Genotype-Environment Interaction Shapes the Microbial Assemblage in Grapevine’s Phyllosphere and Carposphere: An NGS Approach


Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Preparation for PMCs and DNA Extraction
2.2. DNA Amplification and Amplicon Sequence Library Preparation
2.3. Data Processing and Analysis
2.4. Statistical Analysis
3. Results
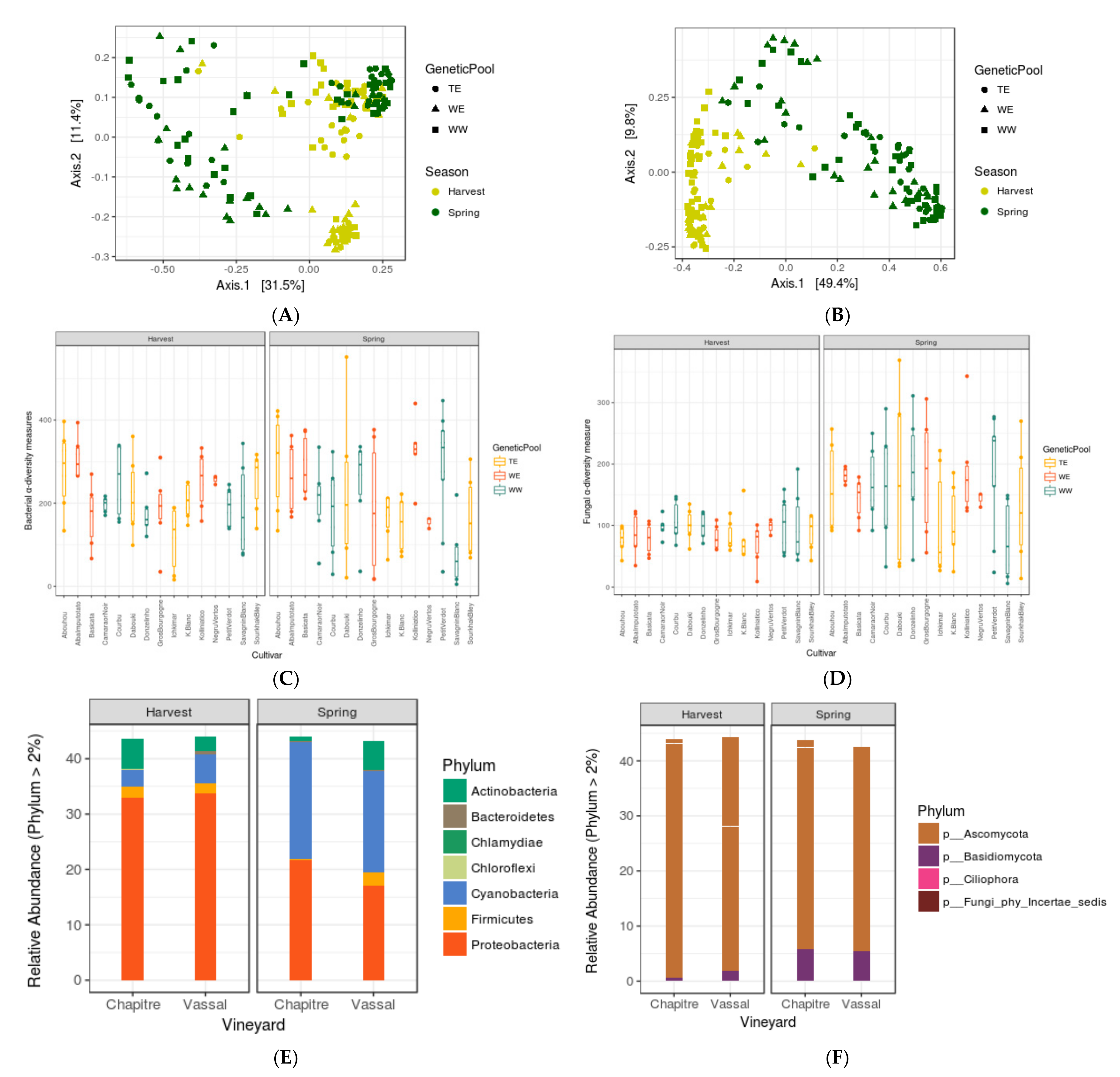
3.1. Seasonal Shifts in Leaf Microbiome Structure
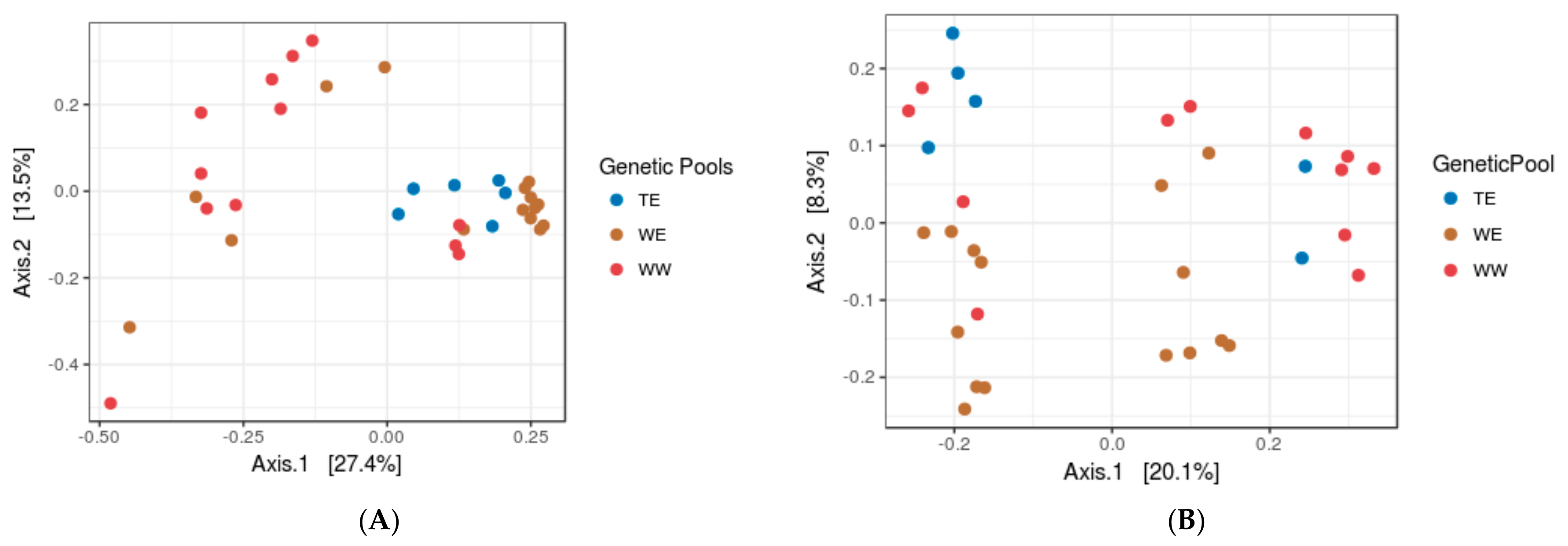
3.2. Assessing the Impacts of Grapevine Cultivars and Genetic Pools
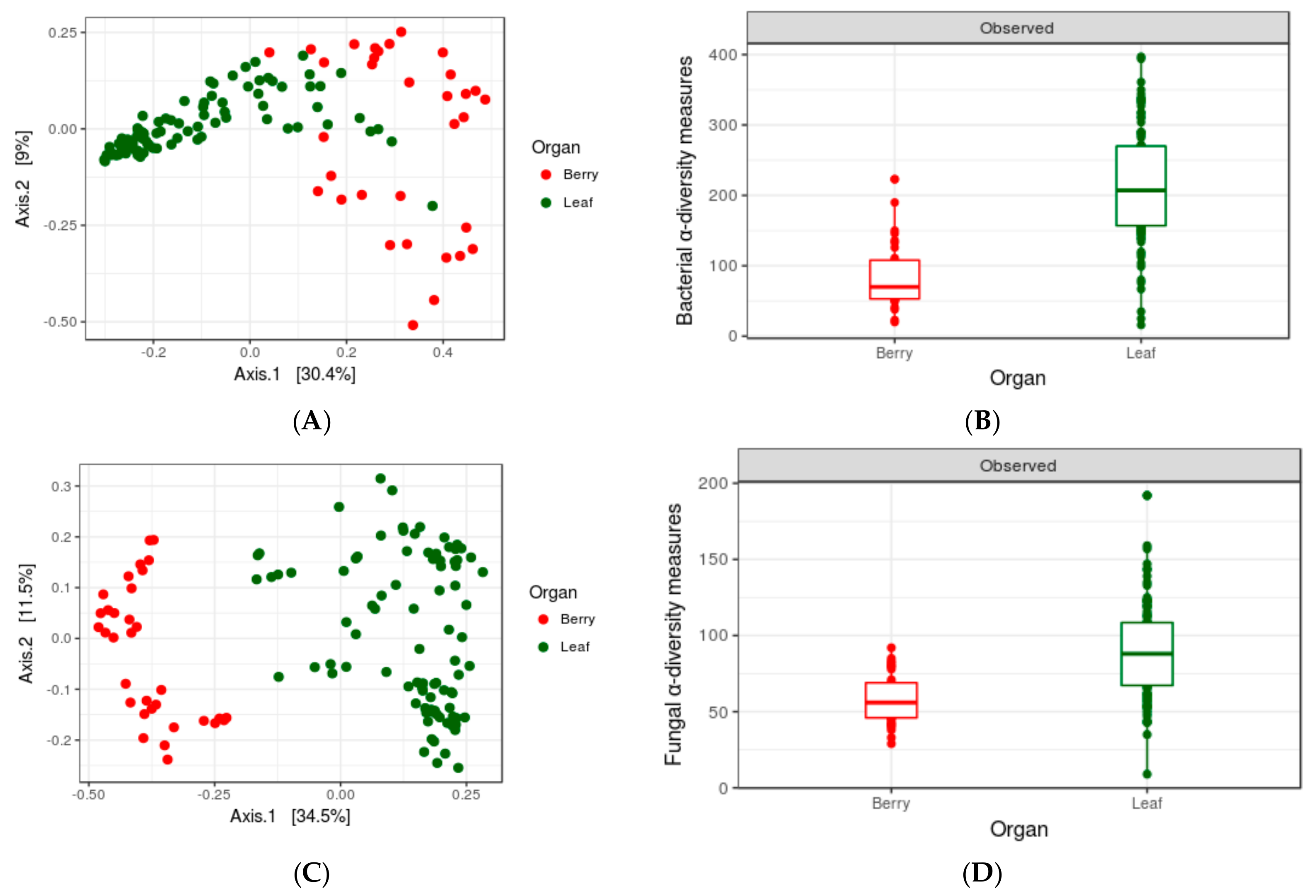
3.3. Impact of Organs
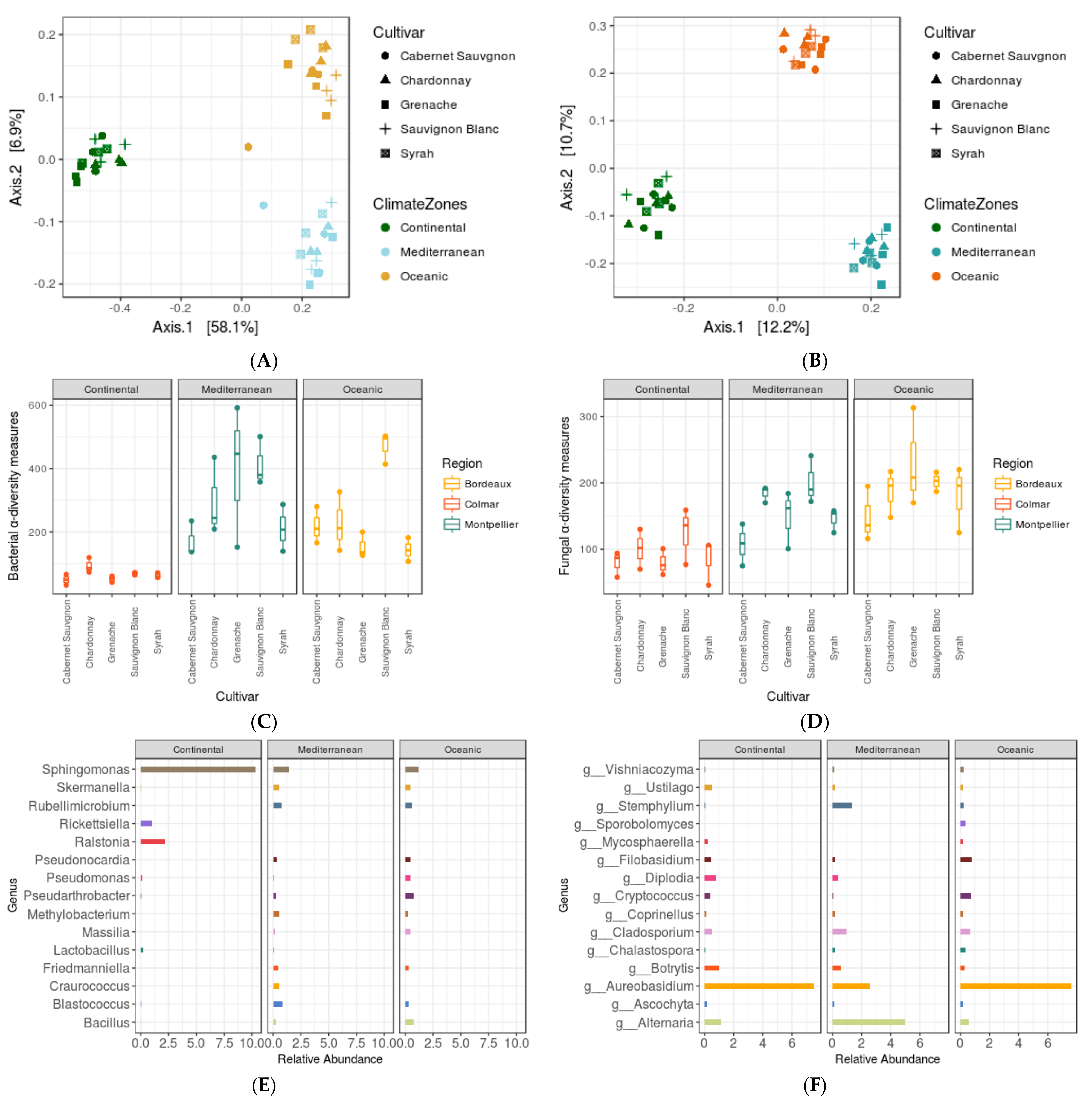
3.4. Impact of Agro-Climate Zones (or Terroir) and Genotype
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Müller, T.; Ruppel, S. Progress in cultivation independent phyllosphere microbiology. FEMS Microbiol. Ecol. 2011, 87, 2–17. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Crowley, D.E.; Borneman, J.; Keen, N.T. Microbial phyllosphere populations are more complex than previously realized. Proc. Natl. Acad. Sci. USA 2001, 98, 3889–3894. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.E. Phyllosphere. In Encyclopedia of Life Sciences; Nature Publishing Group: London, UK, 2001. [Google Scholar]
- Vorholt, J.A. Microbial life in the phyllosphere. Nat. Rev. Microbiol. 2012, 10, 828–840. [Google Scholar] [CrossRef] [PubMed]
- Lindow, S.E.; Brandl, M.T. Microbiology of the phyllosphere. Appl. Environ. Microbiol. 2003, 69, 1875–1883. [Google Scholar] [CrossRef] [PubMed]
