
Changes in Microbial (Bacteria and Archaea) Plankton Community Structure after Artificial Dispersal in Grazer-Free Microcosms

 ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Microcosm Preparation
2.2. Flow Cytometry
2.3. DNA Extraction and Pyrosequencing Analysis
2.4. Analysis of Prokaryoplankton Community Structure
3. Results
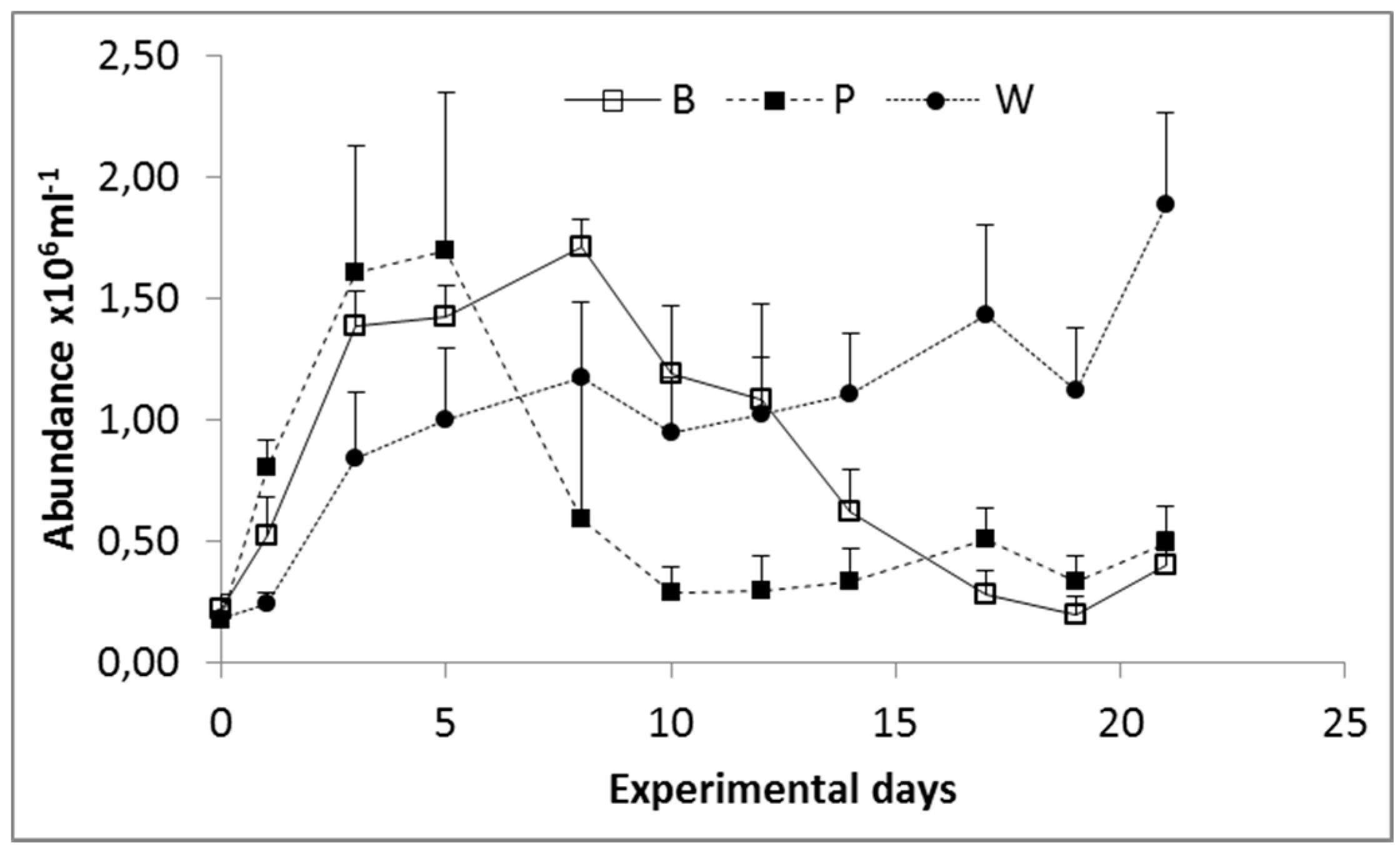
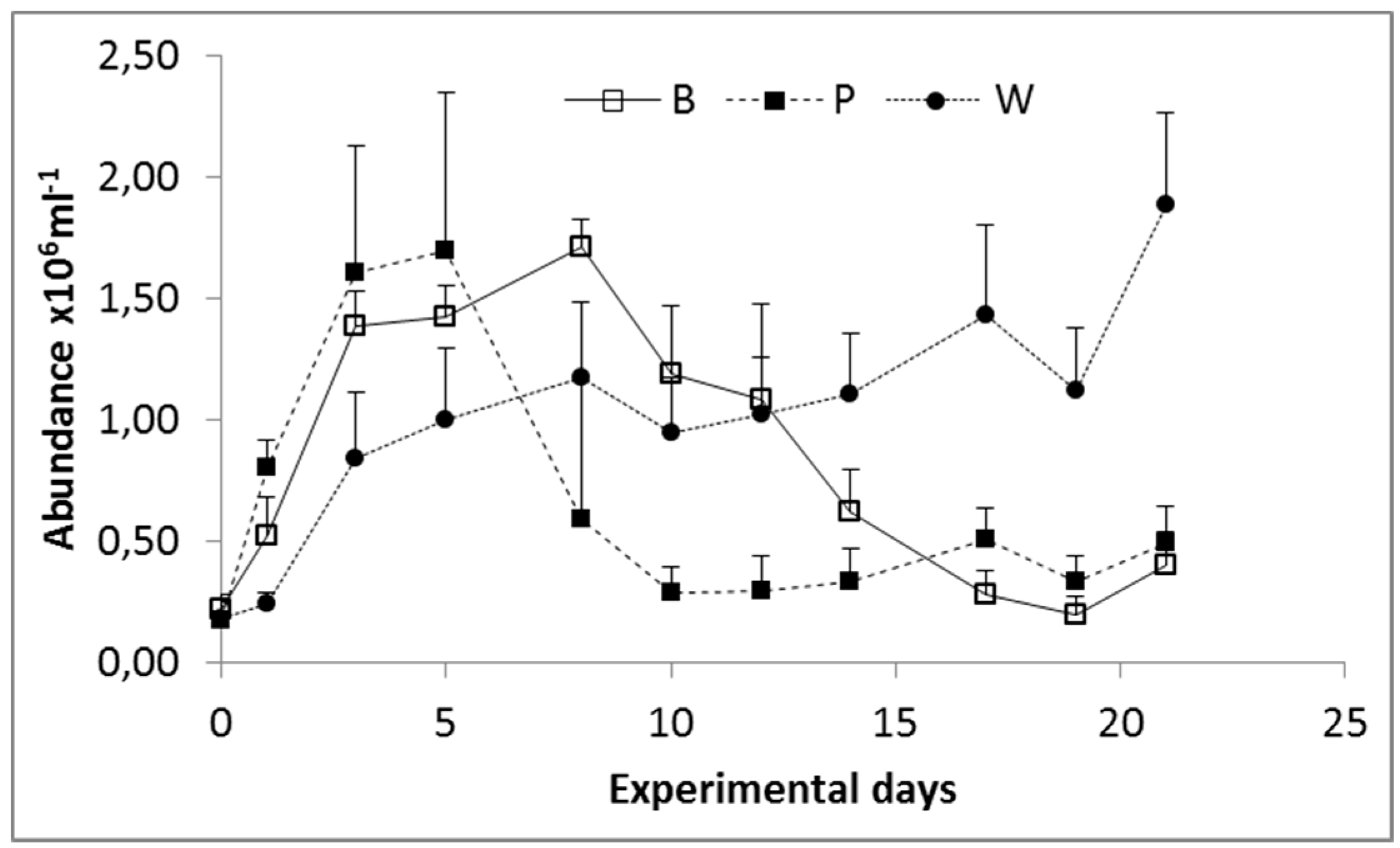
3.1. Prokaryoplankton Abundance
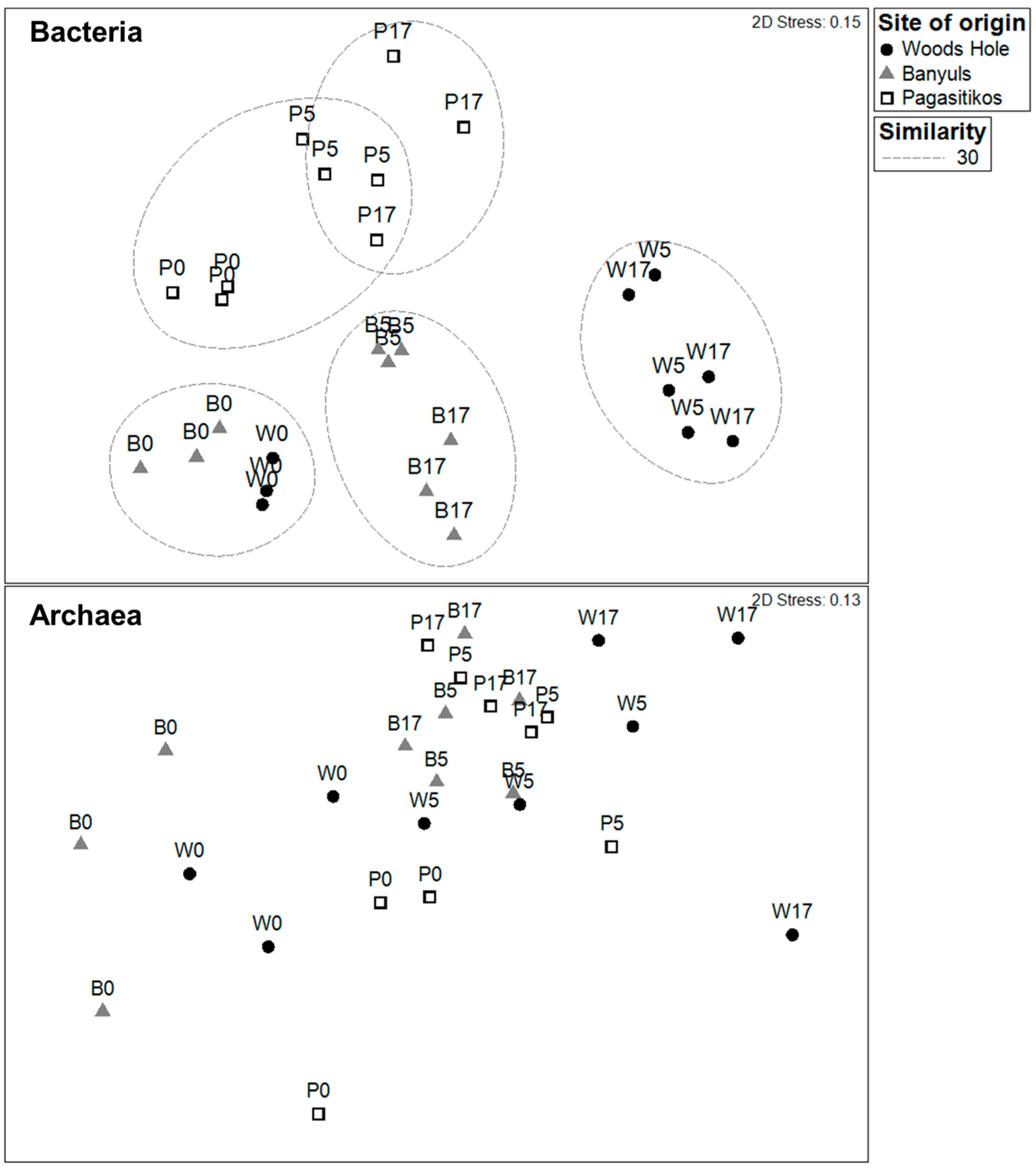
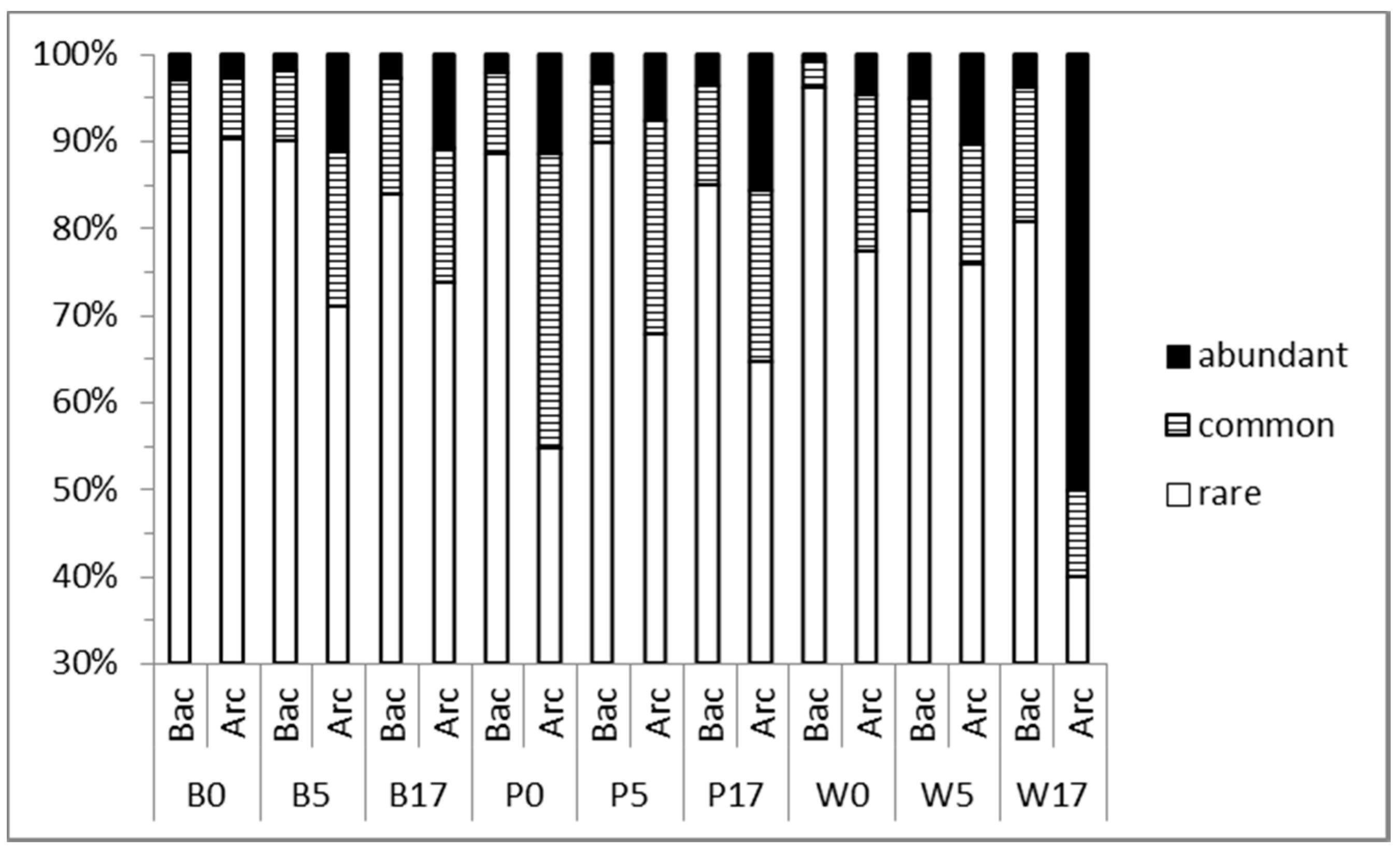
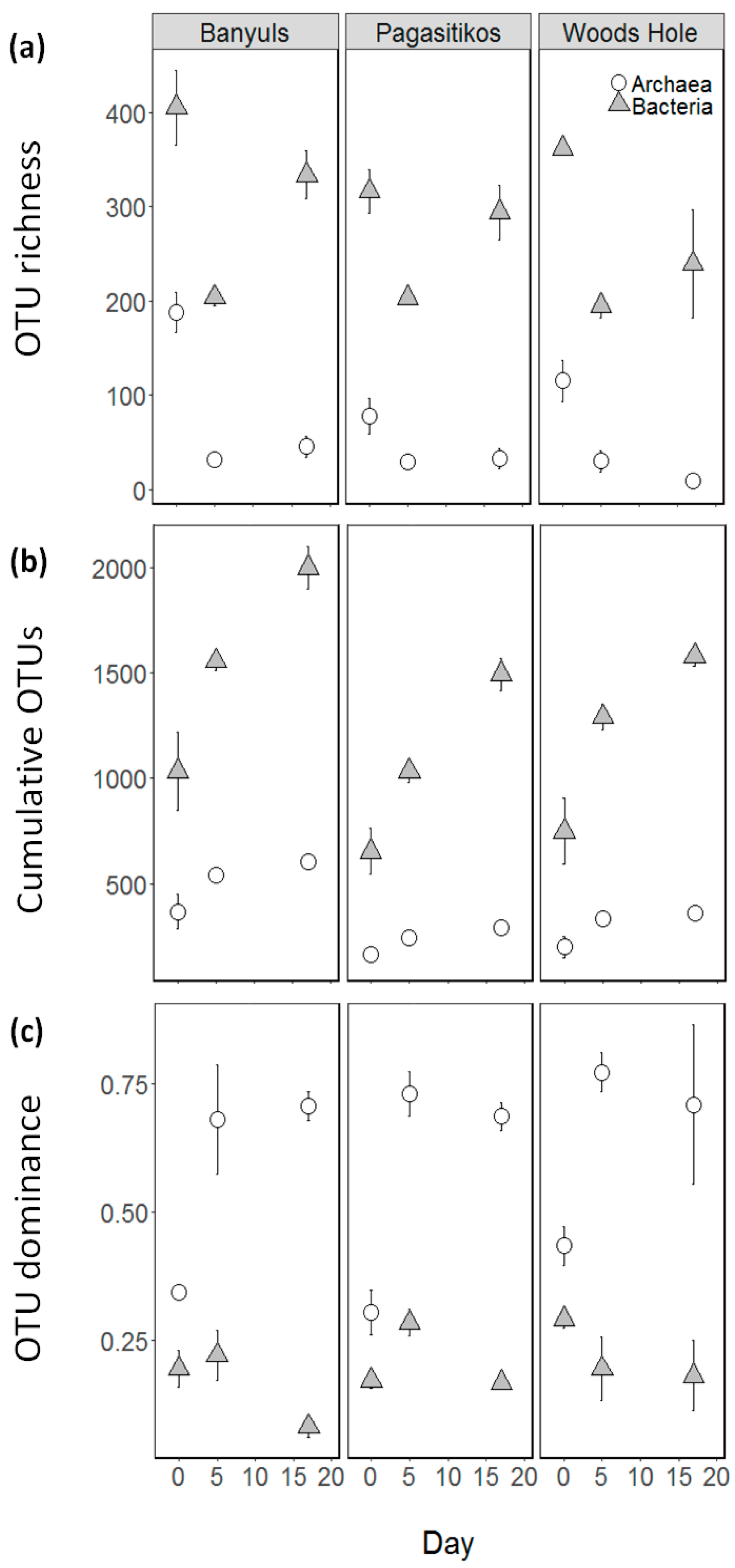
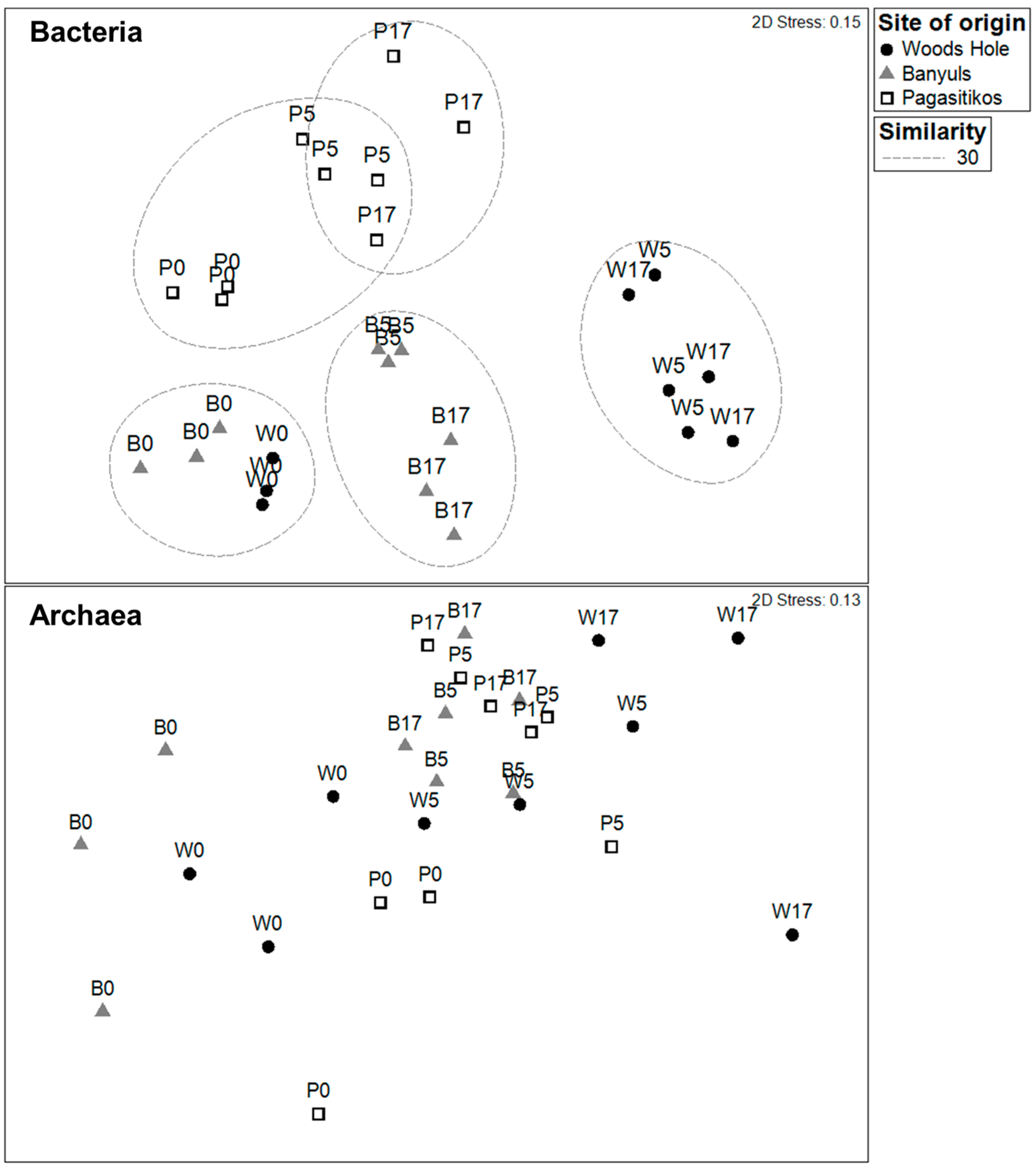
3.2. Prokaryoplankton Community Structure
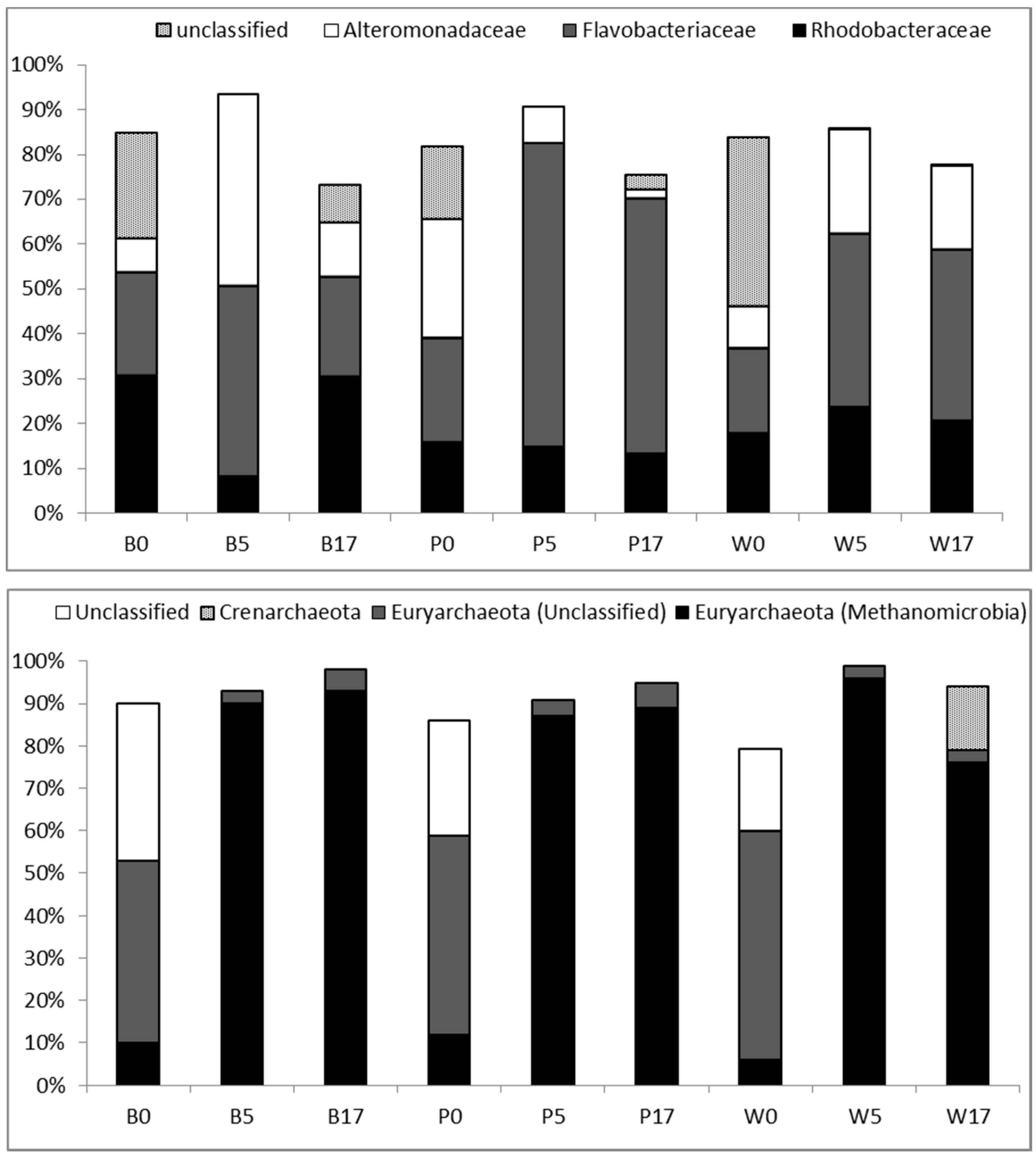
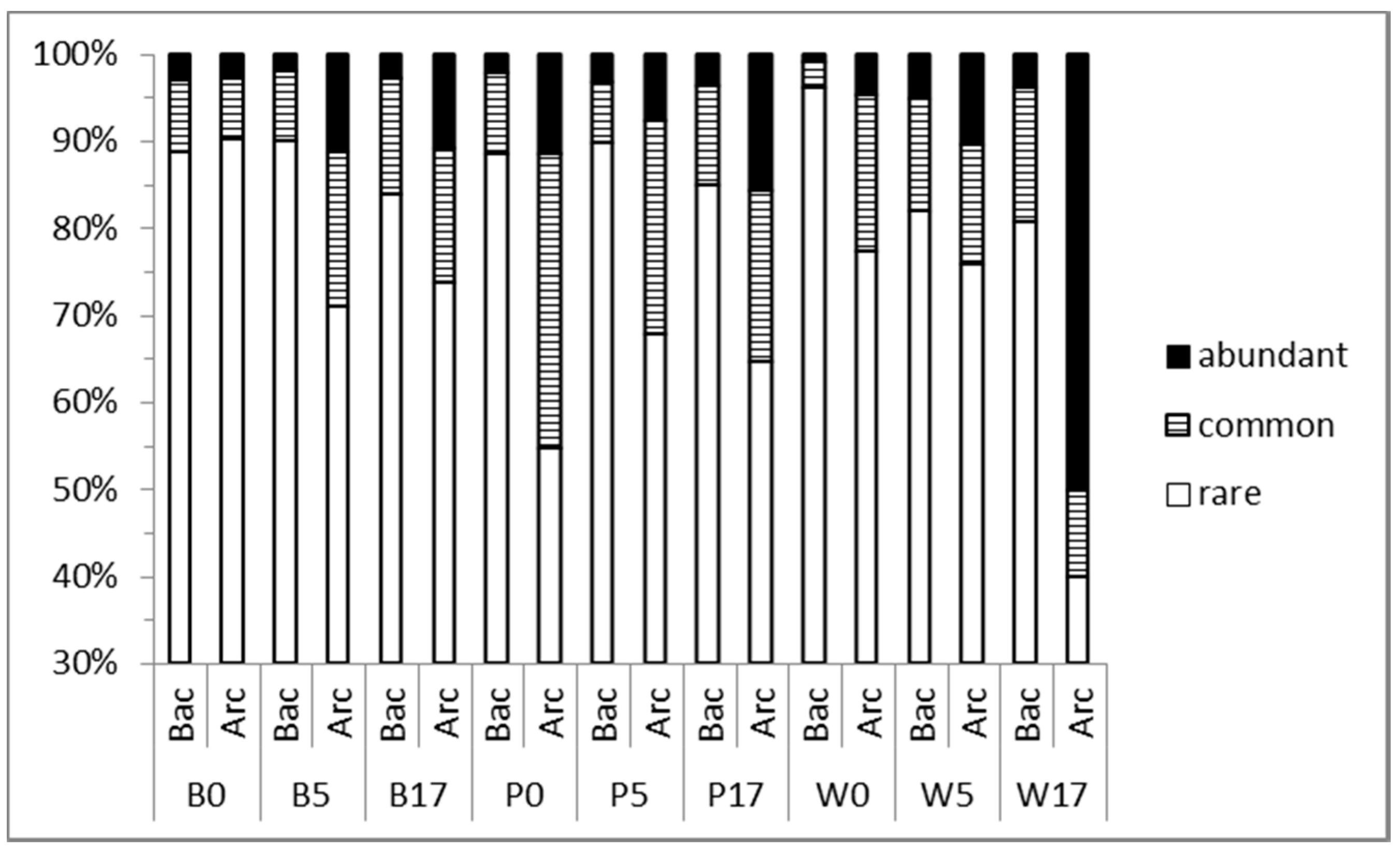
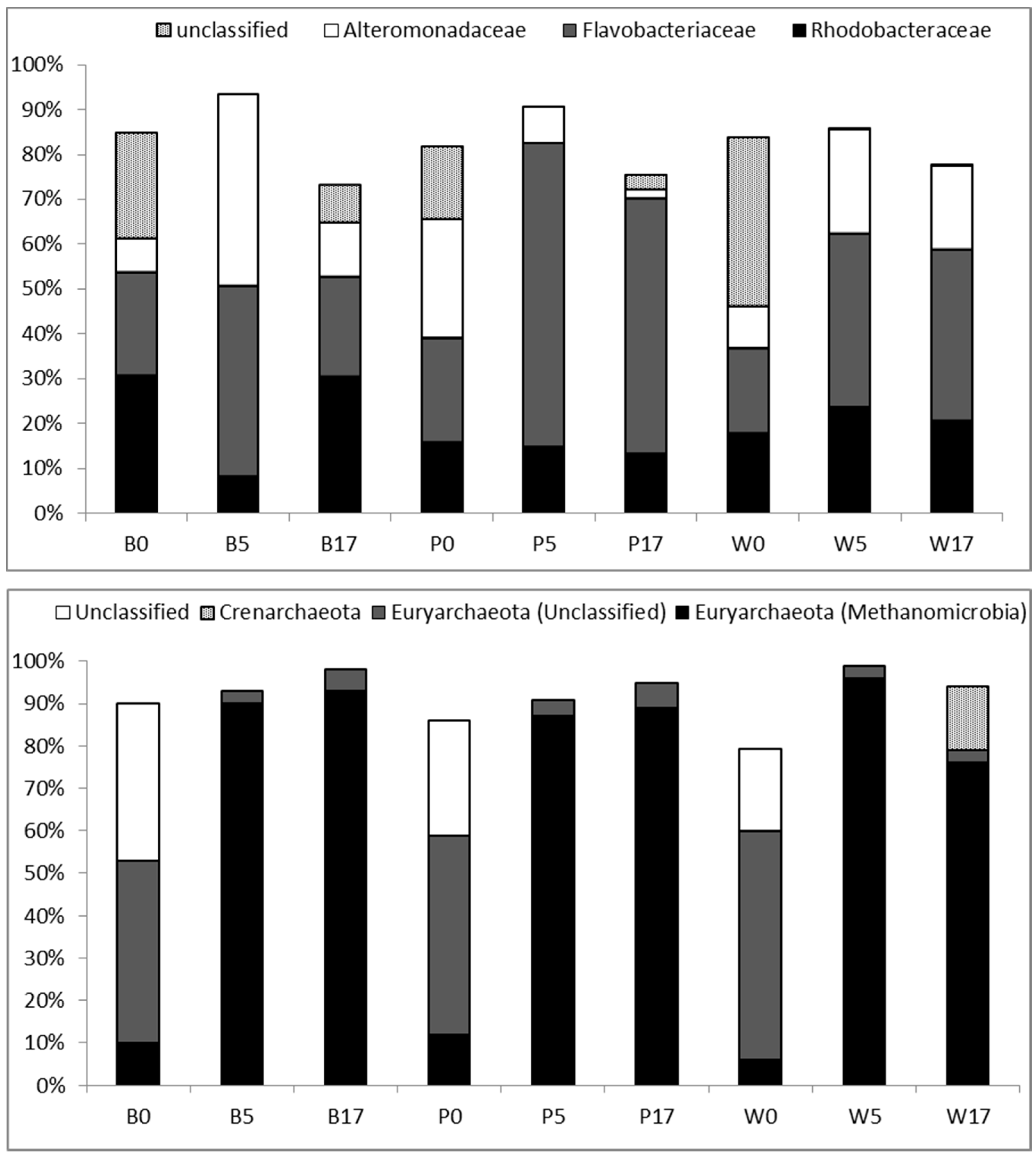
3.3. Taxonomic Diversity at the Family Level
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cermeño, P.; de Vargas, C.; Abrantes, F.; Falkowski, P.G. Phytoplankton biogeography and community stability in the ocean. PLoS ONE 2010, 5, e10037. [Google Scholar] [CrossRef] [PubMed]
- Finlay, B.J.; Esteban, G.F. Ubiquitous microbes and ecosystem function. Limnetica 2001, 20, 31–43. [Google Scholar]
- Pagenkopp Lohan, K.M.; Fleischer, R.C.; Carney, K.J.; Holzer, K.K.; Ruiz, G.M. Amplicon-based pyrosequencing reveals high diversity of protistan parasites in ships’ ballast water: Implications for biogeography and infectious diseases. Microb. Ecol. 2016, 71, 530–542. [Google Scholar] [CrossRef] [PubMed]
- De Wit, R.; Bouvier, T. Everything is everywhere, but, the environment selects’; what did Baas Becking and Beijerinck really say? Environ. Microbiol. 2006, 8, 755–758. