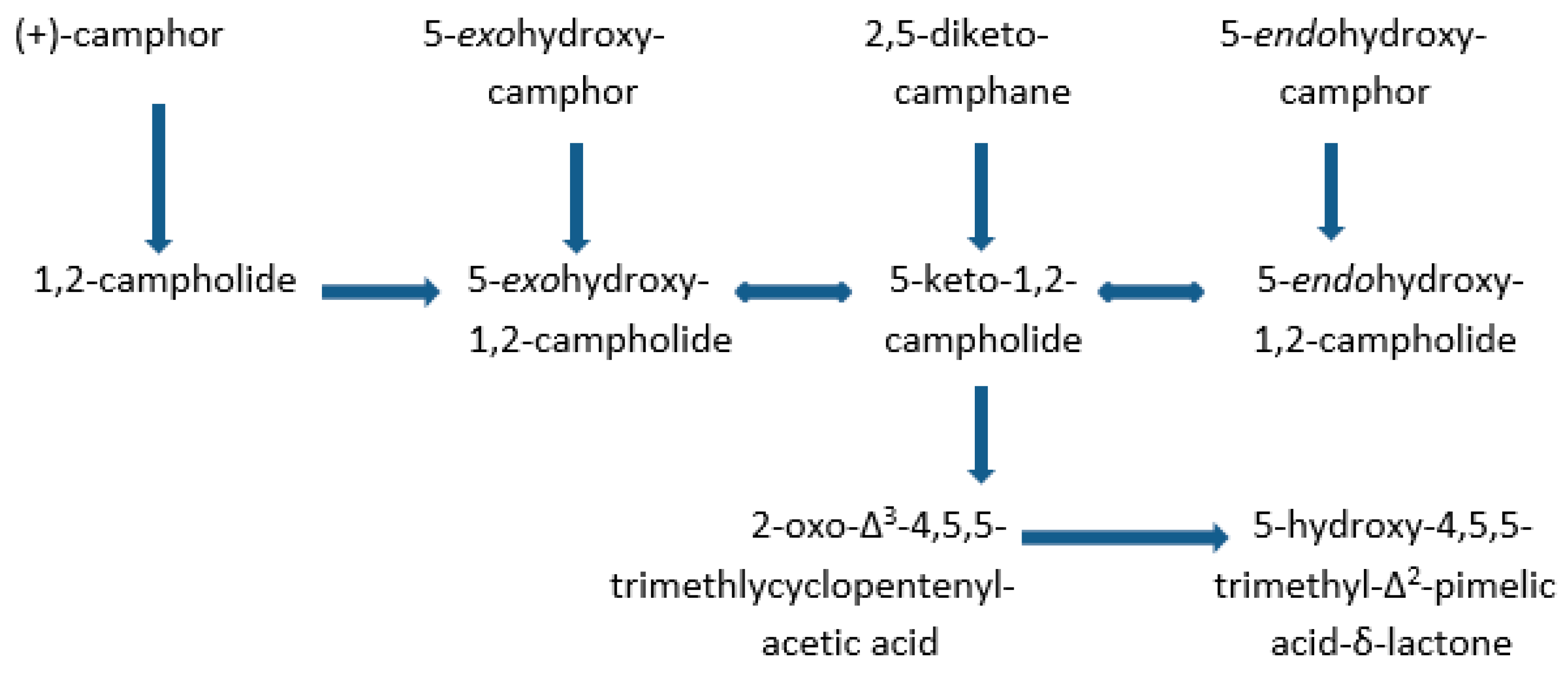
2. Early Research at the University of Illinois
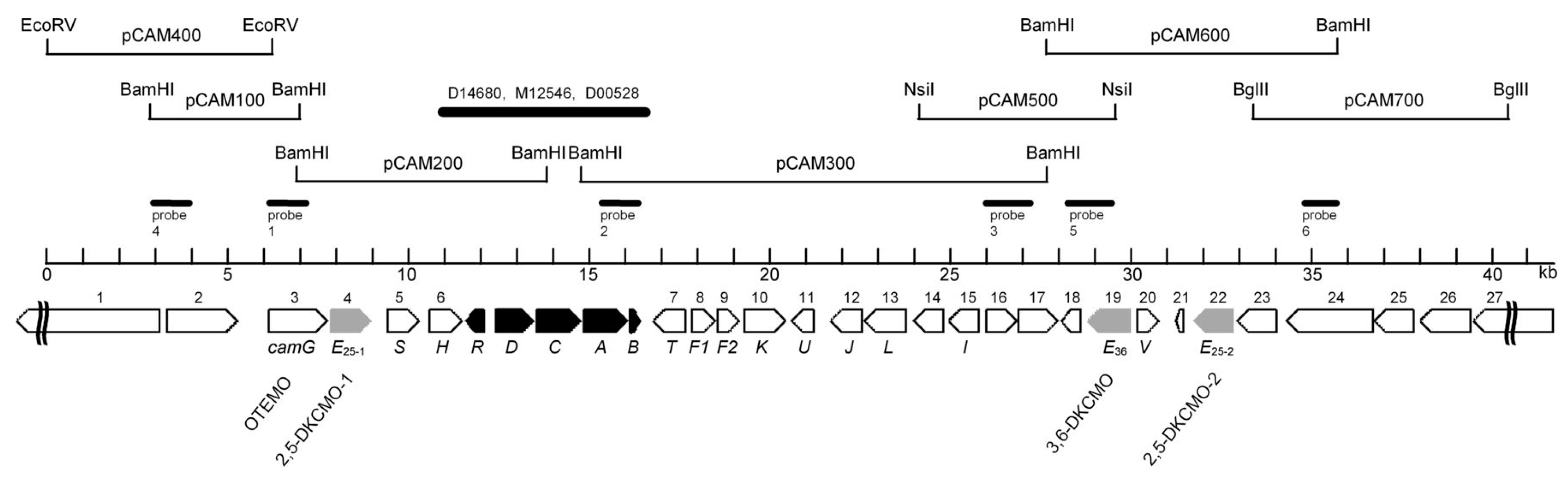
3. Post-Gunsalus Research at the University of Aberystwyth
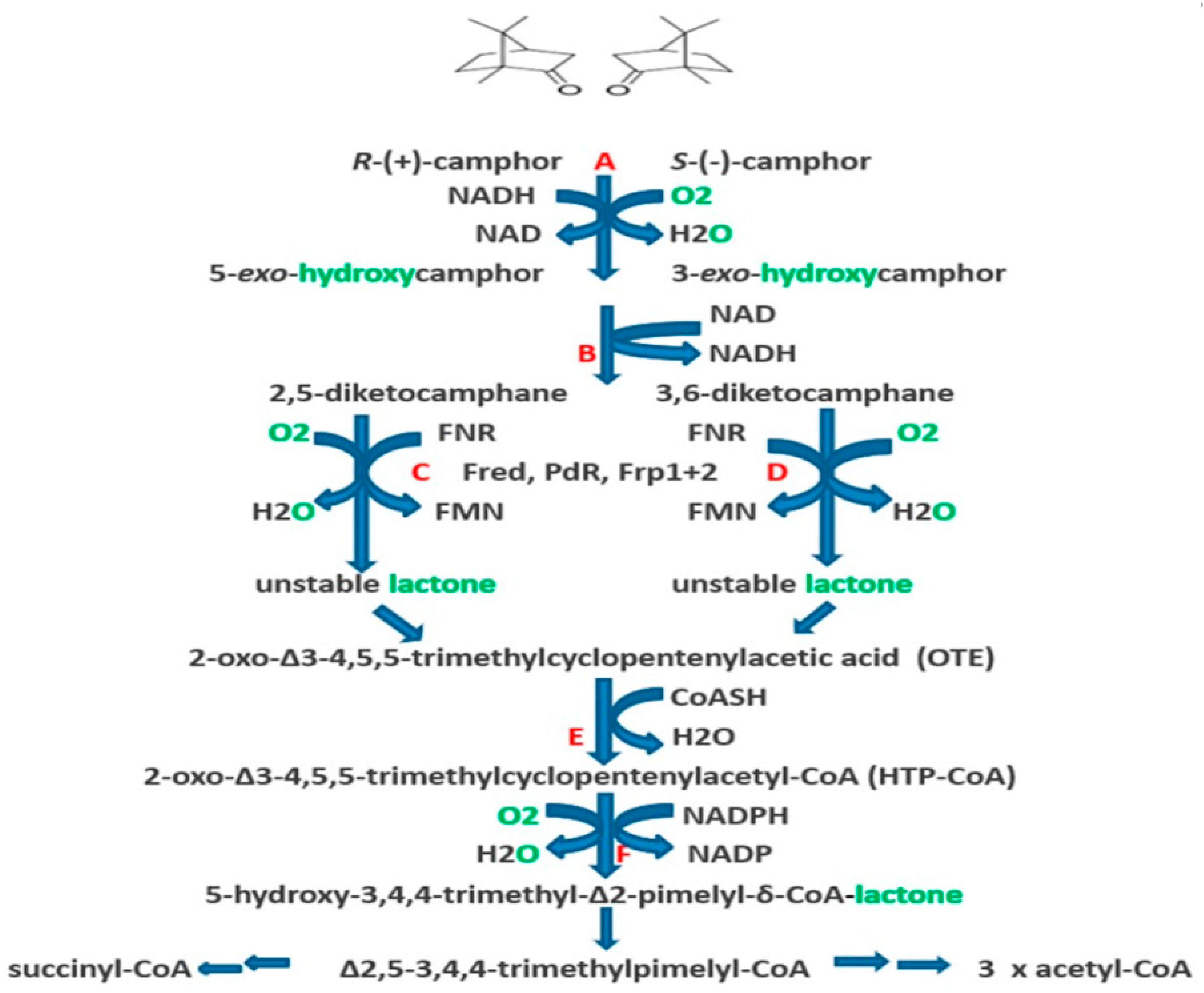
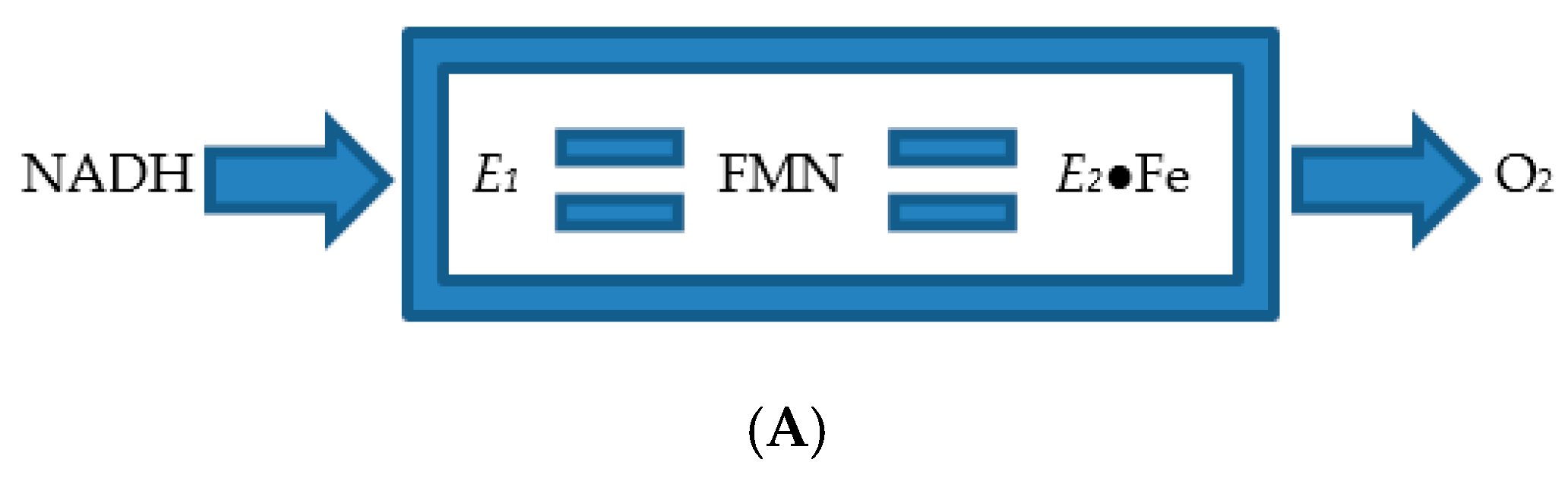
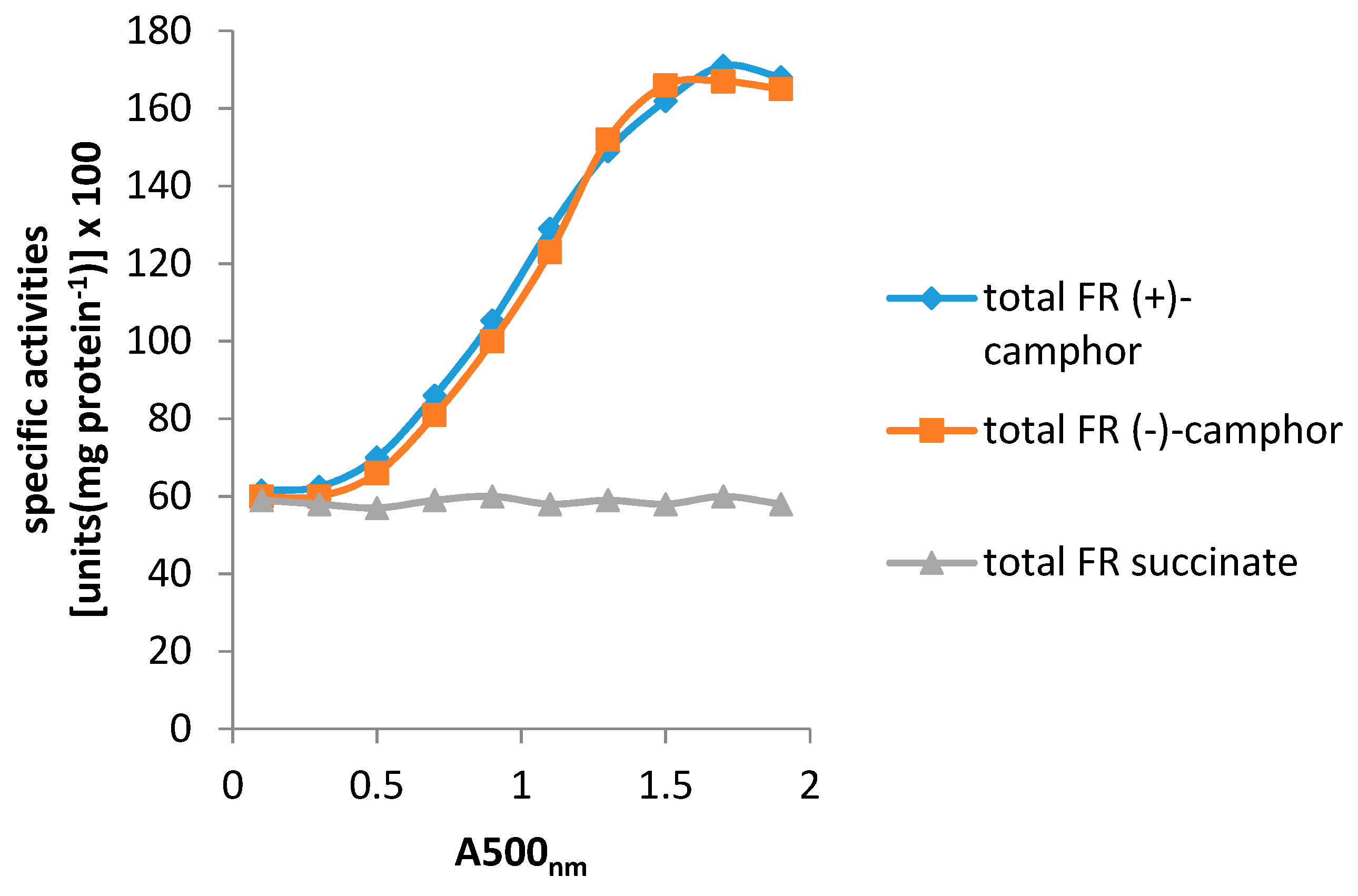
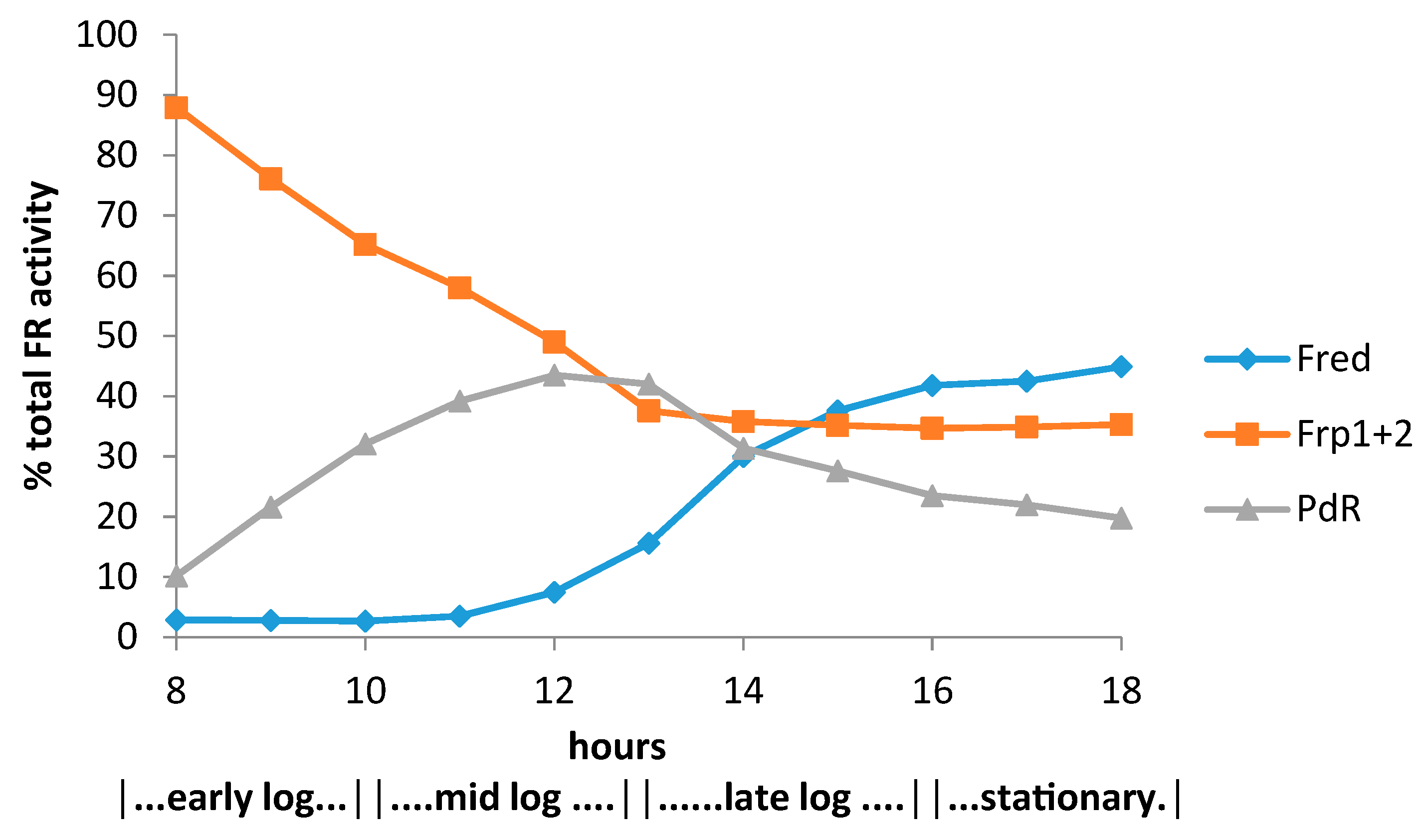
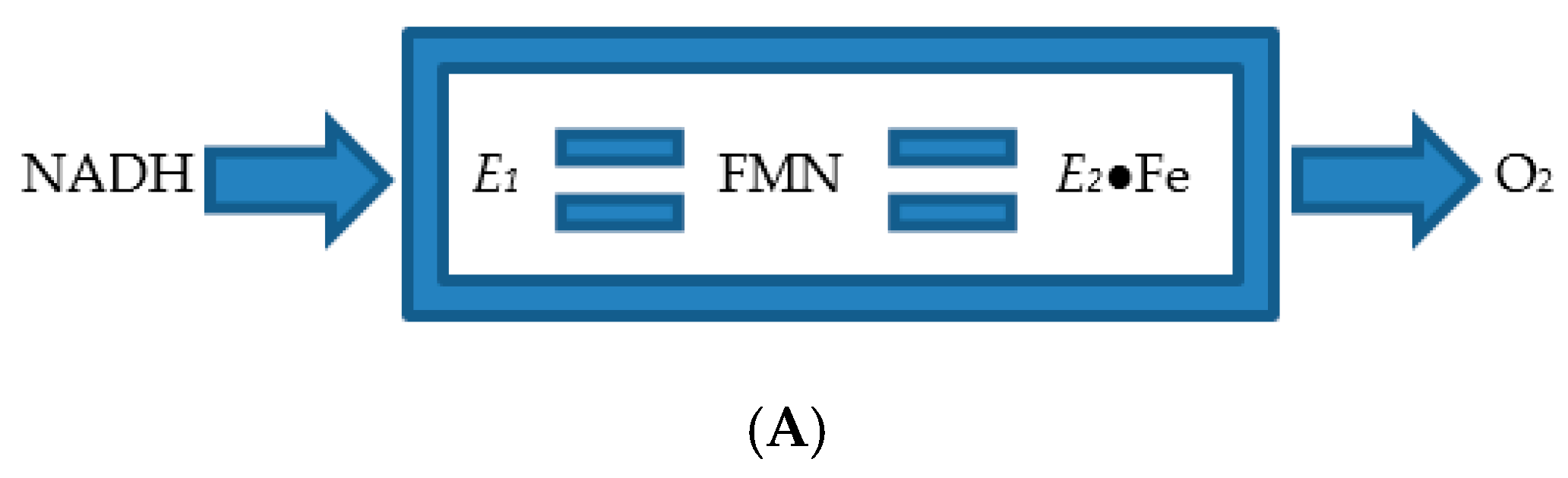
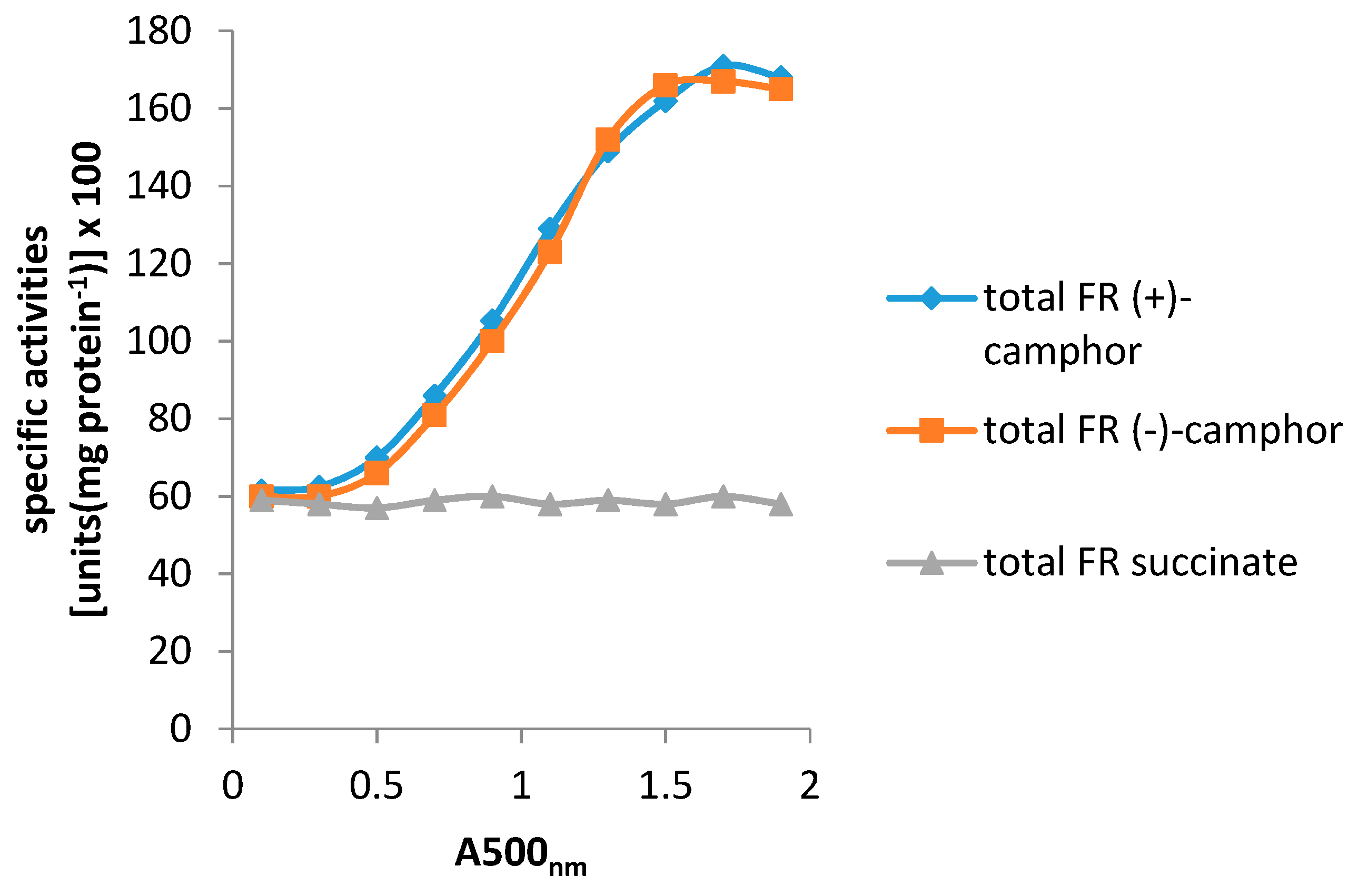
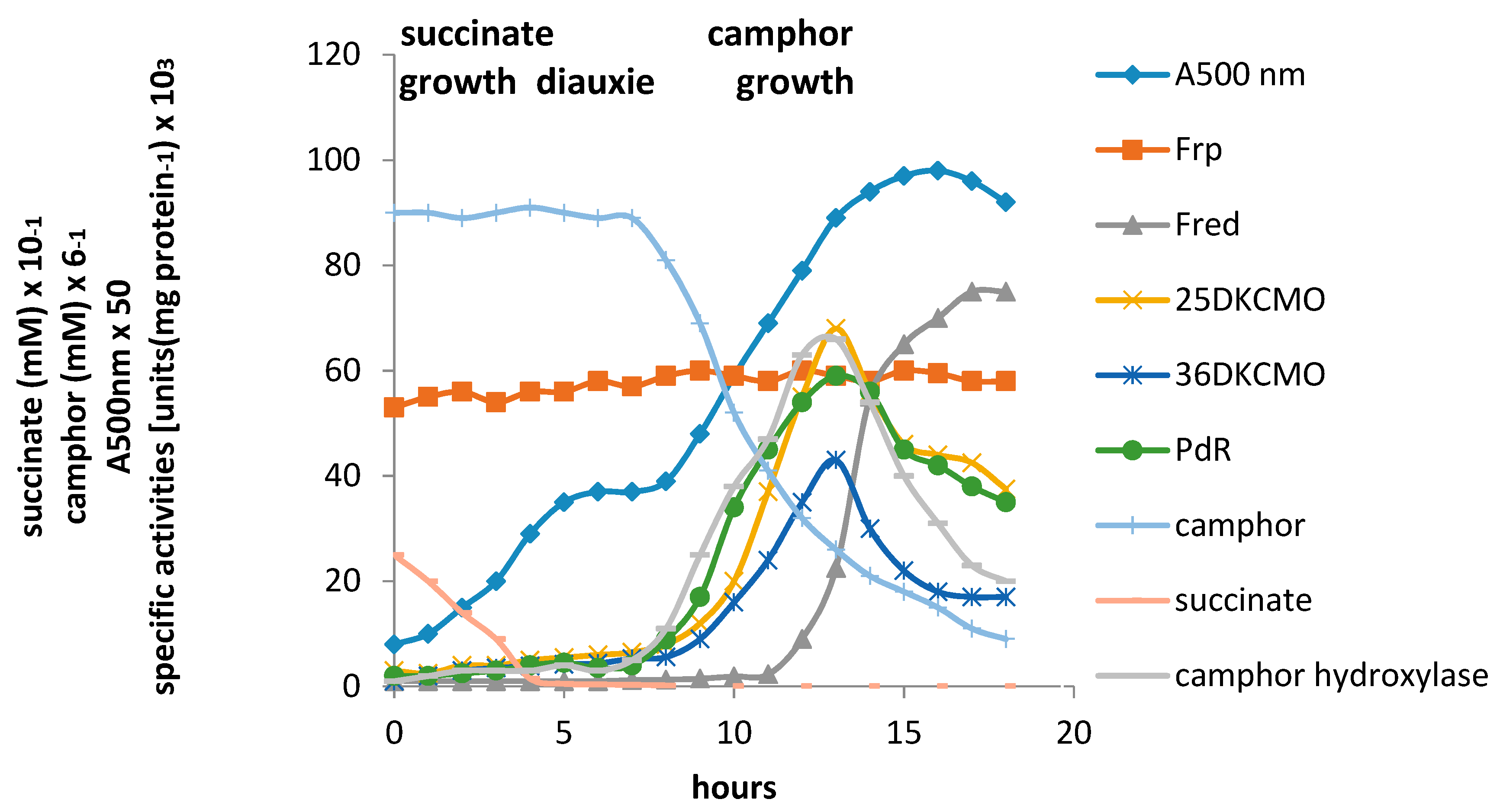
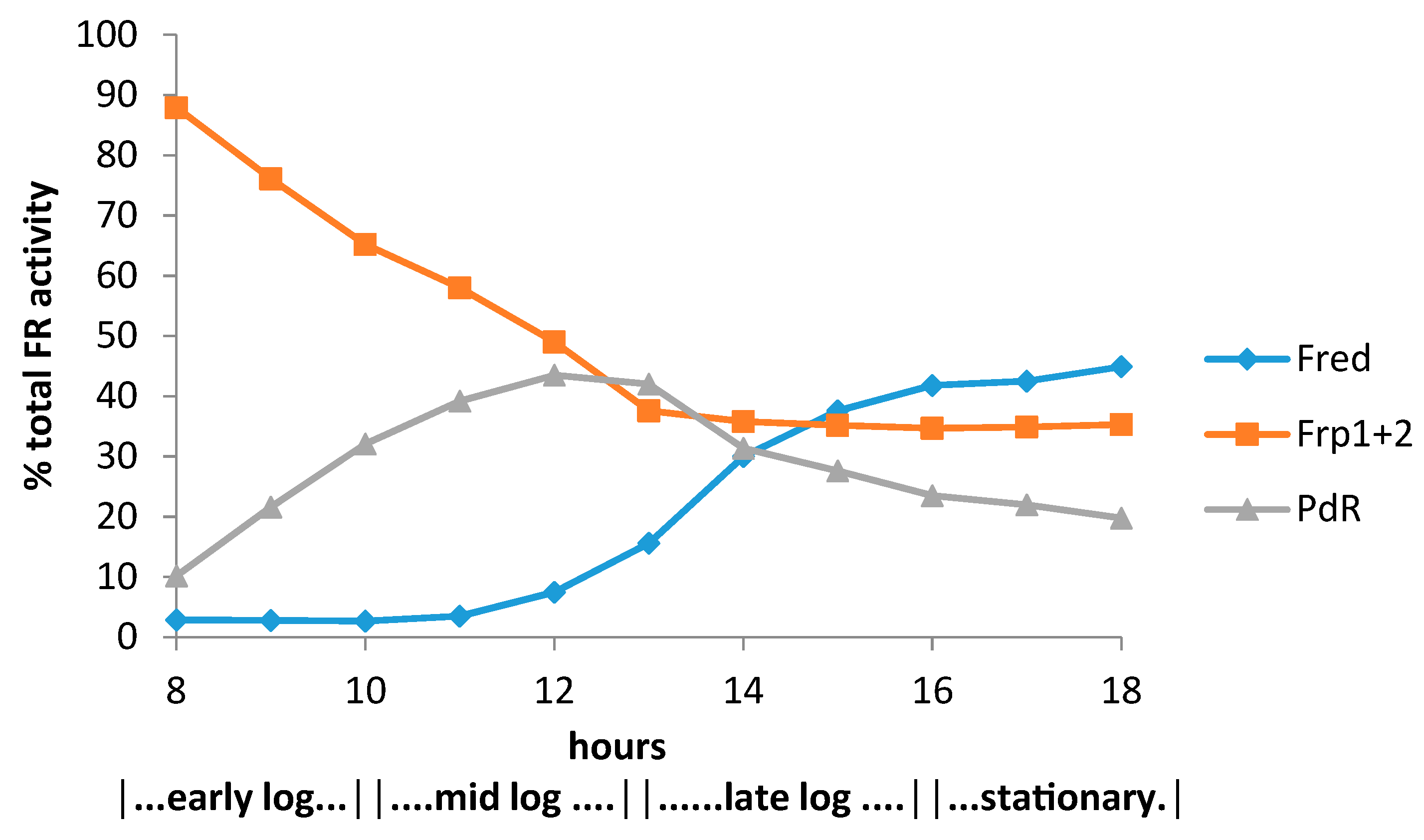
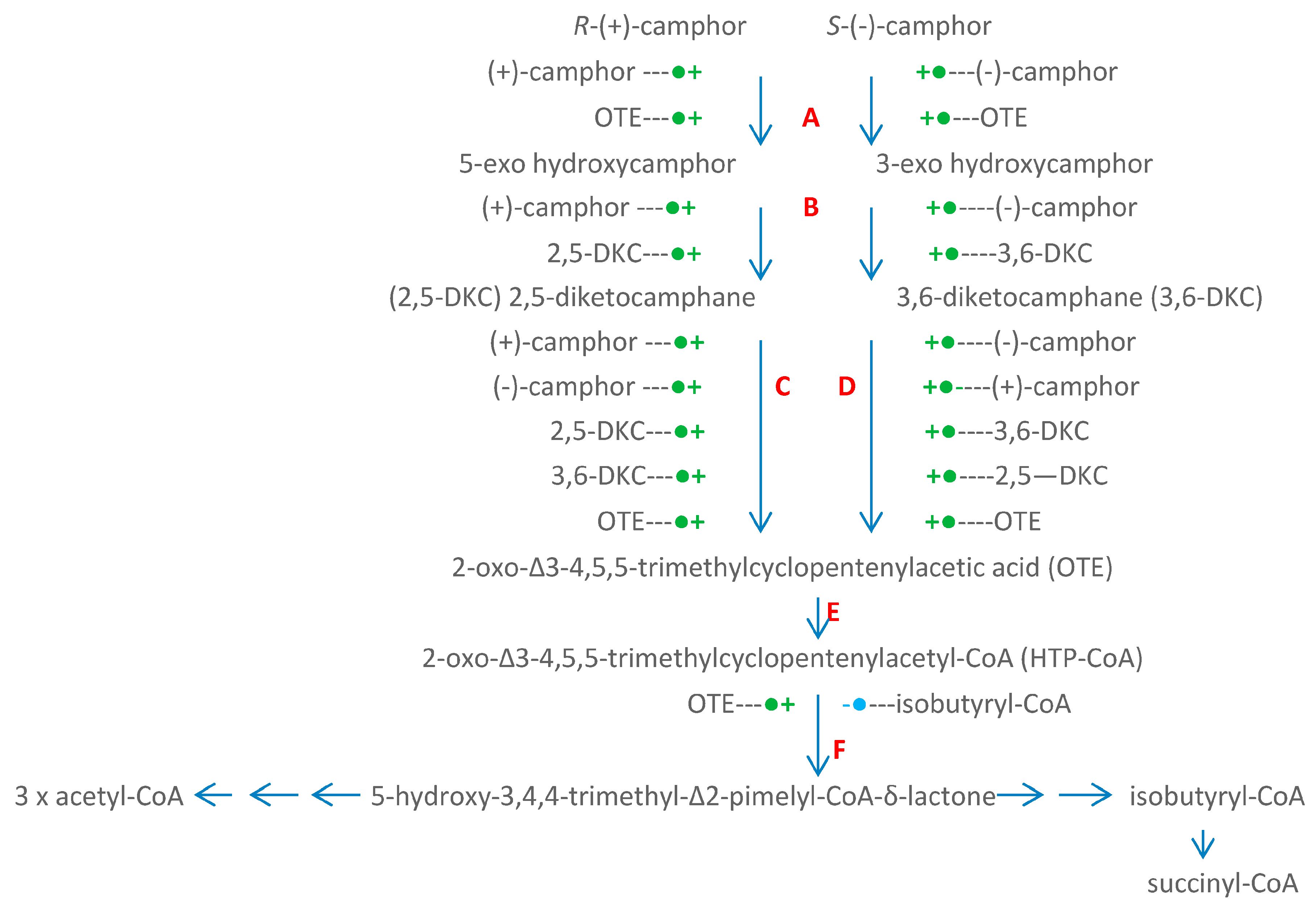
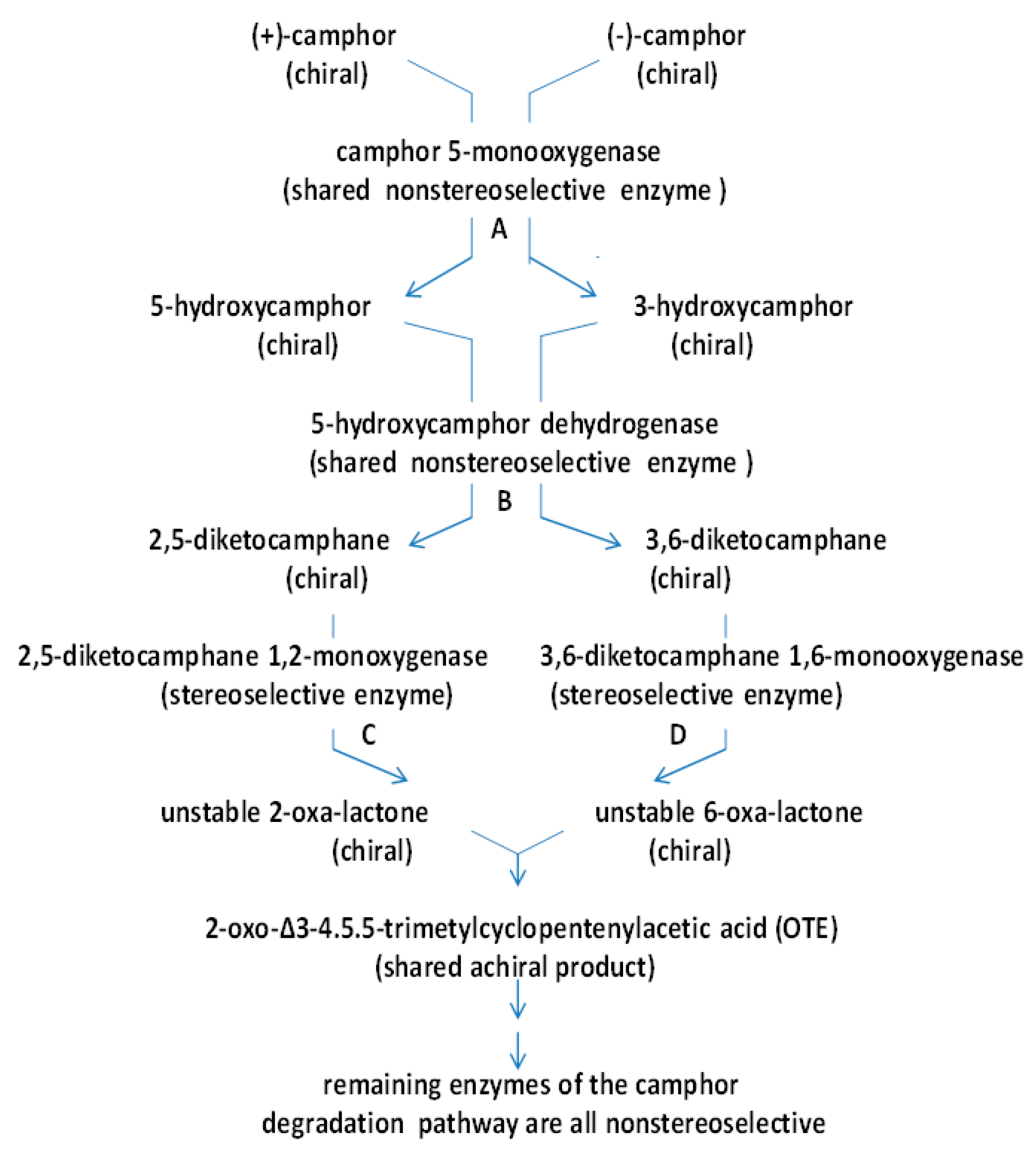
4. A New Millennium and a New Perspective—The Ketolactonases as fd-TCMOs
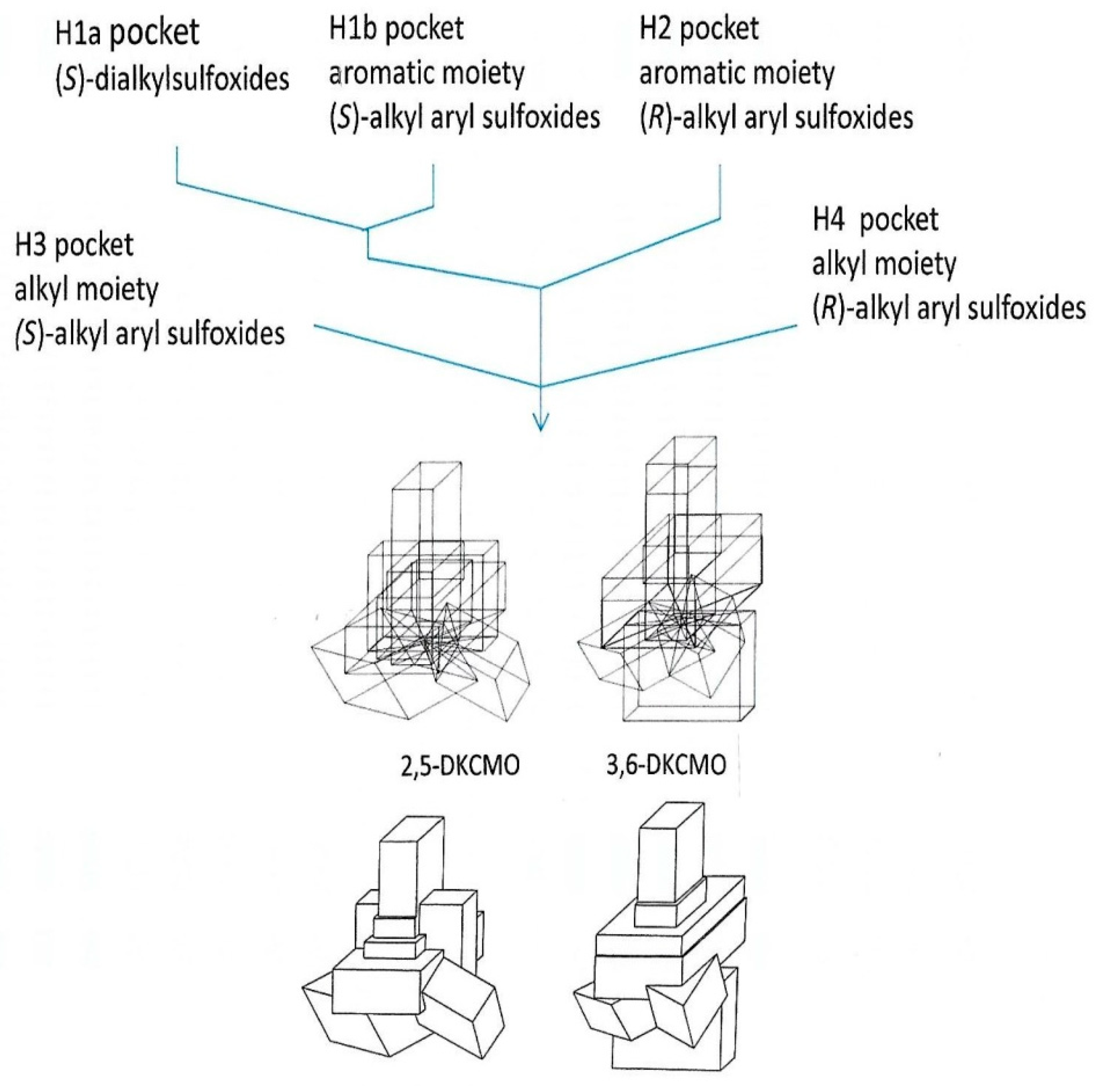
5. Structural Studies of the Ketolactonases
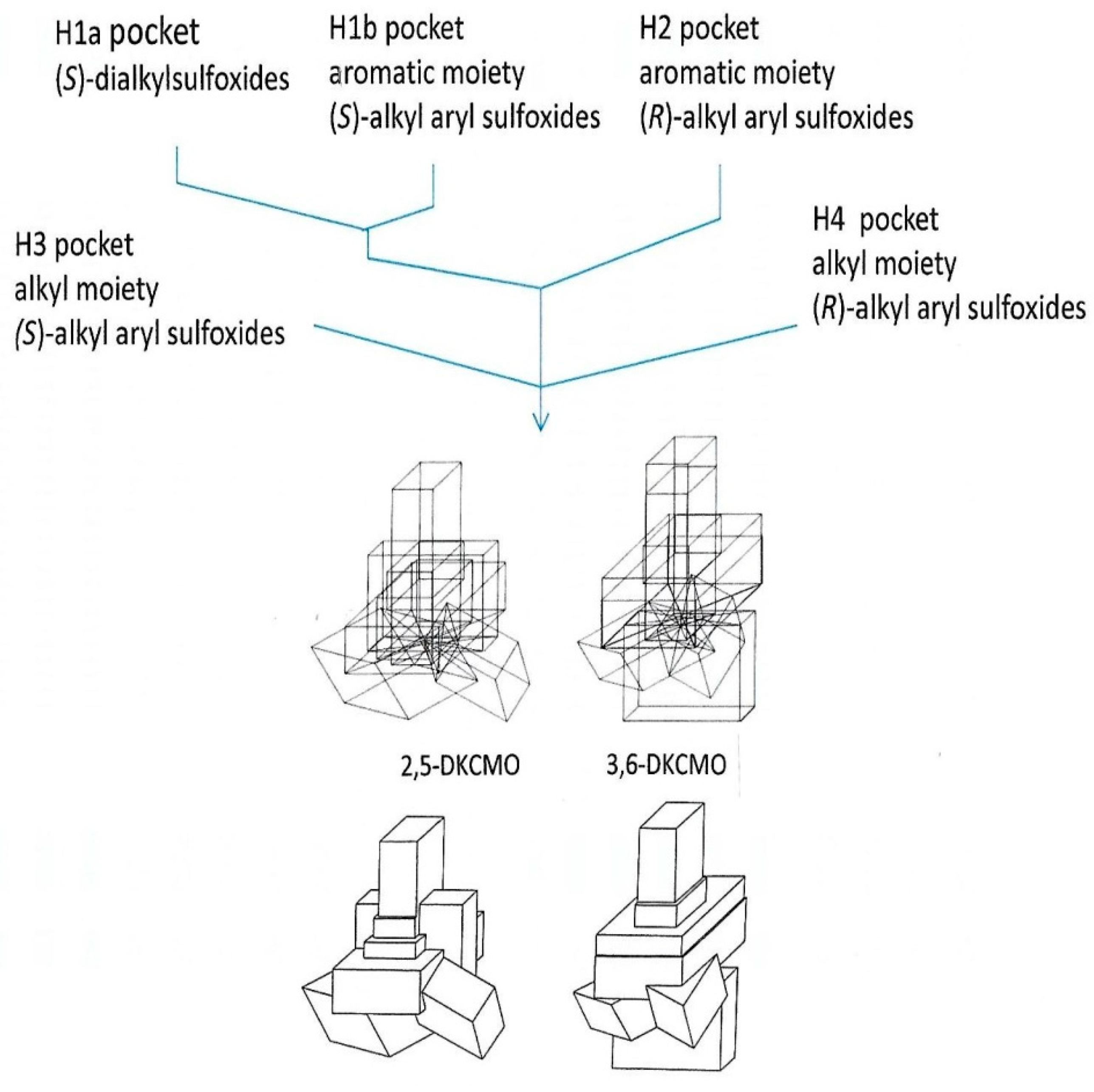
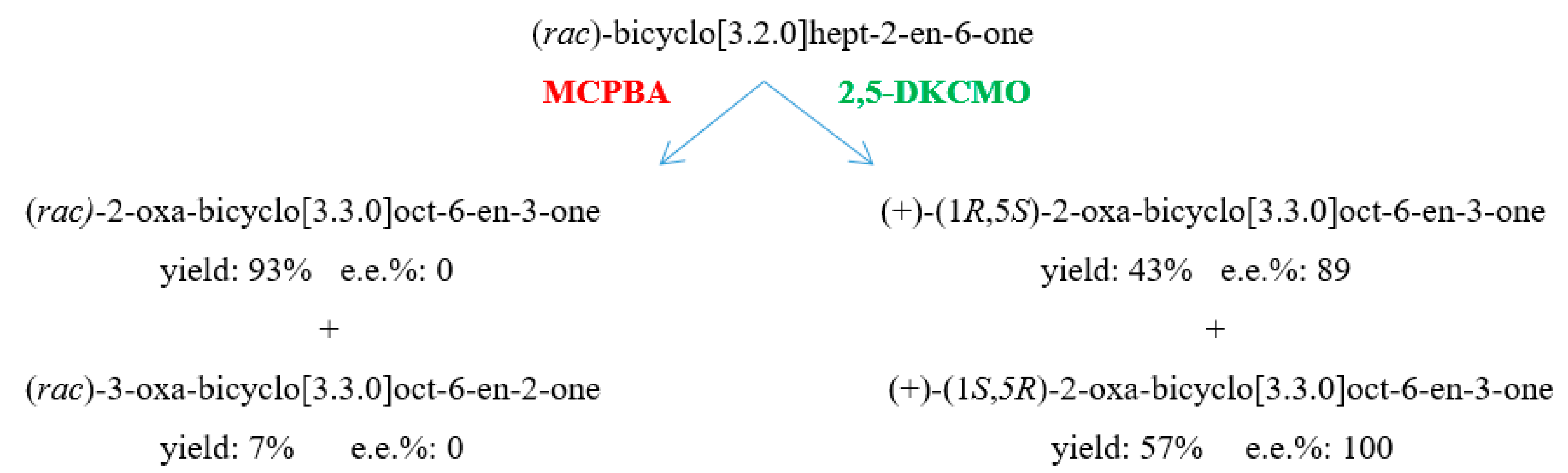
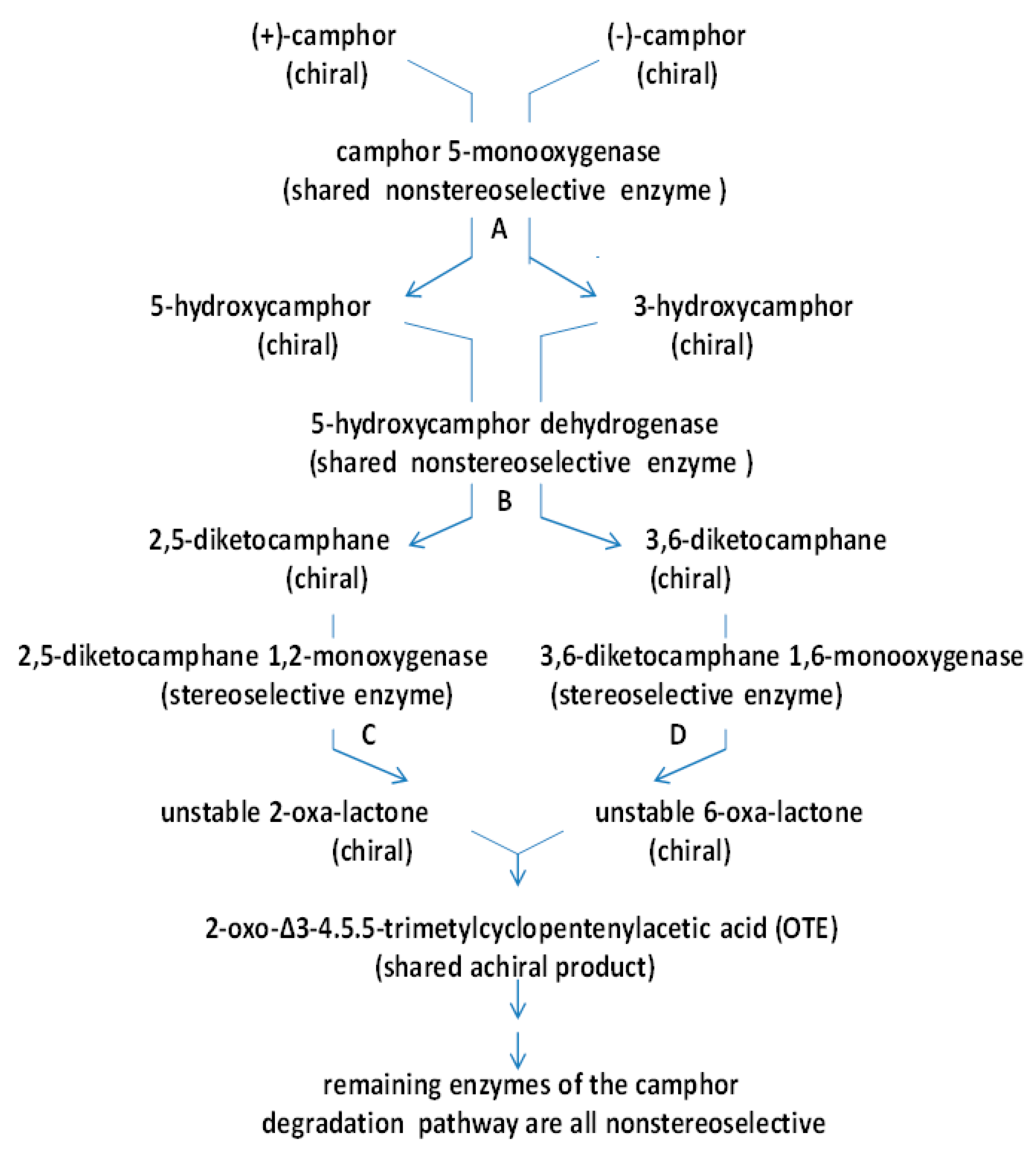
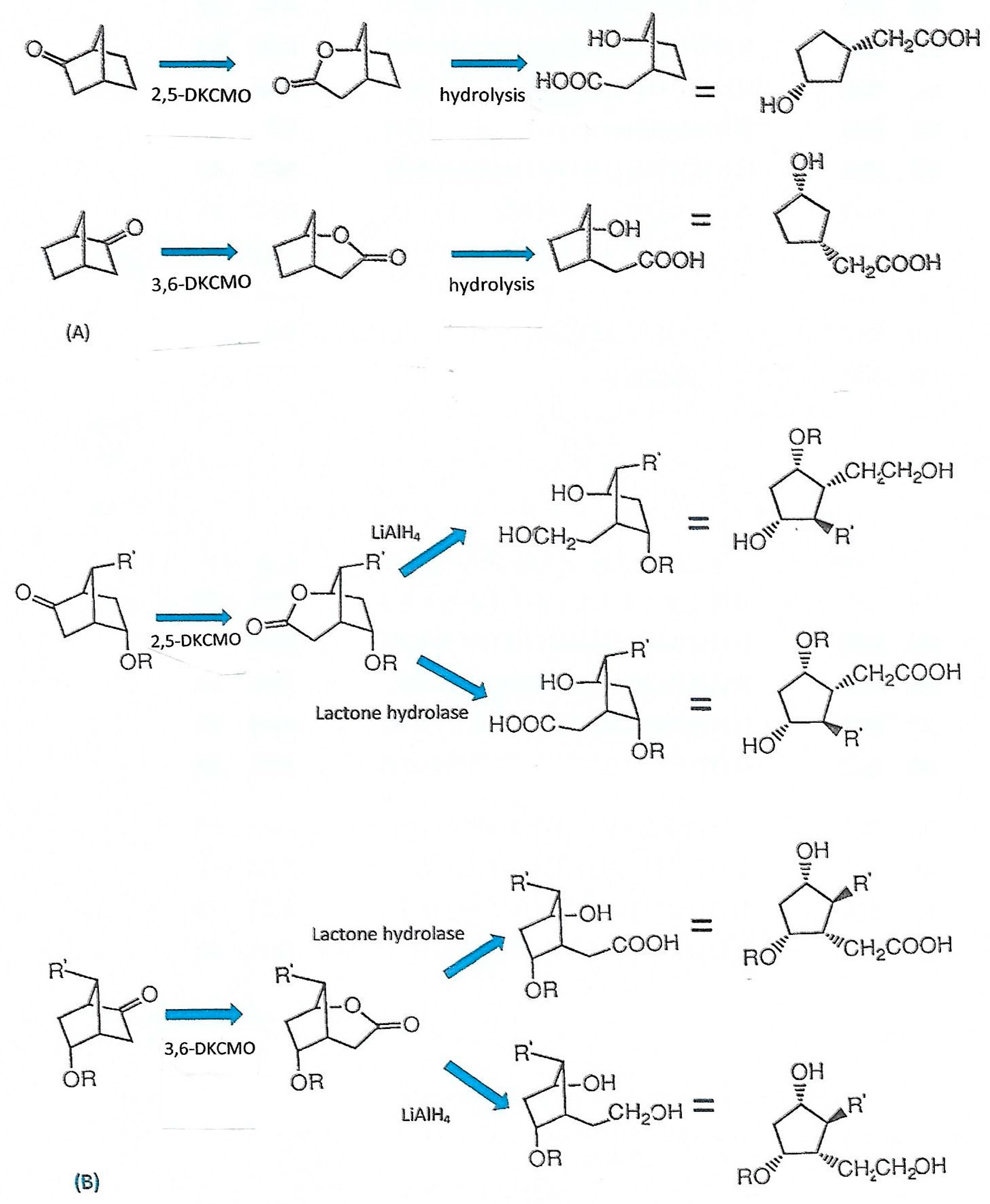
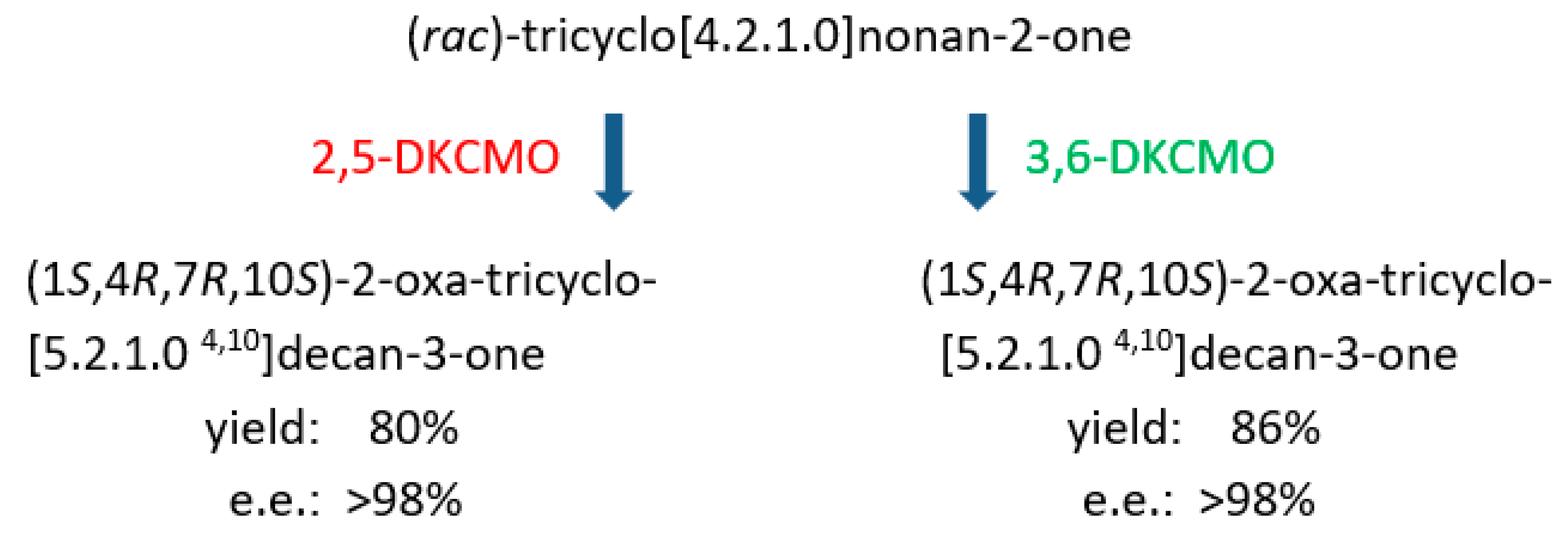
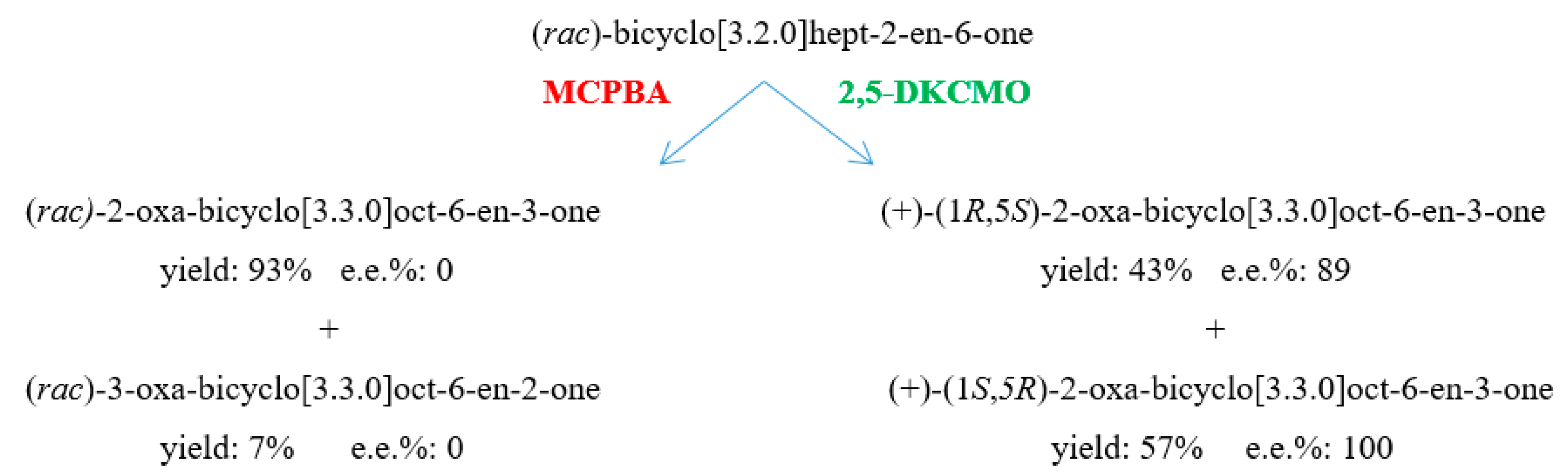
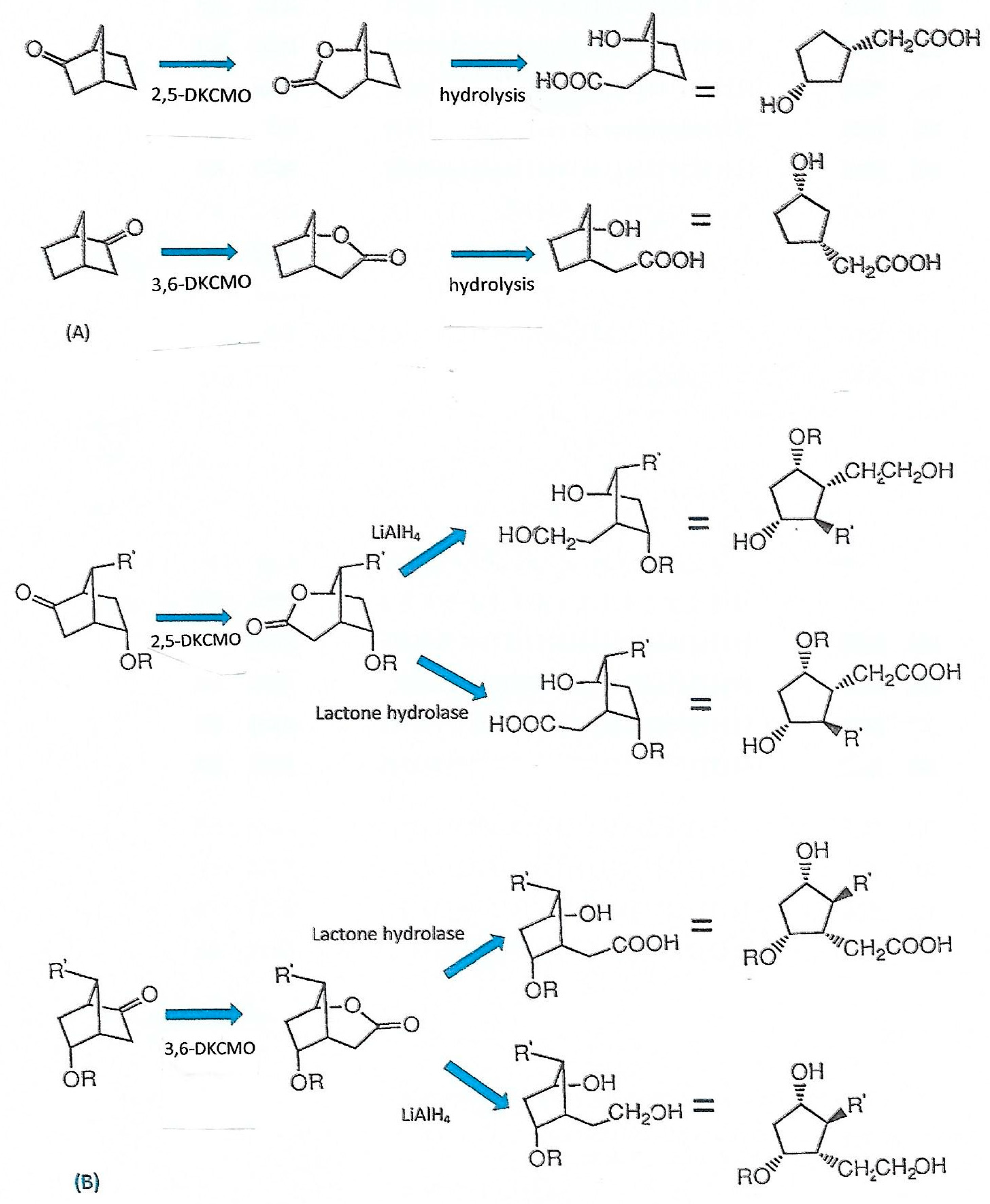
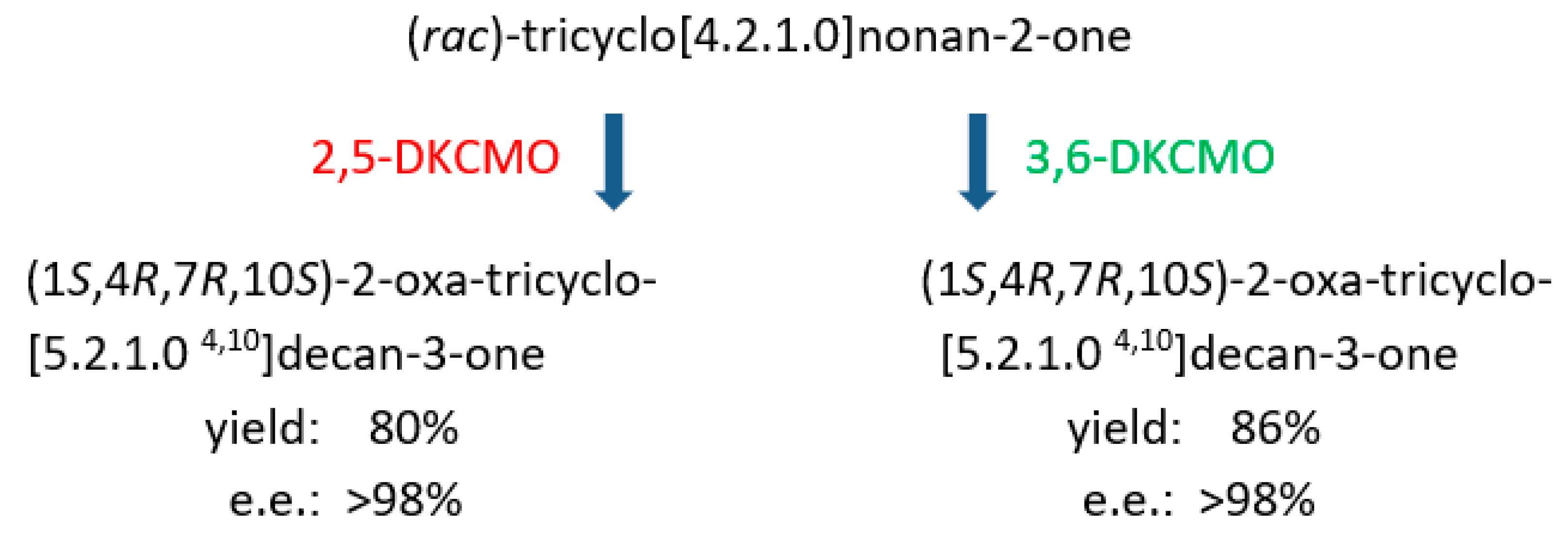
6. Chemo-, Regio-, and Enantioselectivity of the Ketolactonases: Natural and Xenobiotic Substrates, Plus Biocatalytic Applications
Funding
Acknowledgments
Conflicts of Interest
References
- Chakrabarty, A.M.; Gunsalus, C.F.; Gunsalus, I.C. Transduction and the clustering of genes in fluorescent Pseudomonads. Proc. Natl. Acad. Sci. USA 1968, 60, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Rheinwald, J.G.; Chakrabarty, A.M.; Gunsalus, I.C. A transmissible plasmid controlling camphor oxidation in Pseudomonas putida. Proc. Natl. Acad. Sci. USA 1973, 70, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Gunsalus, I.C.; Marshall, V.P. Monoterpene dissimilation. CRC Crit. Rev. Microbiol. 1971, 1, 291–310. [Google Scholar] [CrossRef]
- Iwaki, H.; Grosse, S.; Bergeron, H.; Leisch, H.; Morley, K.; Hasegawa, Y.; Lau, P.C. Camphor pathway redox: Functional recombinant expression of 2,5- and 3,6-diketocamphane monooxygenases in Pseudomonas putida ATCC 17453 with their cognate flavin reductase catalysing Baeyer-Villiger reactions. Appl. Environ. Microbiol. 2013, 79, 3282–3293. [Google Scholar] [CrossRef] [PubMed]
- Willetts, A. Structural studies and synthetic applications of Baeyer-Villiger monooxygenases. Trends Biotechnol. 1998, 15, 55–62. [Google Scholar] [CrossRef]
- Jones, K.H.; Smith, R.T.; Trudgill, P.W. Diketocamphane enantiomer-specific ‘Baeyer-Villiger’ monooxygenases from camphor-grown Pseudomonas putida ATCC 17453. J. Gen. Microbiol. 1993, 139, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Unger, B.P.; Sligar, S.G.; Gunsalus, I.C. Pseudomonas cytochrome P-450. In The Bacteria; Sokatch, J.R., Ed.; Academic Press: New York, NY, USA, 1986; Volume 10, pp. 557–589. [Google Scholar]
- Poulos, T.P. Structural biology of heme monooxygenases. Biochem. Biophys. Res. Commun. 2005, 338, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Leisch, H.; Shi, R.; Grosse, S.; Morley, K.; Bergeron, H.; Cygler, M.; Iwaki, H.; Hasegawa, Y.; Lau, P.C.K. Cloning, Baeyer-Villiger biooxidations, and structures of the camphor pathway 2-oxo-∆3-4,5,5-trimethylcyclopentenylacetyl-Coenzyme A monooxygenase of Pseudomonas putida ATCC 17453. Appl. Environ. Microbiol. 2012, 78, 2200–2212. [Google Scholar] [CrossRef] [PubMed]
- Van Berkel, W.J.H.; Kamerbeek, N.M.; Fraaije, M.W. Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts. J. Biotechnol. 2006, 4, 670–689. [Google Scholar] [CrossRef] [PubMed]
- Gunsalus, I.C.; Conrad, H.E.; Trudgill, P.W.; Jacobson, L.A. Regulation of catabolic metabolism. Israel J. Med. Sci. 1965, 1, 1099–1119. [Google Scholar]
- Trudgill, P.W.; DuBus, R.; Gunsalus, I.C. Mixed function oxidation. VI. Purification of a tightly coupled electron tranbsport complex in camphor lactonization. J. Biol. Chem. 1966, 241, 4288–4290. [Google Scholar] [PubMed]
- Ellis, H.R. The FMN-dependent two-component monooxygenase systems. Arch. Biochem. Biophys. 2010, 497, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Huijbers, M.M.E.; Montersino, S.; Westphal, A.H.; Tischler, D.; van Berkel, W.J.H. Flavin dependent monooxygenases. Arch. Biochem. Biophys. 2014, 544, 2–17. [Google Scholar] [CrossRef] [PubMed]
- Heine, T.; van Berkel, W.J.H.; Gassner, G.; van Pee, K.-H.; Tischler, D. Two-component FAD-dependent monooxygenases: Current knowledge and biotechnological opportunities. Biology 2018, 7, 42. [Google Scholar] [CrossRef] [PubMed]
- Suckaritakul, J.; Tinikul, R.; Chaiyen, P. Mechanisms of reduced flavin transfer in the two-component flavin-dependent monooxygenases. Arch. Biochem. Biophys. 2014, 555–556, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Willetts, A.; Kelly, D.R. Multiple native flavin reductases in camphor-metabolising Pseudomonas putida NCIMB 10007: Functional interaction with two-component diketocamphane monooxygenase isoenzymes. Microbiology 2014, 160, 1784–1794. [Google Scholar] [CrossRef] [PubMed]