- Balint, M.; Tiffin, P.; Hallström, B.; O’Hara, R.B.; Olson, M.S.; Fankhauser, J.D.; Piepenbring, M.; Schmitt, I. Host genotype shapes the foliar fungal microbiome of balsam poplar (Populus balsamifera). PLoS ONE 2013, 8, e53987. [Google Scholar] [CrossRef] [PubMed]
- Balint-Kurti, P.; Simmons, S.J.; Blum, J.E.; Ballaré, C.L.; Stapleton, A.E. Maize leaf epiphytic bacteria diversity patterns are genetically correlated with resistance to fungal pathogen infection. Mol. Plant-Microbe Interact. 2010, 23, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Bodenhausen, N.; Bortfeld-Miller, M.; Ackermann, M.; Vorholt, J.A. A synthetic community approach reveals plant genotypes affecting the phyllosphere microbiota. PLoS Genet. 2014, 10, e1004283. [Google Scholar] [CrossRef] [PubMed]
- Chou, M.Y.; Heuvel, J.V.; Bell, T.H.; Buisse, K.P.; Kniffin, J.K. Vineyard under-vine floor management alters soil microbial composition, while the fruit microbiome shows no corresponding shifts. Sci. Rep. 2018, 8, 11039. [Google Scholar] [CrossRef] [PubMed]
- Compant, S.; van der Heijden, M.G.A.; Sessitsch, A. Climate change effects on beneficial plant microorganism interactions. FEMS Microbiol. Ecol. 2010, 73, 197–214. [Google Scholar] [PubMed]
- Pe nuelas, J.; Rico, L.; Ogaya, R.; Jump, A.S.; Terradas, J. Summer season and long-term drought increase the richness of bacteria and fungi in the foliar phyllosphere of Quercus ilex in a mixed Mediterranean forest. Plant Biol. 2012, 14, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Rico, L.; Ogaya, R.; Terradas, J.; Peñuelas, J. Community structures of N2-fixing bacteria associated with the phyllosphere of a Holm oak forest and their response to drought. Plant Biol. 2014, 16, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Rashidi, F.; Jalili, A.; Kafaki, S.B.; Sagheb-Talebi, K.; Hodgson, J. Anatomical responses of leaves of Black Locust (Robinia pseudoacacia L.) to urban pollutant gases and climatic factors. Trees 2012, 26, 363–375. [Google Scholar]