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, M.A. “Everything is everywhere: But the environment selects”: Ubiquitous distribution and ecological determinism in microbial biogeography. Stud. Hist. Philos. Biol. Biomed. Sci. 2008, 39, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Agogué, H.; Lamy, D.; Neal, P.R.; Sogin, M.L.; Herndl, G.J. Water mass-specificity of bacterial communities in the North Atlantic revealed by massively parallel sequencing. Mol. Ecol. 2011, 20, 258–274. [Google Scholar] [CrossRef] [PubMed]
- Andersson, A.F.; Riemann, L.; Bertilsson, S. Pyrosequencing reveals contrasting seasonal dynamics of taxa within baltic sea bacterioplankton communities. ISME J. 2009, 4, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Meziti, A.; Kormas, K.A.; Moustaka-Gouni, M.; Karayanni, H. Spatially uniform but temporally variable bacterioplankton in a semi-enclosed coastal area. Syst. Appl. Microbiol. 2015, 38, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Lima-Mendez, G.; Faust, K.; Henry, N.; Decelle, J.; Colin, S.; Carcillo, F.; Chaffron, S.; Ignacio-Espinosa, J.C.; Roux, S.; Vincent, F.; et al. Determinants of community structure in the global plankton interactome. Science 2015, 348, 1262073. [Google Scholar] [CrossRef] [PubMed]
- Zinger, L.; Amaral-Zettler, L.A.; Fuhrman, J.A.; Horner-Devine, M.C.; Huse, S.M.; Welch, D.B.M.; Martiny, J.B.H.; Sogin, M.; Boetius, A.; Ramette, A. Global patterns of bacterial beta-diversity in seafloor and seawater ecosystems. PLoS ONE 2011, 6, e24570. [Google Scholar] [CrossRef] [PubMed]
- Shade, A.; Chiu, C.Y.; McMahon, K.D. Differential bacterial dynamics promote emergent community robustness to lake mixing: An epilimnion to hypolimnion transplant experiment. Environ. Microbiol. 2010, 12, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Szekely, A.J.; Berga, M.; Langenheder, S. Mechanisms determining the fate of dispersed bacterial communities in new environments. ISME J. 2013, 7, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Fuhrman, J.A.; Hewson, I.; Schwalbach, M.S.; Steele, J.A.; Brown, M.V.; Naeem, S. Annually reoccurring bacterial communities are predictable from ocean conditions. Proc. Natl. Acad. Sci. USA 2006, 103, 13104–13109. [Google Scholar] [CrossRef] [PubMed]