- Willetts, A.; Kelly, D.R. Flavin-dependent redox transfers by two-component diketocamphane monooxygenases of camphor-grown Pseudomonas putida NCIMB 10007. Microorganism 2016, 4, 38. [Google Scholar] [CrossRef] [PubMed]
- Dagley, S. Our microbial world. In Experiences in Biochemical Perception; Ornston, L.N., Sligar, S.G., Eds.; Academic Press: New York, NY, USA, 1982; p. 46. [Google Scholar]
- Bradshaw, W.H.; Conrad, H.E.; Corey, E.J.; Gunsalus, I.C.; Lednicer, D. Microbial degradation of (+)–camphor. J. Am. Chem. Soc. 1959, 81, 5507. [Google Scholar] [CrossRef]
- Conrad, H.E.; DuBus, R.; Gunsalus, I.C. An enzyme system for cyclic lactonization. Biochem. Biophys. Res. Commun. 1961, 6, 293–297. [Google Scholar] [CrossRef]
- Conrad, H.E.; Hedegaard, J.; Gunsalus, I.C. Lactone intermediates in the microbial oxidation of (+)– camphor. Tet. Lett. 1965, 10, 561–565. [Google Scholar] [CrossRef]
- LeGall, J.; Gunsalus, I.C. Growth of terpene oxidizing pseudomonads. Bacteriol. Proc. 1963, 105. [Google Scholar]
- Conrad, H.E.; DuBus, R.; Namtvedt, M.J.; Gunsalus, I.C. Mixed function oxidation. II. Separation and properties of the enzymes catalysing camphor lactonization. J. Biol. Chem. 1965, 240, 495–503. [Google Scholar] [PubMed]
- Conrad, H.E.; Lieb, K.; Gunsalus, I.C. Mixed function oxidation. III. An electron transport complex in camphor ketolactonization. J. Biol. Chem. 1965, 240, 4029–4037. [Google Scholar] [PubMed]
- Yu, C.A.; Gunsalus, I.C. Monooxygenases. VII. Camphor lactonase I and the role of three protein components. J. Biol. Chem. 1969, 244, 6149–6152. [Google Scholar] [PubMed]
- Gunsalus, I.C.; Conrad, H.E.; Trudgill, P.W. Generation of active oxygen for mixed function oxidation. In Oxidases and Related Redox Systems; King, T.E., Mason, H.S., Morrish, M., Eds.; Wiley: New York, NY, USA, 1965; pp. 417–447. [Google Scholar]
- Hedegaard, J.; Gunsalus, I.C. Mixed function oxidation. IV. An induced methylene hydroxylase in camphor oxidation. J. Biol. Chem. 1965, 240, 4038–4043. [Google Scholar] [PubMed]
- Gunsalus, I.C.; Bertland, A.U.; Jacobson, L.A. Enzyme induction and repression in anabolic and catabolic pathways. Arch. Mikrobiol. 1967, 59, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Trudgill, P.W.; DuBus, R.; Gunsalus, I.C. Mixed function oxidation. V. Flavin interaction with a reduced diphosphopyridine nucleotide dehydrogenase, one of the enzymes participating in camphor lactonization. J. Biol. Chem. 1966, 244, 1194–1205. [Google Scholar]
- Shapiro, A.L.; Vinuela, E.; Maizel, J.V. Molecular weight estimation of polypeptide chains by electrophoresis in SDS-polyacrylamide gels. Biochem. Biophys. Res. Commun. 1967, 28, 815–820. [Google Scholar] [CrossRef]
- Jacobson, L.A. Enzyme Induction and Repression in the Catabolism of (+)–Camphor by Pseudomonas putida. Ph.D. Thesis, University of Illinois, Champaign, IL, USA, December 1967. [Google Scholar]
- Trudgill, P.W. Terpenoid metabolism in Pseudomonas. In The Bacteria; Sokatch, J.R., Ed.; Academic Press: New York, NY, USA, 1986; Volume 10, pp. 483–525. [Google Scholar]
- Taylor, D.G.; Trudgill, P.W. Camphor revisited: Studies of 2,5-diketocamphane 1,2-monooxygenase from Pseudomonas putida ATCC 17453. J. Bacteriol. 1986, 165, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Kadow, M.; Sass, S.; Schmidt, M.; Bornscheuer, U. Recombinant expression and purification of the 2,5-diketocamphane 1,2-monooxygenase from the camphor metabolizing Pseudomonas putida strain NCIMB 10007. AMB Express 2011, 1, 13. [Google Scholar] [CrossRef]
- Kadow, M.; Loschinski, K.; Sass, S.; Schmidt, M.; Bornscheuer, U. Completing the series of BVMOs involved in camphor metabolism of Pseudomonas putida NCIMB 10007 by identification of the two missing genes, their functional expression in E. coli, and biochemical characterization. Appl. Microbiol. Biotechnol. 2012, 96, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Grogan, G. Microbial biotransformations: Oxygenation of Cyclic Ketones by Baeyer-Villiger Monooxygenases from Pseudomonas putida NCIMB 10007. Ph.D. Thesis, University of Exeter, Exeter, UK, October 1995. [Google Scholar]
- Beecher, J.E.; Grogan, G.; Roberts, S.M.; Willetts, A. Enantioselective biooxidations by the enantiocomplementary diketocamphane monooxygenase isoenzymes from Pseudomonas putida NCIMB 10007. Biotechnol. Lett. 1996, 18, 571–576. [Google Scholar] [CrossRef]
- Beecher, J.E.; Willetts, A. Biotransformation of organic sulphides. Predictive active site models for sulfoxidation catalysed by the 2,5-diketocamphane 1,2-monooxygenase and 3,6-diketocamphane 1,6-monooxygenase, enantiocomplementary enzymes from Pseudomonas putida NCIMB 10007. Tetrahedron Asymm. 1998, 9, 1899–1916. [Google Scholar] [CrossRef]