- Jumpponen, A.; Jones, K.L. Seasonally dynamic fungal communities in the Quercus macrocarpa phyllosphere differ between urban and nonurban environments. New Phytol. 2010, 186, 496–513. [Google Scholar] [CrossRef] [PubMed]
- Jochner, S.; Markevych, I.; Beck, I.; Traidl-hoffmann, C.; Heinrich, J.; Menzel, A. The effects of short and long-term air pollutants on plant phenology and leaf characteristics. Environ. Pollut. 2015, 206, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, A.; Halverson, L.J.; Beattie, G.A. Bacterial degradation of airborne phenol in the phyllosphere. Environ. Microbiol. 2007, 9, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Perazzolli, M.; Antonielli, L.; Storari, M.; Puopolo, G.; Pancher, M.; Giovannini, O.; Pindo, M.; Pertot, I. Resilience of the natural phyllosphere microbiota of the grapevine to chemical and biological pesticides. Appl. Environ. Microbiol. 2014, 80, 3585–3596. [Google Scholar] [CrossRef] [PubMed]
- Zarraonaindia, I.; Owens, S.M.; Weisenhorn, P.; West, K.; Hampton-Marcell, J.; Lax, S.; Bokulich, N.A.; Mills, D.A.; Martin, G.; et al. The soil microbiome influences grapevine-associated microbiota. Mbio 2015, 6, e02527-14. [Google Scholar] [CrossRef] [PubMed]
- Pinto, C.; Pinho, D.; Sousa, S.; Pinheiro, M.; Egas, C.; Gomes, A.C. Unravelling the diversity of grapevine microbiome. PLoS ONE 2014, 9, e85622. [Google Scholar] [CrossRef] [PubMed]