- Krause, E.; Wichels, A.; Giménez, L.; Lunau, M.; Schilhabel, M.B.; Gerdts, G. Small changes in pH have direct effects on marine bacterial community composition: A microcosm approach. PLoS ONE 2012, 7, e47035. [Google Scholar] [CrossRef] [PubMed]
- Langenheder, S.; Lindström, E.S.; Tranvik, L.J. Weak coupling between community composition and functioning of aquatic bacteria. Limnol. Oceanogr. 2005, 50, 957–967. [Google Scholar] [CrossRef]
- Galand, P.E.; Gutiérrez-Provecho, C.; Massana, R.; Gasol, J.M.; Casamayor, E.O. Inter-annual recurrence of archaeal assemblages in the coastal NW Mediterranean Sea (Blanes Bay Microbial Observatory). Limnol. Oceanogr. 2010, 55, 2117–2525. [Google Scholar] [CrossRef]
- Hugoni, M.; Taib, N.; Debroas, D.; Domaizon, I.; Dufournel, I.J.; Bronner, G.; Salter, I.; Agogué, H.; Mary, I.; Galand, P.E. Structure of the rare archaeal biosphere and seasonal dynamics of active ecotypes in surface coastal waters. Proc. Natl. Acad. Sci. USA 2013, 110, 6004–6009. [Google Scholar] [CrossRef] [PubMed]
- Smeti, E.; Kormas, K.A.; Spatharis, S. A non-phylogenetic alpha diversity approach on prokaryotic community structure in aquatic systems. Ecol. Indic. 2013, 29, 361–366. [Google Scholar] [CrossRef]
- Dowd, S.; Callaway, T.; Wolcott, R.; Sun, Y.; McKeehan, T.; Hagevoort, R.; Edrington, T. Evaluation of the bacterial diversity in the feces of cattle using 16s rDNA bacterial tag-encoded flx amplicon pyrosequencing (btefap). BMC Microbiol. 2008, 8, 125. [Google Scholar] [CrossRef] [PubMed]