- Gagnon, R.; Grogan, G.; Roberts, S.M.; Villa, R.; Willetts, A. Enzymatic Baeyer-Villiger oxidation of some bicyclo[2.2.1]heptan-2-ones using monooxygenases from Pseudomonas putida NCIMB 10007: Enantioselective preparation of a precursor of azadirachtin. J. Chem. Soc. Perkin Trans. 1995, 12, 1505–1511. [Google Scholar] [CrossRef]
- Gray, K.A.; Mrachko, G.T.; Squires, C.H. Biodesulfurization of fossil fuels. Curr. Opin. Microbiol. 2003, 6, 229–235. [Google Scholar] [CrossRef]
- Meighen, E.A. Molecular biology of bacterial bioluminescence. Microbiol. Rev. 1991, 55, 123–142. [Google Scholar] [PubMed]
- Kadow, M. Baeyer-Villiger Monooxygenases Involved in Camphor Degradation. Ph.D. Thesis, University of Greifswald, Greifswald, Germany, October 2012. [Google Scholar]
- Fontecave, M.; Eliasson, R.; Reichard, P. NAD(P)H-flavin oxidoreductase of Escherichia coli: A ferric iron reductase participating in the generation of the free radical of ribonucleotide reductase. J. Biol. Chem. 1987, 262, 12325–12331. [Google Scholar] [PubMed]
- Leipold, F.; Wardenga, R.; Bornscheuer, U.T. Cloning, expression and characterization of a eukaryotic cycloalkanone monooxygenase from Cylindrocarpon radicicola ATCC 11011. Appl. Microbiol. Biotechnol. 2012, 94, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Russell, T.R.; Tu, S.C. Aminobacter aminovorans NADH:flavin oxidoreductase His 140: A highly conserved residue critical for NADH binding and utilization. Biochemistry 2004, 43, 12887–12893. [Google Scholar] [CrossRef]
- Imagawa, T.; Tsurumura, T.; Sugimoto, Y.; Aki, K.; Ishidoh, K.; Kuramitsuu, S.; Tsuge, H. Structural basis of free reduced flavin generation by flavin reductase from Thermus thermophilus HB8. J. Biol. Chem. 2011, 286, 44078–44085. [Google Scholar] [CrossRef]
- Tu, S.C. Reduced flavin: Donor acceptor enzymes and mechanisms of chanelling. Antiox. Redox Signal. 2001, 3, 881–897. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Edwards, T.E.; Begley, D.W.; Abramov, A.; Thompkins, K.B.; Ferrell, M.; Guo, W.J.; Phan, I.; Olsen, C.; Napuli, A.; et al. Structure of nitrilotriacetate monooxygenase component B from Mycobacterium thermoresistibile. Acta Crystalogr. Sect. F Struct. Biol. Commun. 2011, 67, 1100–1105. [Google Scholar] [CrossRef] [PubMed]
- Yeom, J.; Jeon, C.O.; Madsen, E.L.; Park, W. Ferrodoxin-NADP(+)reductase from Pseudomonas putida functions as a ferric reductase. J. Bacteriol. 2009, 191, 1472–1479. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.A.; Lorence, M.C.; Amarneh, B. Putidaredoxin reductase and putidaredoxin: Cloning, sequence determination and heterologous expression of the proteins. J. Biol. Chem. 1990, 265, 6066–6073. [Google Scholar] [PubMed]
- Tyson, C.A.; Lipscomb, J.A.; Gunsalus, I.C. The roles of putidaredoxin and P450cam in methylene hydroxylation. J. Biol. Chem. 1972, 247, 5777–5784. [Google Scholar] [PubMed]
- Vising, L.C. Roles of secondary metabolism in microbes. Ciba Found. Symp. 1992, 171, 184–194. [Google Scholar]
- Loescheke, A.; Thies, S. Pseudomonas putida—A versatile host for the production of natural products. Appl. Microbiol. Biotechnol. 2015, 99, 6197–6214. [Google Scholar] [CrossRef]
- Gross, H.; Loper, J.E. Genomics of secondary metabolite production by Pseudomonas spp. Nat. Prod. Rep. 2009, 26, 1408–1446. [Google Scholar] [CrossRef]
- Thibaut, D.; Ratet, N.; Bisch, D.; Faucher, D.; Debussch, L.; Blanche, F. Purification of the two-enzyme system catalysing the oxidation of the D-proline residue of pristinamycin IIB during the last step of pristinamycin IIA biosynthesis. J. Bacteriol. 1995, 177, 5199–5205. [Google Scholar] [CrossRef]
- Parry, R.J.; Li, W. An NADPH:FAD oxidoreductase from the valinimycin producer, Streptomyces viridifaciens. J. Biol. Chem. 1997, 272, 23303–23311. [Google Scholar] [CrossRef]
- Valton, J.; Mathevon, C.; Fontecave, M.; Niviere, V.; Ballou, D.P. Mechanism and regulation of the two-component FMN-dependent monooxygenase ActVA-ActVB from Streptomyces coelicolor. J. Biol. Chem. 2008, 283, 10287–10296. [Google Scholar] [CrossRef] [PubMed]
- Willetts, A.; Masters, P.; Steadman, C. Regulation of camphor metabolism: Induction and repression of relevant monooxygenases in Pseudomonas putida NCIMB 10007. Microorganisms 2018, 6, 41. [Google Scholar] [CrossRef] [PubMed]
- Ornston, L.N. Regulation of catabolic pathways in Pseudomonas. Bacteriol. Rev. 1971, 35, 87–116. [Google Scholar] [PubMed]
- Ornston, L.N.; Parke, D. The evolution of induction mechanisms in bacteria: Insights derived from the study of the β-ketoadipate pathway. Curr. Top. Cell Regul. 1977, 12, 209–262. [Google Scholar] [PubMed]
