
Interaction of Candida Species with the Skin

{kind=link}
Abstract
:1. Dermal Candida Ecology and Epidemiology
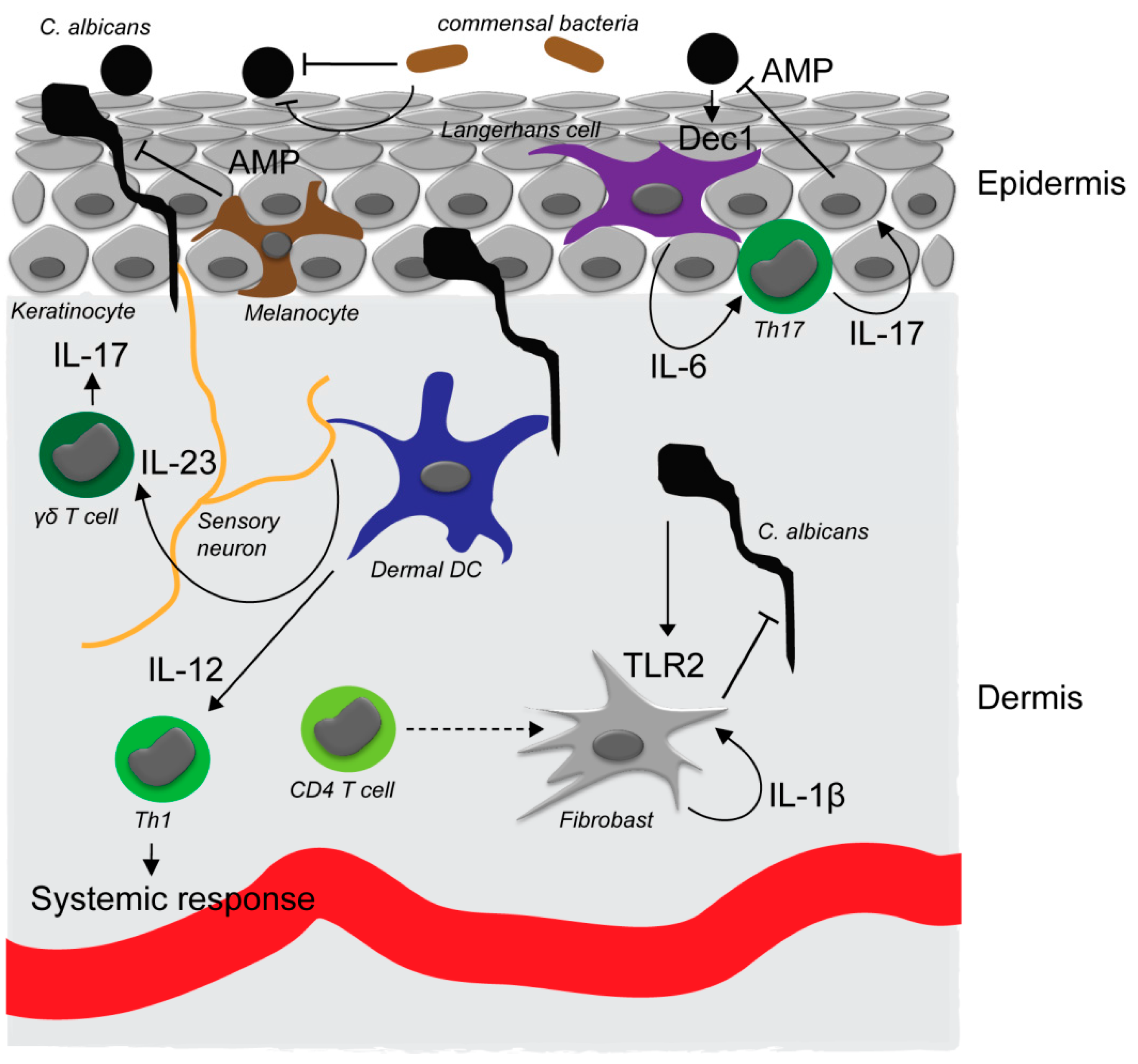
2. Candida Pathogenesis at Epithelial Surfaces
3. Initial Fungal Recognition
4. Immune Networks Involved in Skin Defense against Candida Species
5. Antifungal Defense Execution by Antimicrobial Agents
6. Fungal-Microbiome Interaction
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Findley, K.; Oh, J.; Yang, J.; Conlan, S.; Deming, C.; Meyer, J.A.; Schoenfeld, D.; Nomicos, E.; Park, M.; NIH Intramural Sequencing Center Comparative Sequencing Program; et al. Topographic diversity of fungal and bacterial communities in human skin. Nature 2013, 498, 367–370. [Google Scholar]
- Oh, J.; Freeman, A.F.; Program, N.C.S.; Park, M.; Sokolic, R.; Candotti, F.; Holland, S.M.; Segre, J.A.; Kong, H.H. The altered landscape of the human skin microbiome in patients with primary immunodeficiencies. Genome Res. 2013, 23, 2103–2114. [Google Scholar] [CrossRef] [PubMed]
- Havlickova, B.; Czaika, V.A.; Friedrich, M. Epidemiological trends in skin mycoses worldwide. Mycoses 2008, 51 (Suppl. 4), 2–15. [Google Scholar] [CrossRef] [PubMed]
- Mohandas, V.; Ballal, M. Distribution of Candida species in different clinical samples and their virulence: Biofilm formation, proteinase and phospholipase production: A study on hospitalized patients in southern India. J. Glob. Infect. Dis. 2011, 3, 4–8. [Google Scholar] [CrossRef] [PubMed]
- Kashem, S.W.; Kaplan, D.H. Skin immunity to Candida albicans. Trends Immunol. 2016, 37, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Allen, A.M.; King, R.D. Occlusion, carbon dioxide, and fungal skin infections. Lancet 1978, 1, 360–362. [Google Scholar] [CrossRef]
- Bonifaz, A.; Rojas, R.; Tirado-Sanchez, A.; Chavez-Lopez, D.; Mena, C.; Calderon, L.; Maria, P.O. Superficial mycoses associated with diaper dermatitis. Mycopathologia 2016, 181, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Hofs, S.; Mogavero, S.; Hube, B. Interaction of Candida albicans with host cells: Virulence factors, host defense, escape strategies, and the microbiota. J. Microbiol. 2016, 54, 149–169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.; Tanaka, T.; Tajima, M.; Tsuboi, R.; Nishikawa, A.; Sugita, T. Characterization of the skin fungal microbiota in patients with atopic dermatitis and in healthy subjects. Microbiol. Immunol. 2011, 55, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Taheri Sarvtin, M.; Shokohi, T.; Hajheydari, Z.; Yazdani, J.; Hedayati, M.T. Evaluation of candidal colonization and specific humoral responses against Candida albicans in patients with psoriasis. Int. J. Dermatol. 2014, 53, e555–e560. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.W.; Bukhalo, M.; Blauvelt, A. A clinician’s guide to the diagnosis and treatment of candidiasis in patients with psoriasis. Am. J. Clin. Dermatol. 2016, 17, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Hein, K.Z.; Takahashi, H.; Tsumori, T.; Yasui, Y.; Nanjoh, Y.; Toga, T.; Wu, Z.; Grotzinger, J.; Jung, S.; Wehkamp, J.; et al. Disulphide-reduced psoriasin is a human apoptosis-inducing broad-spectrum fungicide. Proc. Natl. Acad. Sci. USA 2015, 112, 13039–13044. [Google Scholar] [CrossRef] [PubMed]