- Quince, C.; Lanzen, A.; Curtis, T.P.; Davenport, R.J.; Hall, N.; Head, I.M.; Read, L.F.; Sloan, W.T. Accurate determination of microbial diversity from 454 pyrosequencing data. Nat. Meth. 2009, 6, 639–641. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R. Search and clustering orders of magnitude faster than blast. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Kunin, V.; Engelbrektson, A.; Ochman, H.; Hugenholtz, P. Wrinkles in the rare biosphere: Pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ. Microbiol. 2010, 12, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.; Ludwig, W.; Peplies, J.; Glöckner, F. Silva: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef] [PubMed]
- McNaughton, S.J.; Wolf, L.L. Dominance and the niche in ecological systems. Science 1970, 167, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Sjöstedt, J.; Koch-Schmidt, P.; Pontarp, M.; Canbäck, B.; Tunlid, A.; Lundberg, P.; Hagström, Å.; Riemann, L. Recruitment of members from the rare biosphere of marine bacterioplankton communities after an environmental disturbance. Appl. Environ. Microbiol. 2012, 78, 1361–1369. [Google Scholar] [CrossRef] [PubMed]
- Pandit, S.N.; Kolasa, J.; Cottenie, K. Contrasts between habitat generalists and specialists: An empirical extension to the basic metacommunity framework. Ecology 2009, 90, 2253–2262. [Google Scholar] [CrossRef] [PubMed]
- Cury, J.C.; Araujo, F.V.; Coelho-Souza, S.A.; Peixoto, R.S.; Oliveira, J.A.L.; Santos, H.F.; Dávila, A.M.R.; Rosado, A.S. Microbial diversity of a brazilian coastal region influenced by an upwelling system and anthropogenic activity. PLoS ONE 2012, 6, e16553. [Google Scholar] [CrossRef] [PubMed]
- Iverson, V.; Morris, R.M.; Frazar, C.D.; Berthiaume, C.T.; Morales, R.L.; Armbrust, E.V. Untangling genomes from metagenomes: Revealing an uncultured class of marine euryarchaeota. Science 2012, 335, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, K.G.; Albert, D.B.; Biddle, J.F.; Chanton, J.P.; Pizarro, O.; Teske, A. Spatial structure and activity of sedimentary microbial communities underlying a beggiatoa spp. Mat in a gulf of mexico hydrocarbon seep. PLoS ONE 2010, 5, e8738. [Google Scholar] [CrossRef] [PubMed]