- Palleroni, N.J.; Stanier, R.Y. Regulatory mechanisms governing synthesis of the enzymes for tryptophan oxidation in Pseudomonas fluorescens. J. Gen. Microbiol. 1964, 35, 319–334. [Google Scholar] [CrossRef] [PubMed]
- Wheelis, M.L.; Stanier, R.Y. The genetic control of dissimilatory pathways in Pseudomonas putida. Genetics 1970, 66, 245–266. [Google Scholar] [PubMed]
- Newell, C.P.; Lessie, T.G. Induction of histidine-degrading enzymes in Pseudomonas aeruginosa. J. Bacteriol. 1970, 104, 596–598. [Google Scholar]
- Leidigh, B.J.; Wheelis, M.L. Genetic control of the histidine dissimilatory pathway in Pseudomonas putida. Mol. Gen. Genet. 1973, 120, 201–210. [Google Scholar] [CrossRef]
- Parada, J.L.; Magasanik, B. Expression of the hut operon of Salmonella typhimurium in Klebsiella aerogenes and Escherichia coli. J. Bacteriol. 1975, 124, 1263–1269. [Google Scholar]
- Nelson, K.E.; Weinel, C.; Paulsen, I.T.; Dodson, R.J.; Hilbert, H.; Martins dos Santos, V.A.P.; Fouts, D.E.; Gill, S.R.; Pop, M.; Holmes, M.; et al. Complete genome sequence and comparative analysis of the metabolically versatile Pseudomonas putida KT2440. Environ. Microbiol. 2002, 4, 799–808. [Google Scholar] [CrossRef]
- Ahmed, S.N.; Kazlauskas, R.J.; Morinville, A.H.; Grochulski, P.; Schrag, J.D.; Cygler, M. Enantioselectivity of Candida rugose lipase towards carboxylic acids: A predictive rule from substrate mapping and X-ray crystallography. Biocatalysis 1994, 9, 209–225. [Google Scholar] [CrossRef]
- Jones, J.B.; Jakovac, I.J. A new cubic-space section model for predicting the specificity of horse liver alcohol dehydrogenase-catalysed oxidoreductions. Can. J. Chem. 1982, 60, 19–28. [Google Scholar] [CrossRef]
- Faber, K. Active site models. In Biotransformations in Organic Chemistry; Faber, K., Ed.; Springer: Heidelberg, Germany, 2012; p. 86. [Google Scholar]
- Mirza, I.A.; Yachnin, B.J.; Wang, S.; Grosse, S.; Bergeron, H.; Imura, A.; Iwaki, H.; Hasegawa, Y.; Lau, P.C.; Berhuis, A.M. Crystal structures of cyclohexanone monooxygenase reveal complex domain movements and a sliding cofactor. J. Am. Chem. Soc. 2009, 131, 8848–8854. [Google Scholar] [CrossRef] [PubMed]
- Henzler-Wildman, K.; Kern, D. Dynamic personalities of proteins. Nature 2007, 450, 964–972. [Google Scholar] [CrossRef] [PubMed]
- McGhie, E.J. Studies on Monooxygenases from the Camphor Degradation Pathway in Pseudomonas putida NCIMB 10007 that are Important in the Catalysis of Baeyer-Villiger Biotransformation Reactions. Ph.D. Thesis, University of Exeter, Exeter, UK, April 1998. [Google Scholar]
- McGhie, E.J.; Isupov, M.N.; Schroder, E.; Littlechild, J.A. Crystallization and preliminary X-ray diffraction studies of the oxygenating subunit of 3,6-diketocamphane monooxygenase from Pseudomonas putida. Acta Crystalogr. Sect. D 1998, 54, 1035–1038. [Google Scholar] [CrossRef]
- Isupov, M.N.; Lebedev, A.A. NCS-constrained exhaustive search using oligomeric models. Acta Crystalogr. Sect. D 2008, 64, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.J.; Raushel, F.M.; Baldwin, T.O.; Rayment, I. Three-dimensional structure of bacterial luciferase from Vibrio harveyi at 2.4 angstrom resolution. Biochemistry 1995, 34, 6581–6586. [Google Scholar] [CrossRef] [PubMed]
- Campbell, Z.T.; Weichsel, S.; Montfort, W.R.; Baldwin, T.D. Crystal structure of the bacterial luciferase/flavin complex provides insight into the function of the β subunit. Biochemistry 2009, 48, 6085–6094. [Google Scholar] [CrossRef]
- Isupov, M.N.; Schroder, E.; Gibson, R.P.; Beecher, J.; Donadio, G.; Saneei, V.; Dcunha, S.A.; McGhie, E.J.; Sayer, C.; Davenport, C.F.; et al. The oxygenating constituent of 3,6-diketocamphane monooxygenase from the CAM plasmid of Pseudomonas putida: The first crystal structure of a type II Baeyer-Villiger monooxygenase. Acta Crystalog. Sect. D 2015, 71, 2344–2351. [Google Scholar] [CrossRef]
- Mϋller, F.; Hemmerich, P.; Ehrenberg, A. On the molecular and submolecular structure of flavin free radicals and their properties. In Flavins and Flavoproteins; Kamin, H., Ed.; University Park Press: Baltimore, MA, USA, 1971; pp. 253–286. [Google Scholar]
- Willetts, A. Reply to the comment by Littlechild and Isupov. Microorganisms 2017, 5, 55. [Google Scholar] [CrossRef]
- Baeyer, A.; Villiger, V. Einwirkung des caro’schen reagens auf ketone. Ber. Disch. Chem. Ges. 1899, 32, 3625–3633. [Google Scholar] [CrossRef]
- Gusso, A.; Baccin, C.; Pinna, F.; Strukul, G. Platinum-catalysed oxidations with hydrogen peroxide: Enantiospecific Baeyer-Villiger oxidation of cyclic ketones. Organomettalics 1994, 13, 3442–3451. [Google Scholar] [CrossRef]
- Wulgemuth, R. Biocatalysis—Key to sustainable industrial chemistry. Curr. Opin. Biotechnol. 2010, 21, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Reetz, M.T. Bioctalysis in organic chemistry and biotechnology. J. Am. Chem. Soc. 2013, 135, 12480–12496. [Google Scholar] [CrossRef] [PubMed]