- Barata, A.; Malfeito-Ferreira, M.; Loureiro, V. The microbial ecology of wine grape berries. Int. J. Food Microbiol. 2012, 153, 243–259. [Google Scholar] [CrossRef] [PubMed]
- Innerebner, G.; Knief, C.; Vorholt, J.A. Protection of Arabidopsis thaliana against leaf-pathogenic Pseudomonas syringae by Sphingomonas strains in a controlled model system. Appl. Environ. Microbiol. 2011, 77, 3202–3210. [Google Scholar] [CrossRef] [PubMed]
- Ritpitakphong, U.; Falquet, L.; Vimoltust, A.; Berger, A.; Métraux, J.P.; L’Haridon, F. The microbiome of the leaf surface of Arabidopsis protects against a fungal pathogen. New Phytol. 2016, 210, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, S.D.; Péros, J.P.; Lacombe, T.; Launay, A.; Le Paslier, M.C.; Bérard, A.; Mangin, B.; Valière, S.; Martins, F.; Le Cunff, L.; et al. Genetic diversity, linkage disequilibrium and power of a large grapevine (Vitis vinifera L.) diversity panel newly designed for association studies. BMC Plant Biol. 2016, 16, 74. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Innerebner, G.; Zingg, J.; Guder, J.; Vorholt, J.A.A. Forward genetic in planta screen for the identification of plant-protective traits of Sphingomonas sp. Fr1 against Pseudomonas syringae DC3000. Appl. Environ. Microbiol. 2012, 78, 5529–5535. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, D.S.; Yourstone, S.; Mieczkowski, P.; Jones, C.D.; Dangl, J.L. Practical innovations for high-throughput amplicon sequencing. Nat. Methods 2013, 10, 999–1002. [Google Scholar] [CrossRef] [PubMed]
- Souza, R.S.C.; Okura, V.K.; Armanhi, J.S.; Jorrín, B.; Lozano, N.; da Silva, M.J.; González-Guerrero, M.; de Araújo, L.M.; Verza, N.C.; Bagheri, H.C.; et al. Unlocking the bacterial and fungal communities assemblages of sugarcane microbiome. Sci. Rep. 2016, 6, 28774. [Google Scholar] [CrossRef] [PubMed]