- Petihakis, G.; Triantafyllou, G.; Pollani, A.; Koliou, A.; Theodorou, A. Field data analysis and application of a complex water column biogeochemical model in different areas of a semi-enclosed basin: Towards the development of an ecosystem management tool. Mar. Environ. Res. 2005, 59, 493–518. [Google Scholar] [CrossRef] [PubMed]
- Charles, F.; Lantoine, F.; Brugel, S.; Chrétiennot-Dinet, M.-J.; Quiroga, I.; Rivière, B. Seasonal survey of the phytoplankton biomass, composition and production in a littoral NW mediterranean site, with special emphasis on the picoplanktonic contribution. Estuar. Coast. Shelf Sci. 2005, 65, 199–212. [Google Scholar] [CrossRef]
- Alonso-Sáez, L.; Díaz-Pérez, L.; Morán, X.A.G. The hidden seasonality of the rare biosphere in coastal marine bacterioplankton. Environ. Microbiol. 2015, 17, 3766–3780. [Google Scholar] [CrossRef] [PubMed]
- Christian, R.R.; Capone, D.G. Overview of Issues in Aquatic Microbial Ecology. In Manual of Environmental Microbiology, 2nd ed.; Hurst, C.J., Crawford, R.L., Knudsen, G.R., McInerney, M.J., Stetzenbach, L.D., Eds.; ASM Press: Washington, DC, USA, 2002; pp. 323–328. [Google Scholar]
- Marrasé, C.; Lim, E.L.; Caron, D.A. Seasonal and daily changes in bacterivory in a coastal plankton community. Mar. Ecol. Prog. Ser. 1992, 82, 281–289. [Google Scholar]
- Shade, A.; Jones, S.E.; Caporaso, J.G.; Handelsman, J.; Knight, R.; Fierer, N.; Gilbert, J.A. Conditionally rare taxa disproportionately contribute to temporal changes in microbial diversity. mBio 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Langenheder, S.; Lindström, E.S.; Tranvik, L.J. Structure and function of bacterial communities emerging from different sources under identical conditions. Appl. Environ. Microbiol. 2006, 72, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Motegi, C.; Nagata, T.; Miki, T.; Weinbauer, M.G.; Legendre, L.; Rassoulzadegan, F. Interactive effects of viral and bacterial production on marine bacterial diversity. PLoS ONE 2013, 8, e76800. [Google Scholar] [CrossRef] [PubMed]