- Hartline, R.A.; Gunsalus, I.C. Induction specificity and catabolite repression of the early enzymes in camphor degradation by Pseudomonas putida. J. Bacteriol. 1971, 106, 468–478. [Google Scholar] [PubMed]
- Ohno, S. Evolution by Gene Duplication; Springer: New York, NY, USA, 1970. [Google Scholar]
- Mugford, P.F.; Wagner, U.G.; Jiang, Y.; Faber, K.; Kazlauskas, R.J. Enantiocomplementary enzymes: Classification, molecular basis for their enantiopreference, and prospects for mirror-image biotransformations. Angew. Chem. Int. Ed. 2008, 47, 8782–8793. [Google Scholar] [CrossRef] [PubMed]
- Villa, R.; Willetts, A. Oxidations by microbial NADH plus FMN-dependent luciferases from Photobacterium phosphoreum and Vibrio fischeri. J. Mol. Catal. B Enzym. 1997, 2, 193–197. [Google Scholar] [CrossRef]
- Levitt, M.; Sandey, H.; Willetts, A. Regiospecific biotransformations of substituted norbornanones by microorganisms. Biotechnol. Lett. 1990, 12, 197–200. [Google Scholar] [CrossRef]
- Levitt, M.; Newton, R.F.; Roberts, S.M.; Willetts, A. Preparation of optically active 6’-fluorocarbocyclic nucleosides utilizing an enantiospecific enzyme-catalysed Baeyer-Villiger type oxidation. J. Chem. Soc. Chem. Commun. 1990, 619–620. [Google Scholar] [CrossRef]
- Gagnon, R.; Grogan, G.; Levitt, M.; Roberts, S.M.; Wan, P.W.H.; Willetts, A. Biological Baeyer-Villiger oxidations of some monocyclic ans bicyclic ketones using monooxygenases from Acinetobacter calcoaceticus NCIMB 9871 and Pseudomonas putida NCIMB 10007. J. Chem. Soc. Perkin Trans. 1994, 1, 2537–2543. [Google Scholar] [CrossRef]
- Veitch, G.E.; Boyer, A.; Ley, S.V. The azadirachtin story. Angew. Che. Int. Ed. 2008, 47, 9402–9429. [Google Scholar] [CrossRef] [PubMed]
- Carnell, A.J.; Roberts, S.M.; Sik, V.; Willetts, A. Enzyme-catalysed Baeyer-Villiger oxidations of some substituted bicyclo[3.2.0]heptanones. J. Chem. Soc. Chem. Commun. 1990, 1438–1439. [Google Scholar] [CrossRef]
- Carnell, A.; Roberts, S.M.; Sik, V.; Willetts, A. Microbial oxidation of 7-endo-methylbicyclo[3.2.0]hept-2-en-6-one, 7,7-dimethylbicyclo[3.2.0]hept-2-en-6-one, and 2-exo-bromo-3-endo-hydroxy-7,7-dimethylbicyclo- [3.2.0]hept-2-en-6-one using Acinetobacter NCIMB 9871. J. Chem. Soc. Perkin Trans. 1991, 2385–2389. [Google Scholar] [CrossRef]
- Adger, B.; Bes, T.; Grogan, G.; McCague, R.; Pedragosa-Moreau, S.; Roberts, S.M.; Villa, R.; Wan, P.H.; Willetts, A. Applications of enzymic Baeyer-Villiger oxidations of 2-substituted cycloalkanones to the total synthesis of R-(+)−lipoic acid. J. Chem. Soc. Chem. Commun. 1995, 1563–1564. [Google Scholar] [CrossRef]
- Bes, T.; Roberts, S.M.; Villa, R.; Wan, P.W.H.; Willetts, A. Oxidation biotransformations by microorganisms: Production of chiral lactones by cyclopentanone monooxygenase from Pseudomonas sp. NCIMB 9872. J. Mol. Catal. B Enzym. 1996, 1, 127–134. [Google Scholar] [CrossRef]
- Gagnon, R.; Grogan, G.; Groussain, E.; Pedragosa-Moreau, S.; Richardson, P.; Roberts, S.M.; Willetts, A.; Alphand, V.; Lebreton, J.; Furstoss, R. Oxidation of some prochiral 3-substituted cyclobutanones using monooxygenase enzymes: A single-step method for the synthesis of optically enriched 3-substituted ϒ-lactones. J. Chem. Soc. Perkin Trans. 1995, 2527–2528. [Google Scholar] [CrossRef]
- Torres Pazmino, D.E.; Winkler, M.; Glieder, A.; Fraaije, M.W. Monooxygenases as biocatalysts: Classification, mechanistic aspects and biotechnological applications. J. Biotechnol. 2010, 146, 9–24. [Google Scholar] [CrossRef] [PubMed]
- De Gonzalo, G.; Mihovilovic, M.D.; Fraaije, M.W. Recent development in the application of Baeyer-Villiger monooxygenases as biocatalysts. ChemBioChem. 2010, 11, 2209–2231. [Google Scholar] [CrossRef] [PubMed]
- Leisch, H.; Morley, K.; Lau, P.C. Baeyer-Villiger monooxygenases: More than just green chemistry. Chem. Rev. 2012, 111, 4165–4222. [Google Scholar] [CrossRef]
- Balke, K.; Kadow, M.; Mallin, H.; Sass, S.; Bornscheuer, U.T. Discovery, application, and protein engineering of Baeyer-Villiger monooxygenases for organic synthesis. Org. Biomol. Chem. 2012, 10, 6249–6265. [Google Scholar] [CrossRef]
- Bucho, M.; Gemeiner, P.; Schenkmayerova, A.; Krajcovoc, T.; Rudroff, F.; Mihovilovic, M.D. Baeyer-Villiger oxidations: Biotechnological approach. Appl. Microbiol. Biotechnol. 2016, 100, 6585–6599. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.R.; Knowles, C.J.; Madhi, J.G.; Wright, M.A.; Taylor, I.N.; Roberts, S.M.; Wan, P.W.H.; Grogan, G.; Pedragosa-Moreau, S.; Willetts, A. Stereochemical congruency between the Baeyer-Villiger oxidations of bicyclic and tricyclic ketones. J. Chem. Soc. Chem. Commun. 1996, 2333–2334. [Google Scholar] [CrossRef]