- Pirofski, L.A.; Casadevall, A. The damage-response framework of microbial pathogenesis and infectious diseases. Adv. Exp. Med. Biol. 2008, 635, 135–146. [Google Scholar] [PubMed]
- Gow, N.A.; Hube, B. Importance of the Candida albicans cell wall during commensalism and infection. Curr. Opin. Microbiol. 2012, 15, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, L.L.; Cota, E. Candida albicans agglutinin-like sequence (als) family vignettes: A review of als protein structure and function. Front. Microbiol. 2016, 7, 280. [Google Scholar] [CrossRef] [PubMed]
- Gabaldon, T.; Martin, T.; Marcet-Houben, M.; Durrens, P.; Bolotin-Fukuhara, M.; Lespinet, O.; Arnaise, S.; Boisnard, S.; Aguileta, G.; Atanasova, R.; et al. Comparative genomics of emerging pathogens in the Candida glabrata clade. BMC Genom. 2013, 14, 623. [Google Scholar] [CrossRef] [PubMed]
- Maestre-Reyna, M.; Diderrich, R.; Veelders, M.S.; Eulenburg, G.; Kalugin, V.; Bruckner, S.; Keller, P.; Rupp, S.; Mosch, H.U.; Essen, L.O. Structural basis for promiscuity and specificity during Candida glabrata invasion of host epithelia. Proc. Natl. Acad. Sci. USA 2012, 109, 16864–16869. [Google Scholar] [CrossRef] [PubMed]
- Diderrich, R.; Kock, M.; Maestre-Reyna, M.; Keller, P.; Steuber, H.; Rupp, S.; Essen, L.O.; Mosch, H.U. Structural hot spots determine functional diversity of the Candida glabrata epithelial adhesin family. J. Biol. Chem. 2015, 290, 19597–19613. [Google Scholar] [CrossRef] [PubMed]
- Urban, C.; Xiong, X.; Sohn, K.; Schroppel, K.; Brunner, H.; Rupp, S. The moonlighting protein tsa1p is implicated in oxidative stress response and in cell wall biogenesis in Candida albicans. Mol. Microbiol. 2005, 57, 1318–1341. [Google Scholar] [CrossRef] [PubMed]
- Lopez, C.M.; Wallich, R.; Riesbeck, K.; Skerka, C.; Zipfel, P.F. Candida albicans uses the surface protein gpm1 to attach to human endothelial cells and to keratinocytes via the adhesive protein vitronectin. PLoS ONE 2014, 9, e90796. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Ruiz, E.; Galan-Diez, M.; Zhu, W.; Fernandez-Ruiz, E.; d'Enfert, C.; Filler, S.G.; Cossart, P.; Veiga, E. Candida albicans internalization by host cells is mediated by a clathrin-dependent mechanism. Cell Microbiol. 2009, 11, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.N.; Solis, N.V.; Phan, Q.T.; Bajwa, J.S.; Kashleva, H.; Thompson, A.; Liu, Y.; Dongari-Bagtzoglou, A.; Edgerton, M.; Filler, S.G. Host cell invasion and virulence mediated by Candida albicans ssa1. PLoS Pathog. 2010, 6, e1001181. [Google Scholar] [CrossRef] [PubMed]