- Steele, J.A.; Countway, P.D.; Xia, L.; Vigil, P.D.; Beman, J.M.; Kim, D.Y.; Chow, C.-E.T.; Sachdeva, R.; Jones, A.C.; Schwalbach, M.S.; et al. Marine bacterial, archaeal and protistan association networks reveal ecological linkages. ISME J. 2011, 5, 1414–1425. [Google Scholar] [CrossRef] [PubMed]
- Pande, S.; Kaftan, F.; Lang, S.; Svatos, A.; Germerodt, S.; Kost, C. Privatization of cooperative benefits stabilizes mutualistic cross-feeding interactions in spatially structured environments. ISME J. 2016, 10, 1413–1423. [Google Scholar] [CrossRef] [PubMed]
- Widder, S.; Allen, R.J.; Pfeiffer, T.; Curtis, T.P.; Wiuf, C.; Sloan, W.T.; Cordero, O.X.; Brown, S.P.; Momeni, B.; Shou, W.; et al. Challenges in microbial ecology: Building predictive understanding of community function and dynamics. ISME J. 2016, 10, 2557–2568. [Google Scholar] [CrossRef] [PubMed]
- Pedler, B.E.; Aluwihare, L.I.; Azam, F. Single bacterial strain capable of significant contribution to carbon cycling in the surface ocean. Proc. Natl. Acad. Sci. USA 2014, 111, 7202–7207. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.E.; Wear, E.K. Microbial diversity and the lability of dissolved organic carbon. Proc. Natl. Acad. Sci. USA 2014, 111, 7166–7167. [Google Scholar] [CrossRef] [PubMed]
- Yokokawa, T.; Nagata, T. Growth and grazing mortality rates of phylogenetic groups of bacterioplankton in coastal marine environments. Appl. Environ. Microbiol. 2005, 71, 6799–6807. [Google Scholar] [CrossRef] [PubMed]