- Ottolina, G.; Carrea, G.; Colonna, S.; Ruckemann, A. A predictive active site model for cyclohexanone monooxygenase catalyzed Baeyer-Villiger oxidations. Tetrahedron Asymm. 1996, 7, 1123–1136. [Google Scholar] [CrossRef]
- Branchaud, B.P.; Walsh, C.T. Functional group diversity in enzymic oxygenation reactions catalyzed by bacterial flavin-containing cyclohexanone oxygenase. J. Am. Chem. Soc. 1985, 107, 2153–2161. [Google Scholar] [CrossRef]
- Van Hellemond, E.W.; Janssen, D.B.; Fraaije, M.W. Discovery of novel styrene monooxygenase originating from the metagenome. Appl. Environ. Microbiol. 2007, 73, 5832–5839. [Google Scholar] [CrossRef] [PubMed]
- Tischler, D.; Kermer, R.; Groning, J.A.D.; Kaschabek, S.R.; van Berkel, W.J.H.; Schlomann, M. StyA1 and StyA2B from Rhodococcus opacus 1CP: A multifunctional styrene monooxygenase system. J. Bacteriol. 2010, 192, 5220–5227. [Google Scholar] [CrossRef] [PubMed]
- Nikodinovic-Runic, J.; Coulombel, L.; Francuski, D.; Sharma, N.D.; Boyd, D.R.; Ferrall, R.M.O.; O’Connor, K.E. The oxidation of alkylaryl sulfides and benzothiophenes by Escherichia coli cells expressing wild-type and engineered styrene monoooxygenase from Pseudomonas putida CA-3. Appl. Microbiol. Biotechnol. 2013, 97, 4849–4858. [Google Scholar] [CrossRef] [PubMed]
- Tischler, D.; Schwabe, R.; Siegel, L.; Joffroy, K.; Kaschabek, S.; Scholtissek, A.; Heine, T. VpStyA1/YpStyA2B of Variovorax paradoxus EPS: Aaryl alkyl sulfoxidase rather than a styrene epoxidising monooxygenase. Molecules 2018, 23, 809. [Google Scholar] [CrossRef]
- Beecher, J.E. Sulfoxidation by Microbial Monooxygenases. Ph.D. Thesis, University of Exeter, Exeter, UK, March 1998. [Google Scholar]
- Solladie, G. Asymmetric synthesis using reagents containing a chiral sulfoxide group. Synthesis 1981, 3, 185–196. [Google Scholar] [CrossRef]
- Solladie, G.; Moine, G. Applications of chiral sulfoxides in asymmetric synthesis: The enantiospecific synthesis of the chroman ring of alpha-tocopherol (vitamin E). J. Am. Chem. Soc. 1984, 106, 6097–6098. [Google Scholar] [CrossRef]
- Roiz-Martinez, A.; de Gonzalo, G.; Torres Pazmino, D.E.; Fraaije, M.W.; Gotor, V. Enzymatic synthesis of novel chiral sulfoxides employing Baeyer-Villiger monooxygenases. Eur. J. Org. Chem. 2010, 6409–6416. [Google Scholar] [CrossRef]
- Dover, L.G.; Alahari, A.; Gratraud, P.; Gomes, J.M.; Bhowruth, V.; Reynolds, R.C.; Besra, G.S.; Kremer, I. EthA, a common activator of thiocarbamide-containing drugs acting on different mycological targets. Antimicrob. Agents Chemother. 2007, 51, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Nishida, C.R.; Ortiz de Montellano, P.R. Bioactivation of antituberculosis thioamide and thiourea prodrugs by bacterial and mammalian flavin monooxygenases. Chem. Biol. Interact. 2011, 192, 21–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bong, Y.K.; Clay, M.D.; Collier, S.J.; Mijts, B.; Vogel, M.; Zhang, X.; Zhu, J.; Nazor, J.; Smith, D.; Song, S. Synthesis of Prazole Compounds. WO/2011/071982A3, 22 December 2011. [Google Scholar]
- Camm, T. Sulfoxidation of Isothiocyanates by Camphor-Induced Monooxygenases. Master’s Thesis, University of Exeter, Exeter, UK, November 1998. [Google Scholar]
- Zhang, Y.; Talalay, P.; Cho, C.-G.; Posner, G.H. A major inducer oif anticarcinogenic protective enzymes from broccoli: Isolation and elucidation of structure. Proc. Natl. Acad. Sci. USA 1992, 89, 2399–2403. [Google Scholar] [CrossRef] [PubMed]
- Houghton, C.A.; Fassen, R.G.; Coombes, J.S. Sulforaphane: Translational research from laboratory bench to clinic. Nutr. Rev. 2013, 71, 709–726. [Google Scholar] [CrossRef] [PubMed]
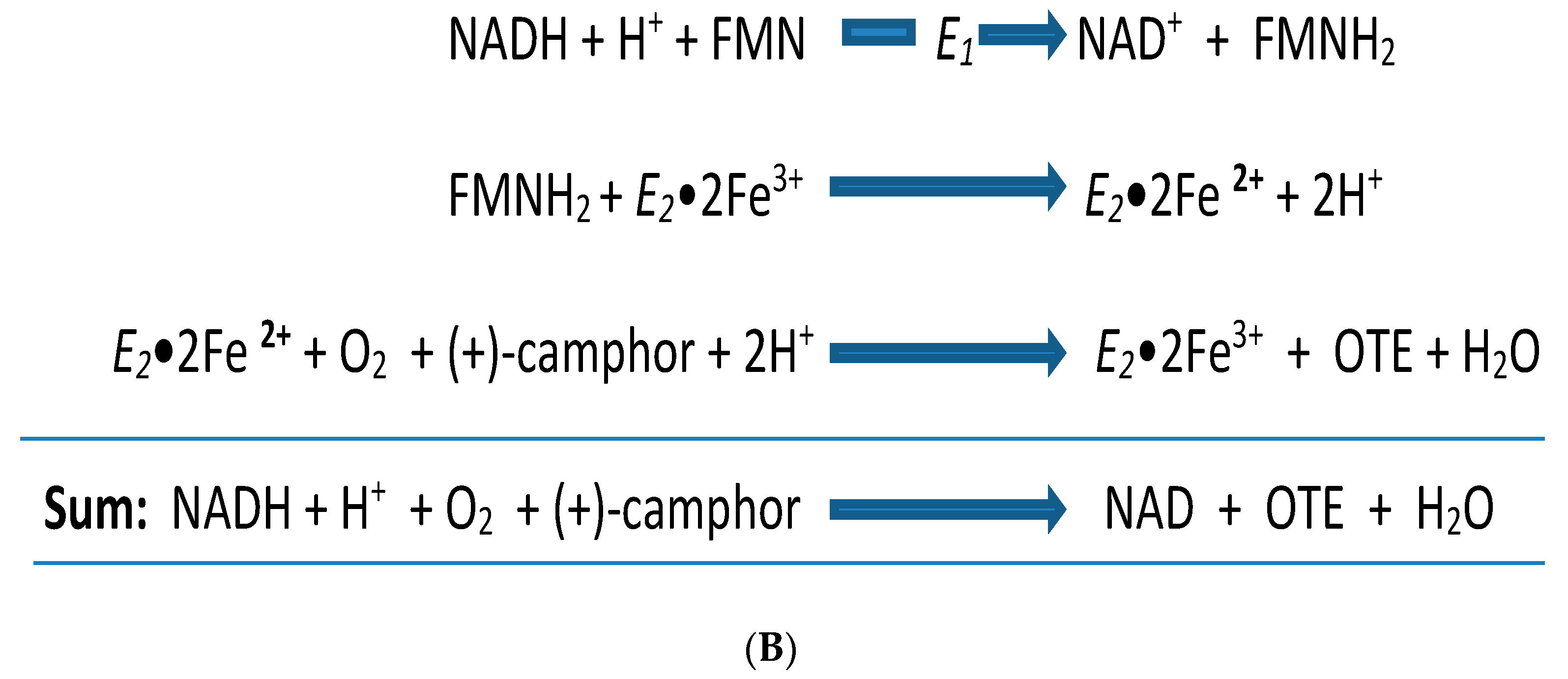




{kind=link}
{kind=link}
{kind=link}
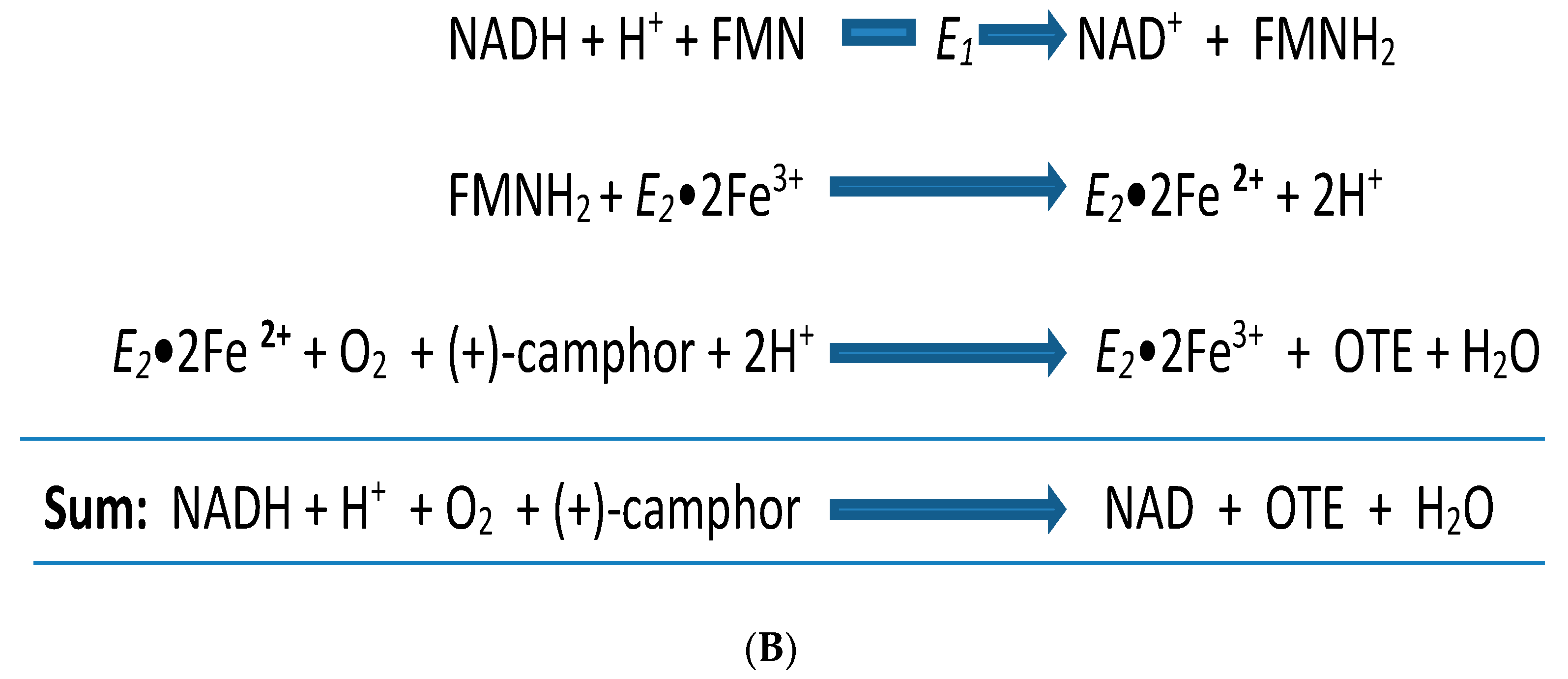{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cofactor | Moles/Mole Enzyme |
|---|---|
| Total iron | 1.01 |
| Heme iron | 0.015 |
| Flavin (FMN) | 0.094 |
| Molybdenum | <0.001 |
| Copper | 0.076 |
| Substrate | 2,5-DKCMO-Generated Oxygenated Product(s) | 3,6-DKCMO-Generated Oxygenated Product(s) | ||
|---|---|---|---|---|
| 7-endo-methylbicyclo[3.2.0]hept-2-en-6-one | (1S,5R)-3-oxa-lactone | (1R,5S)-2-oxa-lactone | (1S,5R)-3-oxa-lactone | (1R,5S)-2-oxa-lactone |
| >95%, e.e. | >80%, e.e. | 5%, e.e. | 10%, e.e. | |
| 100% conversion | 100% conversion | |||
| 7,7-dimethylbicyclo[3.2.0]hept-2-en-6-one | (1S,5R)-3-oxa-lactone | (1R,5S)-2-oxa-lactone | racemic 3-oxa-lactone | no 2-oxa-lactone detected |
| >80%, e.e. | >95%, e.e. | |||
| 100% conversion | 100% conversion | |||
| methyl-para-tolyl sulfide | (S)-sulfoxide | (S)-sulfoxide | ||
| 75%, e.e. | 40%, e.e. | |||
| 20% conversion | 30% conversion | |||
| Substrate | Enzyme | |||
|---|---|---|---|---|