- Grube, M.; Schmid, F.; Berg, G. Black fungi and associated bacterial communities in the phyllosphere of grapevine. Fungal Biol. 2011, 115, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- Abarenkov, K.; Henrik Nilsson, R.; Larsson, K.H.; Alexander, I.J.; Eberhardt, U.; Erland, S.; Høiland, K.; Kjøller, R.; Larsson, E.; Pennanen, T.; et al. The UNITE database for molecular identification of fungi—Recent updates and future perspectives. New Phytol. 2010, 186, 281–285. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.F.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P. Vegan: Community Ecology Package. R Package Version 2.4-3. 2017. Available online: https://CRAN.R-project.org/package=vegan (accessed on 11 June 2018).
- Whittaker, R.H. Evolution and Measurement of Species Diversity. Taxon 1972, 21, 213–251. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar]
- Wickham, H. The Split-Apply-Combine Strategy for Data Analysis. J. Stat. Softw. 2011, 40, 1–29. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar]
- Chambers, J.M.; Freeny, A.E.; Heiberger, R.M. Analysis of Variance; Designed Experiments. In Statistical Models in S, 1st ed.; Tatlor & Francis Group: New York, NY, USA, 1992. [Google Scholar]
- Bokulich, N.A.; Collins, T.S.; Masarweh, C.; Allen, G.; Heymann, H.; Ebeler, S.E.; Mills, D.A. Associations among Wine Grape Microbiome, Metabolome, and Fermentation Behavior Suggest Microbial Contribution to Regional Wine Characteristics. Mbio 2016, 7, e00631-16. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Thorngate, J.H.; Richardson, P.M.; Mills, D.A. Microbial biogeography of wine grapes is conditioned by cultivar, vintage, and climate. Proc. Natl. Acad. Sci. USA 2014, 111, E139–E148. [Google Scholar] [CrossRef] [PubMed]
- Garijo, P.; López, R.; Santamaría, P.; Ocón, E.; Olarte, C.; Sanz, S.; Gutiérrez, A.R. Presence of enological microorganisms in the grapes and the air of a vineyard during the ripening period. Eur. Food Res. Technol. 2011, 233, 359–365. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genetic Pools | |||
|---|---|---|---|
| WW | WE | TE | |
| Donzelinho | Basicata | Ichkimar | |
| Cultivars of Vitis Vinifera | Petit Verdot | Negru Vertos | Khoussaïné blanc |
| Camaraou Noir | Alba Imputotato | Sourkhak Biley | |
| Courbu | Gros Bourgogne | Abouhu | |
| Savagnin Blanc | Koilliniatico | Dabouki |
| Data | Number of Samples | Input Reads | Filtered Reads | Denoised and Merged | OTUs |
|---|---|---|---|---|---|
| 16S data | |||||
| Set1 | 213 | 16113978 | 10874688 | 7795650 | 13521 |
| Set2 | 45 | 7460569 | 5294234 | 3866297 | 4581 |
| ITS data | |||||
| Set1 | 213 | 14780926 | 13600570 | 9900482 | 10162 |
| Set2 | 45 | 6683219 | 4564572 | 2450315 | 3164 |
| Spring | Harvest | |||
|---|---|---|---|---|
| Factors | ANOVA (on α-Diversity Measures) | PERMANOVA on PCoA Clusters | ANOVA (on α-Diversity Measures) | PERMANOVA on PCoA Clusters |
| 16S data | ||||
| Cultivars | F = 2.361, p = 0.0009 | F = 1.129, p = 0.002 | F = 2.837, p = 0.002 | F = 2.737, p = 0.001 |
| Genetic Pool | F = 1.54, p = 0.221 | F = 1.178, p = 0.082 | F = 1.189, p = 0.308 | F = 2.617, p = 0.001 |
| ITS data | ||||