- Phan, Q.T.; Myers, C.L.; Fu, Y.; Sheppard, D.C.; Yeaman, M.R.; Welch, W.H.; Ibrahim, A.S.; Edwards, J.E., Jr.; Filler, S.G. Als3 is a Candida albicans invasin that binds to cadherins and induces endocytosis by host cells. PLoS Biol. 2007, 5, e64. [Google Scholar] [CrossRef] [PubMed]
- Wachtler, B.; Citiulo, F.; Jablonowski, N.; Forster, S.; Dalle, F.; Schaller, M.; Wilson, D.; Hube, B. Candida albicans-epithelial interactions: Dissecting the roles of active penetration, induced endocytosis and host factors on the infection process. PLoS ONE 2012, 7, e36952. [Google Scholar] [CrossRef] [PubMed]
- Dalle, F.; Wachtler, B.; L’Ollivier, C.; Holland, G.; Bannert, N.; Wilson, D.; Labruere, C.; Bonnin, A.; Hube, B. Cellular interactions of Candida albicans with human oral epithelial cells and enterocytes. Cell Microbiol. 2010, 12, 248–271. [Google Scholar] [CrossRef] [PubMed]
- Sohn, K.; Senyurek, I.; Fertey, J.; Konigsdorfer, A.; Joffroy, C.; Hauser, N.; Zelt, G.; Brunner, H.; Rupp, S. An in vitro assay to study the transcriptional response during adherence of Candida albicans to different human epithelia. FEMS Yeast Res. 2006, 6, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Dieterich, C.; Schandar, M.; Noll, M.; Johannes, F.J.; Brunner, H.; Graeve, T.; Rupp, S. In vitro reconstructed human epithelia reveal contributions of Candida albicans efg1 and cph1 to adhesion and invasion. Microbiology 2002, 148, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Schaller, M.; Korting, H.C.; Schafer, W.; Bastert, J.; Chen, W.; Hube, B. Secreted aspartic proteinase (sap) activity contributes to tissue damage in a model of human oral candidosis. Mol. Microbiol. 1999, 34, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Schaller, M.; Bein, M.; Korting, H.C.; Baur, S.; Hamm, G.; Monod, M.; Beinhauer, S.; Hube, B. The secreted aspartyl proteinases sap1 and sap2 cause tissue damage in an in vitro model of vaginal candidiasis based on reconstituted human vaginal epithelium. Infect. Immun. 2003, 71, 3227–3234. [Google Scholar] [CrossRef] [PubMed]
- Schaller, M.; Schackert, C.; Korting, H.C.; Januschke, E.; Hube, B. Invasion of Candida albicans correlates with expression of secreted aspartic proteinases during experimental infection of human epidermis. J. Investig. Dermatol. 2000, 114, 712–717. [Google Scholar] [CrossRef] [PubMed]
- Lermann, U.; Morschhauser, J. Secreted aspartic proteases are not required for invasion of reconstituted human epithelia by Candida albicans. Microbiology 2008, 154, 3281–3295. [Google Scholar] [CrossRef] [PubMed]
- Correia, A.; Lermann, U.; Teixeira, L.; Cerca, F.; Botelho, S.; da Costa, R.M.; Sampaio, P.; Gartner, F.; Morschhauser, J.; Vilanova, M.; et al. Limited role of secreted aspartyl proteinases sap1 to sap6 in Candida albicans virulence and host immune response in murine hematogenously disseminated candidiasis. Infect. Immun. 2010, 78, 4839–4849. [Google Scholar] [CrossRef] [PubMed]