| 2,5-DKCMO-1 a | 2,5-DKCMO-2 b | 2,5-DKCMO-1 + 2 c | 3,6-DKCMO d | |
| (A) | ||||
| (+)-camphor product(s) | 2-oxa-lactone | 2-oxa-lactone | 2-oxa-lactone | none |
| conversion % | 100 | 100 | 100 | 0 |
| e.e.% | >97 | >97 | >95 | n.a. |
| (−)-camphor product(s) | none | none | none | 6-oxa-lactone |
| conversion % | n.a. | n.a | n.a | 100 |
| ee% | n.a. | n.a | n.a | >97 |
| 2,5-diketocamphane product(s) | n.t | n.t | OTE(indirect) | none |
| conversion% | 100 | n.a. | ||
| e.e.% | n.a. | n.a. | ||
| 3,6-diketocamphane product(s) | n.t | n.t | none | OTE(indirect) |
| conversion % | n.a. | 100 | ||
| e.e.% | n.a. | n.a. | ||
| (+)-fenchone product(s) | none | none | none | none |
| conversion % | n.a. | n.a | n.a | n.a |
| ee% | n.a. | n.a. | n.a. | n.a. |
| (−)-fenchone product(s) | none | none | none | none |
| conversion % | n.a | n.a | n.a | n.a |
| e.e.% | n.a. | n.a. | n.a | n.a. |
| (+)-nopinone product(s) | none | none | n, t | none |
| conversion % | n.a. | n.a | n.a | |
| ee% | n.a. | n.a | n.a. | |
| (rac)-bicyclo[2.2.1]heptan-2,5-dione product(s): | 2-oxa-lactone | n.t | n.t. | 2-oxa-lactone |
| conversion % | 94 | 26 | ||
| e.e.% | n.t | n.t. | ||
| 6-oxo-cineole product(s) | n.t | n.t | 6-oxa-lactone | n.t |
| conversion% | 25 | |||
| e.e.% | n.t | |||
| (rac)-bicyclo[3.2.0]hept-2-en-6-one product(s) | i. (+)-2-oxa-lactone (1R,5S) | i. (+)-2-oxa-lactone (1R,5S) | i. (+)-2-oxa-lactone (1R,5S) | i. (+)-2-oxa-lactone (1R,5S) |
| ii. (+)-3-oxa-lactone (1S,5R) | ii. (+)-3-oxa-lactone (1S,5R) | ii. (+)-3-oxa-lactone (1S,5R) | ii. (+)-3-oxa-lactone (1S,5R) | |
| conversion % | i. 55 | i. 50 | i. 43 | i. 18 |
| ii. 35 | ii.50 | ii. 57 | ii. 22 | |
| e.e.% | i. 77 | i. 87 | i. 89 | i. 33 |
| ii. 99 | ii. 97 | ii. 100 | ii. 82 | |
| (rac)- bicyclo[2.2.1]heptan-2-one(norcamphor) product(s) | i. (−)-2-oxa-lactone (1R,5S) | i. (−)-2-oxa-lactone (1R,5S) | (−)-2-oxa-lactone (1R,5S) | (−)-2-oxa-lactone (1R,5S) |
| ii (−)-3-oxa-lactone (1R,5S) | ii (−)-3-oxa-lactone (1R,5S) | |||
| conversion % | i. 48 | i. 35 | 20 | 37 |
| ii. 16 | ii. 25 | |||
| e.e.% | i. 40 | i. 58 | 60 | >90 |
| ii. 57 | ii. 20 | |||
| (rac)-5-exo-hydroxybicyclo[2.2.1]heptan-2-one product(s) | n.t. | n.t. | none | (−)-2-oxa-lactone (1R,5S) |
| conversion % | n.a | 33 | ||
| ee | n.a. | >95 | ||
| (rac)-5-exo-acetoxybicyclo[2.2.1]heptan-2-one product(s) | n.t. | n.t | (−)-2-oxa-lactone (1R,5S) | none |
| conversion % | 35 | n.a. | ||
| e.e.% | >95 | n.a | ||
| (B) | ||||
| Cyclobutanone product(s) | none | n.t. | n.t. | 2-oxa-lactone |
| conversion % | n.a. | 13 | ||
| e.e.% | n.a. | n.a. | ||
| Cyclopentanone product(s) | none | n.t | n.t | 2-oxa-lactone |
| conversion % | n.a. | 24 | ||
| e.e.% | n.a. | n.a. | ||
| 2-methylcyclopentanone product(s) | none | none | n.t. | none |
| conversion % | n.a. | n.a. | n.a. | |
| e.e.% | n.a. | n.a. | n.a. | |
| 2-n-hexylcyclopentanone product(s) | (+)-2-oxa-lactone | (+)-2-oxa-lactone | n.t. | none |
| conversion % | 6 | 3 | ||
| E. | 12 | 5 | ||
| 2-cyclopenten-1-one product(s) | n.t. | n.t. | n.t. | none |
| conversion % | n.a. | |||
| e.e.% | n.a. | |||
| 3-methyl-2-cyclopenten-1-one product(s) | n.t. | n.t. | n.t. | none |
| conversion % | n.a. | |||
| e.e.% | n.a | |||
| 2,3,4,5-tetramethyl-2-cyclopenten-1-one product(s) | n.t. | n.t. | n.t. | none |
| conversion % | n.a. | |||
| e.e.% | n.a. | |||
| Cyclohexanone product(s) | none | n.t. | n.t. | 2-oxa-lactone |
| conversion % | n.a. | 3 | ||
| e.e.% | n.a. | n.a. | ||
| 2-cyclohexen-1-one product(s) | n.t. | n.t. | n.t. | none |
| conversion % | n.a. | |||
| e.e.% | n.a. | |||
| 2-methylcyclohexanone product(s) | (+)-2-oxa-lactone | (+)-2-oxa-lactone | n.t. | (+)-2-oxa-lactone |
| conversion % | 8 | 4 | 2 | |
| E | 3.2 | 3.1 | 3.2 | |
| 2-ethylcyclohexanone product(s) | (+)-2-oxa-lactone | (+)-2-oxa-lactone | n.t. | none |
| conversion % | 11 | 6 | n.a. | |
| E | 43 | 22 | n.a. | |
| 2-n-propylcyclohexanone product(s) | (+)-2-oxa-lactone | (+)-2-oxa-lactone | n.t. | none/trace |
| conversion % | 18 | 13 | n.a. | |
| E | 19 | 8.5 | n.a. | |
| 2-phenylcyclohexanone product(s) | (−)-2-oxa-lactone | (−)-2-oxa-lactone | n.t. | (rac)-2-oxa-lactone |
| conversion % | 11 | 6 | 2 | |
| E | 43 | 22 | n.a. | |
| 4-methylcyclohexanone product(s) | (−)-2-oxa-lactone | (−)-2-oxa-lactone | n.t. | trace |
| conversion % | 8 | 5 | n.a. | |
| e.e.% | 27 | 55 | n.a. | |
| 4-ethylcyclohexanone product(s) | (−)-2-oxa-lactone | (−)-2-oxa-lactone | n.t. | (+)-2-oxa-lactone |
| conversion % | 29 | 10 | 3 | |
| e.e.% | 71 | 89 | 87 | |
| 4-n-pentylcyclohexanone product(s) | (−)-2-oxa-lactone | trace | n.t. | trace |
| conversion % | 2 | n.a. | n.a | |
| e.e.% | 26 | n.a. | n.a. | |
| 4-tert-butylcyclohexanone product(s) | (+)-2-oxa-lactone | trace | n.t. | none |