- Allers, E.; Gómez-Consarnau, L.; Pinhassi, J.; Gasol, J.M.; Šimek, K.; Pernthaler, J. Response of alteromonadaceae and rhodobacteriaceae to glucose and phosphorus manipulation in marine mesocosms. Environ. Microbiol. 2007, 9, 2417–2429. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, B.M.; Zubkov, M.V.; Sahm, K.; Burkill, P.H.; Amann, R. Changes in community composition during dilution cultures of marine bacterioplankton as assessed by flow cytometric and molecular biological techniques. Environ. Microbiol. 2000, 2, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Cottrel, M.T.; Kirchman, D.L. Natural Assemblages of Marine Proteobacteria and Members of the Cytophaga-Flavobacter Cluster Consuming Low- and High-Molecular-Weight Dissolved Organic Matter. Appl. Environ. Microbiol. 2000, 66, 1692–1697. [Google Scholar] [CrossRef]
- Suzuki, M.; Nakagawa, Y.; Harayama, S.; Yamamoto, S. Phylogenetic analysis and taxonomic study of marine cytophaga-like bacteria: Proposal for tenacibaculum gen. Nov. With tenacibaculum maritimum comb. Nov. In addition, tenacibaculum ovolyticum comb. Nov., and description of tenacibaculum mesophilum sp. Nov. In addition, tenacibaculum amylolyticum sp. Nov. Int. J. Syst. Evol. Microbiol. 2001, 51, 1639–1652. [Google Scholar] [PubMed]
- Elifantz, H.; Horn, G.; Ayon, M.; Cohen, Y.; Minz, D. Rhodobacteraceae are the key members of the microbial community of the initial biofilm formed in eastern mediterranean coastal seawater. FEMS Microbiol. Ecol. 2013, 85, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Kostka, J.E.; Prakash, O.; Overholt, W.A.; Green, S.J.; Freyer, G.; Canion, A.; Delgardio, J.; Norton, N.; Hazen, T.C.; Huettel, M. Hydrocarbon-degrading bacteria and the bacterial community response in gulf of mexico beach sands impacted by the deepwater horizon oil spill. Appl. Environ. Microbiol. 2011, 77, 7962–7974. [Google Scholar] [CrossRef] [PubMed]
- Lefort, T.; Gasol, J.M. Global-scale distributions of marine surface bacterioplankton groups along gradients of salinity, temperature, and chlorophyll: A meta-analysis of fluorescence in situ hybridization studies. Aquat. Microb. Ecol. 2013, 70, 111–130. [Google Scholar] [CrossRef]