| Cultivars | F = 1.17, p = 0.315 | F = 1.583, p = 0.006 | F = 0.752, p = 0.715 | F = 2.098, p = 0.001 |
| Genetic Pool | F = 1.384, p = 0.255 | F = 2.218, p = 0.015 | F = 3.368, p = 0.038 | F = 2.764, p = 0.001 |
| Factors | ANOVA (on α-Diversity Measures) | PERMANOVA on PCoA Clusters |
|---|---|---|
| 16S data | ||
| Cultivars | F = 2.546, p = 0.002 | F = 2.598, p = 0.001 |
| Genetic Pool | F = 4.261, p = 0.023 | F = 4.612, p = 0.001 |
| ITS data | ||
| Cultivars | F = 4.575, p = 0.00142 | F = 3.169, p = 0.001 |
| Genetic Pool | F = 2.739, p = 0.07 | F = 4.612, p = 0.003 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, P.; Santoni, S.; This, P.; Péros, J.-P. Genotype-Environment Interaction Shapes the Microbial Assemblage in Grapevine’s Phyllosphere and Carposphere: An NGS Approach. Microorganisms 2018, 6, 96. https://doi.org/10.3390/microorganisms6040096
Singh P, Santoni S, This P, Péros J-P. Genotype-Environment Interaction Shapes the Microbial Assemblage in Grapevine’s Phyllosphere and Carposphere: An NGS Approach. Microorganisms. 2018; 6(4):96. https://doi.org/10.3390/microorganisms6040096
Chicago/Turabian StyleSingh, Prashant, Sylvain Santoni, Patrice This, and Jean-Pierre Péros. 2018. "Genotype-Environment Interaction Shapes the Microbial Assemblage in Grapevine’s Phyllosphere and Carposphere: An NGS Approach" Microorganisms 6, no. 4: 96. https://doi.org/10.3390/microorganisms6040096
APA StyleSingh, P., Santoni, S., This, P., & Péros, J.-P. (2018). Genotype-Environment Interaction Shapes the Microbial Assemblage in Grapevine’s Phyllosphere and Carposphere: An NGS Approach. Microorganisms, 6(4), 96. https://doi.org/10.3390/microorganisms6040096