- Mayer, F.L.; Wilson, D.; Hube, B. Candida albicans pathogenicity mechanisms. Virulence 2013, 4, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Moyes, D.L.; Wilson, D.; Richardson, J.P.; Mogavero, S.; Tang, S.X.; Wernecke, J.; Hofs, S.; Gratacap, R.L.; Robbins, J.; Runglall, M.; et al. Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature 2016, 532, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Brown, G.D.; Kullberg, B.J.; Gow, N.A. An integrated model of the recognition of Candida albicans by the innate immune system. Nat. Rev. Microbiol. 2008, 6, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Willment, J.A.; Marshall, A.S.; Reid, D.M.; Williams, D.L.; Wong, S.Y.; Gordon, S.; Brown, G.D. The human beta-glucan receptor is widely expressed and functionally equivalent to murine dectin-1 on primary cells. Eur J. Immunol. 2005, 35, 1539–1547. [Google Scholar] [CrossRef] [PubMed]
- LeibundGut-Landmann, S.; Gross, O.; Robinson, M.J.; Osorio, F.; Slack, E.C.; Tsoni, S.V.; Schweighoffer, E.; Tybulewicz, V.; Brown, G.D.; Ruland, J.; et al. Syk- and card9-dependent coupling of innate immunity to the induction of t helper cells that produce interleukin 17. Nat. Immunol. 2007, 8, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Gantner, B.N.; Simmons, R.M.; Underhill, D.M. Dectin-1 mediates macrophage recognition of Candida albicans yeast but not filaments. EMBO J. 2005, 24, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Patlan, A.; Campillo-Navarro, M.; Rodriguez-Cortes, O.; Munoz-Cruz, S.; Wong-Baeza, I.; Estrada-Parra, S.; Estrada-Garcia, I.; Serafin-Lopez, J.; Chacon-Salinas, R. Recognition of Candida albicans by dectin-1 induces mast cell activation. Immunobiology 2015, 220, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- De Jong, M.A.; Vriend, L.E.; Theelen, B.; Taylor, M.E.; Fluitsma, D.; Boekhout, T.; Geijtenbeek, T.B. C-type lectin langerin is a beta-glucan receptor on human langerhans cells that recognizes opportunistic and pathogenic fungi. Mol. Immunol. 2010, 47, 1216–1225. [Google Scholar] [CrossRef] [PubMed]
- Joly, S.; Ma, N.; Sadler, J.J.; Soll, D.R.; Cassel, S.L.; Sutterwala, F.S. Cutting edge: Candida albicans hyphae formation triggers activation of the nlrp3 inflammasome. J. Immunol. 2009, 183, 3578–3581. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, M.; van der Lee, R.; Cheng, S.C.; Johnson, M.D.; Kumar, V.; Ng, A.; Plantinga, T.S.; Smeekens, S.P.; Oosting, M.; Wang, X.; et al. The rig-i-like helicase receptor mda5 (ifih1) is involved in the host defense against candida infections. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Gow, N.A.; Munro, C.A.; Bates, S.; Collins, C.; Ferwerda, G.; Hobson, R.P.; Bertram, G.; Hughes, H.B.; Jansen, T.; et al. Immune sensing of Candida albicans requires cooperative recognition of mannans and glucans by lectin and toll-like receptors. J. Clin. Investig. 2006, 116, 1642–1650. [Google Scholar] [CrossRef] [PubMed]