| conversion % | 5 | n.a. | n.a | |
| e.e.% | 61 | n.a. | n.a. | |
| 3-methyl-2-cyclohexene-1-one product(s) | none | n.t. | n.t. | none |
| conversion % | n.a. | n.a. | ||
| e.e.% | n.a. | n.a. | ||
| 3,5,5-trimethyl-2-cyclohexene-1-one product(s) | none | n.t. | n.t. | none |
| conversion % | n.a. | n.a. | ||
| e.e.% | n.a. | n.a | ||
| (C) | ||||
| Progesterone product(s) | none | n.t. | n.t. | n.t |
| conversion % | 0 | |||
| e.e.% | n.a. | |||
| 2-decanone product(s) | none | n.t. | n.t. | n.c. |
| conversion % | 0 | 11 | ||
| e.e.% | n.a. | n.a. | ||
| Acetophenone product(s) | none | n.t. | n.t. | n.c. |
| conversion % | 0 | 80 | ||
| e.e.% | n.a. | n.a. | ||
| 4-phenyl-2-butanone | none | n.t. | n.t. | n.c. |
| conversion % | 0 | 48 | ||
| e.e.% | n.a. | n.a. | ||
| (D) | ||||
| Methyl-p-tolyl sulphide product(s) | n.t. | n.t. | (S)-sulfoxide | (S)-sulfoxide |
| conversion % | 12 | 57 | ||
| e.e.% | 62 | 32 | ||
| p-methoxyphenyl methyl sulphide product(s) | n.t. | n.t. | (S)-sulfoxide | (S)-sulfoxide |
| conversion % | 29 | 65 | ||
| e.e.% | 71 | 25 | ||
| p-bromophenyl methyl sulphide product(s) | n.t. | n.t. | (S)-sulfoxide | (S)-sulfoxide |
| conversion % | 29 | 65 | ||
| e.e.% | 71 | 25 | ||
| p-chlorophenyl methyl sulphide product(s) | n.t. | n.t. | (S)-sulfoxide | (S)-sulfoxide |
| conversion % | 5 | 15 | ||
| e.e.% | 45 | 27 | ||
| p-fluorophenyl methyl sulphide product(s) | n.t. | n.t. | (S)-sulfoxide | (R)-sulfoxide |
| conversion % | 9 | 72 | ||
| e.e.% | 39 | 30 | ||
| n-pentyl methyl sulphide product(s) | n.t. | n.t. | (S)-sulfoxide | (rac)-sulfoxide |
| conversion % | 28 | 90 | ||
| e.e.% | 32 | n.a. | ||
| n-hexyl methyl sulphide product(s) | n.t. | n.t. | (S)-sulfoxide | (S)-sulfoxide |
| conversion % | 19 | 50 | ||
| e.e.% | 37 | 19 | ||
| n-heptyl methyl sulphide product(s) | n.t. | n.t. | (S)-sulfoxide | (S)-sulfoxide |
| conversion % | 10 | 43 | ||
| e.e.% | 30 | 20 | ||
| n-octyl methyl sulphide product(s) | n.t. | n.t. | (S)-sulfoxide | (S)-sulfoxide |
| conversion % | 6 | 20 | ||
| e.e.% | 36 | 16 | ||
| n-nonyl methyl sulphide product(s) | n.t. | n.t. | (S)-sulfoxide | (R)-sulfoxide |
| conversion % | 2 | 9 | ||
| e.e.% | 16 | 8 | ||
| n-decyl methyl sulphide product(s) | n.t. | n.t. | (S)-sulfoxide | (R)-sulfoxide |
| conversion % | 1 | 9 | ||
| e.e.% | 5 | 9 | ||
| n-octyl ethyl sulphide product(s) | n.t. | n.t. | (S)-sulfoxide | (S)-sulfoxide |
| conversion % | 3 | 14 | ||
| e.e.% | 21 | 14 | ||
| methyl phenyl sulphide product(s) | n.t. | n.t. | (S)-sulfoxide | (S)-sulfoxide |
| conversion % | 9 | 53 | ||
| e.e.% | 35 | 9 | ||
| ethyl phenyl sulphide product(s) | n.t. | n.t. | (R)-sulfoxide | (rac)-sulfoxide |
| conversion % | 5 | 25 | ||
| e.e.% | 2 | n.a. | ||
| n-propyl phenyl sulphide product(s) | n.t. | n.t. | (R)-sulfoxide | (S)-sulfoxide |
| conversion % | 8 | 20 | ||
| e.e.% | 8 | 19 | ||
| isopropyl phenyl sulphide product(s) | n.t. | n.t. | (S)-sulfoxide | (R)-sulfoxide |
| conversion % | 11 | 21 | ||
| e.e.% | 20 | 24 | ||
| n-butyl phenyl sulphide product(s) | n.t. | n.t. | (R)-sulfoxide | (S)-sulfoxide |
| conversion % | 5 | 14 | ||
| e.e.% | 11 | 43 | ||
| benzyl methyl sulphide product(s) | n.t. | n.t. | (S)-sulfoxide | (R)-sulfoxide |
| conversion % | 29 | 85 | ||
| e.e.% | 30 | 4 | ||
| benzyl ethyl sulphide product(s) | n.t. | n.t. | (S)-sulfoxide | (R)-sulfoxide |
| conversion % | 20 | 41 | ||
| e.e.% | 2 | 9 | ||
| 2,3-dihydrobenzo-thiophene product(s) | n.t. | n.t. | (S)-sulfoxide | (R)-sulfoxide |
| conversion % | 18 | 24 | ||
| e.e.% | 38 | 41 | ||
| 1-thiatetrahydro-naphthalene product(s) | n.t. | n.t. | (S)-sulfoxide | (R)-sulfoxide |
| conversion % | 15 | 28 | ||
| e.e.% | 17 | 7 | ||
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Willetts, A. Characterised Flavin-Dependent Two-Component Monooxygenases from the CAM Plasmid of Pseudomonas putida ATCC 17453 (NCIMB 10007): ketolactonases by Another Name. Microorganisms 2019, 7, 1. https://doi.org/10.3390/microorganisms7010001
Willetts A. Characterised Flavin-Dependent Two-Component Monooxygenases from the CAM Plasmid of Pseudomonas putida ATCC 17453 (NCIMB 10007): ketolactonases by Another Name. Microorganisms. 2019; 7(1):1. https://doi.org/10.3390/microorganisms7010001
Chicago/Turabian StyleWilletts, Andrew. 2019. "Characterised Flavin-Dependent Two-Component Monooxygenases from the CAM Plasmid of Pseudomonas putida ATCC 17453 (NCIMB 10007): ketolactonases by Another Name" Microorganisms 7, no. 1: 1. https://doi.org/10.3390/microorganisms7010001
APA StyleWilletts, A. (2019). Characterised Flavin-Dependent Two-Component Monooxygenases from the CAM Plasmid of Pseudomonas putida ATCC 17453 (NCIMB 10007): ketolactonases by Another Name. Microorganisms, 7(1), 1. https://doi.org/10.3390/microorganisms7010001