- Jouault, T.; Ibata-Ombetta, S.; Takeuchi, O.; Trinel, P.A.; Sacchetti, P.; Lefebvre, P.; Akira, S.; Poulain, D. Candida albicans phospholipomannan is sensed through toll-like receptors. J. Infect. Dis. 2003, 188, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Pivarcsi, A.; Bodai, L.; Rethi, B.; Kenderessy-Szabo, A.; Koreck, A.; Szell, M.; Beer, Z.; Bata-Csorgoo, Z.; Magocsi, M.; Rajnavolgyi, E.; et al. Expression and function of toll-like receptors 2 and 4 in human keratinocytes. Int. Immunol. 2003, 15, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, Q.; Shen, Y.; Liu, W. Candida albicans phospholipomannan triggers inflammatory responses of human keratinocytes through toll-like receptor 2. Exp. Dermatol. 2009, 18, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Haley, K.; Igyarto, B.Z.; Ortner, D.; Bobr, A.; Kashem, S.; Schenten, D.; Kaplan, D.H. Langerhans cells require myd88-dependent signals for Candida albicans response but not for contact hypersensitivity or migration. J. Immunol. 2012, 188, 4334–4339. [Google Scholar] [CrossRef] [PubMed]
- Kuhbacher, A.; Sohn, K.; Burger-Kentischer, A.; Rupp, S. Immune cell-supplemented human skin model for studying fungal infections. Methods Mol. Biol. 2017, 1508, 439–449. [Google Scholar] [PubMed]
- Kuhbacher, A.; Henkel, H.; Stevens, P.; Grumaz, C.; Finkelmeier, D.; Burger-Kentischer, A.; Sohn, K.; Rupp, S. Dermal fibroblasts play a central role in skin model protection against C. albicans invasion. J. Infect. Dis. 2017. [Google Scholar] [CrossRef] [PubMed]
- Nahum, A.; Dadi, H.; Bates, A.; Roifman, C.M. The biological significance of tlr3 variant, l412f, in conferring susceptibility to cutaneous candidiasis, CMV and autoimmunity. Autoimmun. Rev. 2012, 11, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Nahum, A.; Dadi, H.; Bates, A.; Roifman, C.M. The l412f variant of toll-like receptor 3 (tlr3) is associated with cutaneous candidiasis, increased susceptibility to cytomegalovirus, and autoimmunity. J. Allergy Clin. Immunol. 2011, 127, 528–531. [Google Scholar] [CrossRef] [PubMed]
- Weindl, G.; Naglik, J.R.; Kaesler, S.; Biedermann, T.; Hube, B.; Korting, H.C.; Schaller, M. Human epithelial cells establish direct antifungal defense through tlr4-mediated signaling. J. Clin. Investig. 2007, 117, 3664–3672. [Google Scholar] [CrossRef] [PubMed]
- Rosentul, D.C.; Delsing, C.E.; Jaeger, M.; Plantinga, T.S.; Oosting, M.; Costantini, I.; Venselaar, H.; Joosten, L.A.; van der Meer, J.W.; Dupont, B.; et al. Gene polymorphisms in pattern recognition receptors and susceptibility to idiopathic recurrent vulvovaginal candidiasis. Front. Microbiol. 2014, 5, 483. [Google Scholar] [CrossRef] [PubMed]
- Pappu, R.; Ramirez-Carrozzi, V.; Sambandam, A. The interleukin-17 cytokine family: Critical players in host defence and inflammatory diseases. Immunology 2011, 134, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Puel, A.; Cypowyj, S.; Bustamante, J.; Wright, J.F.; Liu, L.Y.; Lim, H.K.; Migaud, M.; Israel, L.; Chrabieh, M.; Audry, M.; et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science 2011, 332, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.; Cypowyj, S.; Aytekin, C.; Galicchio, M.; Camcioglu, Y.; Nepesov, S.; Ikinciogullari, A.; Dogu, F.; Belkadi, A.; Levy, R.; et al. Inherited il-17rc deficiency in patients with chronic mucocutaneous candidiasis. J. Exp. Med. 2015, 212, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Boisson, B.; Wang, C.; Pedergnana, V.; Wu, L.; Cypowyj, S.; Rybojad, M.; Belkadi, A.; Picard, C.; Abel, L.; Fieschi, C.; et al. An act1 mutation selectively abolishes interleukin-17 responses in humans with chronic mucocutaneous candidiasis. Immunity 2013, 39, 676–686. [Google Scholar] [CrossRef] [PubMed]
- Boisson-Dupuis, S.; Kong, X.F.; Okada, S.; Cypowyj, S.; Puel, A.; Abel, L.; Casanova, J.L. Inborn errors of human stat1: Allelic heterogeneity governs the diversity of immunological and infectious phenotypes. Curr. Opin. Immunol. 2012, 24, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Johansen, C.; Usher, P.A.; Kjellerup, R.B.; Lundsgaard, D.; Iversen, L.; Kragballe, K. Characterization of the interleukin-17 isoforms and receptors in lesional psoriatic skin. Br. J. Dermatol. 2009, 160, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Conti, H.R.; Whibley, N.; Coleman, B.M.; Garg, A.V.; Jaycox, J.R.; Gaffen, S.L. Signaling through il-17c/il-17re is dispensable for immunity to systemic, oral and cutaneous candidiasis. PLoS ONE 2015, 10, e0122807. [Google Scholar] [CrossRef] [PubMed]
- Saunte, D.M.; Mrowietz, U.; Puig, L.; Zachariae, C. Candida infections in psoriasis and psoriatic arthritis patients treated with il-17 inhibitors and their practical management. Br. J. Dermatol. 2016. [Google Scholar] [CrossRef]
- Zielinski, C.E.; Mele, F.; Aschenbrenner, D.; Jarrossay, D.; Ronchi, F.; Gattorno, M.; Monticelli, S.; Lanzavecchia, A.; Sallusto, F. Pathogen-induced human th17 cells produce ifn-gamma or il-10 and are regulated by il-1beta. Nature 2012, 484, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Igyarto, B.Z.; Haley, K.; Ortner, D.; Bobr, A.; Gerami-Nejad, M.; Edelson, B.T.; Zurawski, S.M.; Malissen, B.; Zurawski, G.; Berman, J.; et al. Skin-resident murine dendritic cell subsets promote distinct and opposing antigen-specific t helper cell responses. Immunity 2011, 35, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Kashem, S.W.; Igyarto, B.Z.; Gerami-Nejad, M.; Kumamoto, Y.; Mohammed, J.; Jarrett, E.; Drummond, R.A.; Zurawski, S.M.; Zurawski, G.; Berman, J.; et al. Candida albicans morphology and dendritic cell subsets determine t helper cell differentiation. Immunity 2015, 42, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Martin, B.; Hirota, K.; Cua, D.J.; Stockinger, B.; Veldhoen, M. Interleukin-17-producing gammadelta t cells selectively expand in response to pathogen products and environmental signals. Immunity 2009, 31, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Maher, C.O.; Dunne, K.; Comerford, R.; O’Dea, S.; Loy, A.; Woo, J.; Rogers, T.R.; Mulcahy, F.; Dunne, P.J.; Doherty, D.G. Candida albicans stimulates il-23 release by human dendritic cells and downstream il-17 secretion by vdelta1 t cells. J. Immunol. 2015, 194, 5953–5960. [Google Scholar] [CrossRef] [PubMed]
- Sutton, C.E.; Lalor, S.J.; Sweeney, C.M.; Brereton, C.F.; Lavelle, E.C.; Mills, K.H. Interleukin-1 and il-23 induce innate il-17 production from gammadelta t cells, amplifying th17 responses and autoimmunity. Immunity 2009, 31, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Kashem, S.W.; Riedl, M.S.; Yao, C.; Honda, C.N.; Vulchanova, L.; Kaplan, D.H. Nociceptive sensory fibers drive interleukin-23 production from cd301b+ dermal dendritic cells and drive protective cutaneous immunity. Immunity 2015, 43, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Smeekens, S.P.; Plantinga, T.S.; van de Veerdonk, F.L.; Heinhuis, B.; Hoischen, A.; Joosten, L.A.; Arkwright, P.D.; Gennery, A.; Kullberg, B.J.; Veltman, J.A.; et al. Stat1 hyperphosphorylation and defective il12r/il23r signaling underlie defective immunity in autosomal dominant chronic mucocutaneous candidiasis. PLoS ONE 2011, 6, e29248. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.A.; Schlapbach, C. Th9 cells in skin disorders. Semin. Immunopathol. 2017, 39, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Swidergall, M.; Ernst, J.F. Interplay between Candida albicans and the antimicrobial peptide armory. Eukaryot. Cell 2014, 13, 950–957. [Google Scholar] [CrossRef] [PubMed]
- Sohnle, P.G.; Hahn, B.L.; Santhanagopalan, V. Inhibition of Candida albicans growth by calprotectin in the absence of direct contact with the organisms. J. Infect. Dis. 1996, 174, 1369–1372. [Google Scholar] [CrossRef] [PubMed]
- Sohnle, P.G.; Hunter, M.J.; Hahn, B.; Chazin, W.J. Zinc-reversible antimicrobial activity of recombinant calprotectin (migration inhibitory factor-related proteins 8 and 14). J. Infect. Dis. 2000, 182, 1272–1275. [Google Scholar] [CrossRef] [PubMed]
- Salazar, V.A.; Arranz-Trullen, J.; Navarro, S.; Blanco, J.A.; Sanchez, D.; Moussaoui, M.; Boix, E. Exploring the mechanisms of action of human secretory rnase 3 and rnase 7 against Candida albicans. Microbiologyopen 2016, 5, 830–845. [Google Scholar] [CrossRef] [PubMed]
- Ali, R.S.; Falconer, A.; Ikram, M.; Bissett, C.E.; Cerio, R.; Quinn, A.G. Expression of the peptide antibiotics human beta defensin-1 and human beta defensin-2 in normal human skin. J. Investig. Dermatol. 2001, 117, 106–111. [Google Scholar] [PubMed]
- Liang, S.C.; Tan, X.Y.; Luxenberg, D.P.; Karim, R.; Dunussi-Joannopoulos, K.; Collins, M.; Fouser, L.A. Interleukin (il)-22 and il-17 are coexpressed by th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 2006, 203, 2271–2279. [Google Scholar] [CrossRef] [PubMed]
- Hau, C.S.; Tada, Y.; Kanda, N.; Watanabe, S. Immunoresponses in dermatomycoses. J. Dermatol. 2015, 42, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Eyerich, S.; Wagener, J.; Wenzel, V.; Scarponi, C.; Pennino, D.; Albanesi, C.; Schaller, M.; Behrendt, H.; Ring, J.; Schmidt-Weber, C.B.; et al. Il-22 and tnf-alpha represent a key cytokine combination for epidermal integrity during infection with Candida albicans. Eur J. Immunol. 2011, 41, 1894–1901. [Google Scholar] [CrossRef] [PubMed]
- Kagami, S.; Rizzo, H.L.; Kurtz, S.E.; Miller, L.S.; Blauvelt, A. Il-23 and il-17a, but not il-12 and il-22, are required for optimal skin host defense against Candida albicans. J. Immunol. 2010, 185, 5453–5462. [Google Scholar] [CrossRef] [PubMed]
- Holdren, G.O.; Rosenthal, D.J.; Yang, J.; Bates, A.M.; Fischer, C.L.; Zhang, Y.; Brogden, N.K.; Brogden, K.A. Antimicrobial activity of chemokine cxcl10 for dermal and oral microorganisms. Antibiotics (Basel) 2014, 3, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Branzk, N.; Lubojemska, A.; Hardison, S.E.; Wang, Q.; Gutierrez, M.G.; Brown, G.D.; Papayannopoulos, V. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat. Immunol. 2014, 15, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Urban, C.F.; Reichard, U.; Brinkmann, V.; Zychlinsky, A. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell Microbiol. 2006, 8, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Trautwein-Weidner, K.; Gladiator, A.; Nur, S.; Diethelm, P.; LeibundGut-Landmann, S. Il-17-mediated antifungal defense in the oral mucosa is independent of neutrophils. Mucosal Immunol. 2015, 8, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Coughlin, L.A.; Neubauer, M.M.; Kim, J.; Kim, M.S.; Zhan, X.; Simms-Waldrip, T.R.; Xie, Y.; Hooper, L.V.; Koh, A.Y. Activation of hif-1alpha and ll-37 by commensal bacteria inhibits Candida albicans colonization. Nat. Med. 2015, 21, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Wanke, I.; Steffen, H.; Christ, C.; Krismer, B.; Gotz, F.; Peschel, A.; Schaller, M.; Schittek, B. Skin commensals amplify the innate immune response to pathogens by activation of distinct signaling pathways. J. Investig. Dermatol. 2011, 131, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Naik, S.; Bouladoux, N.; Linehan, J.L.; Han, S.J.; Harrison, O.J.; Wilhelm, C.; Conlan, S.; Himmelfarb, S.; Byrd, A.L.; Deming, C.; et al. Commensal-dendritic-cell interaction specifies a unique protective skin immune signature. Nature 2015, 520, 104–108. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kühbacher, A.; Burger-Kentischer, A.; Rupp, S. Interaction of Candida Species with the Skin. Microorganisms 2017, 5, 32. https://doi.org/10.3390/microorganisms5020032
Kühbacher A, Burger-Kentischer A, Rupp S. Interaction of Candida Species with the Skin. Microorganisms. 2017; 5(2):32. https://doi.org/10.3390/microorganisms5020032
Chicago/Turabian StyleKühbacher, Andreas, Anke Burger-Kentischer, and Steffen Rupp. 2017. "Interaction of Candida Species with the Skin" Microorganisms 5, no. 2: 32. https://doi.org/10.3390/microorganisms5020032
APA StyleKühbacher, A., Burger-Kentischer, A., & Rupp, S. (2017). Interaction of Candida Species with the Skin. Microorganisms, 5(2), 32. https://doi.org/10.3390/microorganisms5020032