- Stoica, E.; Herndl, G.J. Contribution of crenarchaeota and euryarchaeota to the prokaryotic plankton in the coastal northwestern black sea. J. Plankton Res. 2007, 29, 699–706. [Google Scholar] [CrossRef]
- Crump, B.C.; Baross, J.A. Archaeaplankton in the Columbia River, its estuary and the adjacent coastal ocean, USA. FEMS Microbiol. Ecol. 2000, 31, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Wuchter, C.; Abbas, B.; Coolen, M.J.L.; Herfort, L.; Van Bleijswijk, J.; Timmers, P.; Strous, M.; Teira, E.; Herndl, G.J.; Middelburg, J.J.; et al. Archaeal nitrification in the ocean. Proc. Natl. Acad. Sci. USA 2006, 103, 12317–12322. [Google Scholar] [CrossRef] [PubMed]
- Bogard, M.J.; del Giorgio, P.A.; Boutet, L.; Chaves, M.C.G.; Prairie, Y.T.; Merante, A.; Derry, A.M. Oxic water column methanogenesis as a major coponent of aquatic CH4 fluxes. Nat. Commun. 2014, 5, 5350. [Google Scholar] [CrossRef] [PubMed]
- Fiegna, F.; Moreno-Letelier, A.; Bell, T.; Barraclough, T.G. Evolution of species interactions determines microbial community productivity in new environments. ISME J. 2015, 9, 1235–1245. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| F-Ratio | |||
|---|---|---|---|
| Site | Day | Interaction of Site with Day | |
| Richness—all | 6.89 * | 48.93 ** | 3.40 * |
| Richness—Archaea | 8.02 ** | 48.95 ** | 5.01 ** |
| Richness—Bacteria | 2.68 | 24.42 ** | 1.34 |
| Dominance—Archaea | 1.696 | 25.73 ** | 0.27 |
| Dominance—Bacteria | 0.83 | 4.56 ** | 2.12 |
| Cumulative OTUs—all | 4.66 * | 16.12 *** | 0.08 |
| Cumulative OTUs—Archaea | 35.76 *** | 22.68 *** | 0.67 |
| Cumulative OTUs—Bacteria | 11.99 *** | 74.08 *** | 0.21 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karayanni, H.; Meziti, A.; Spatharis, S.; Genitsaris, S.; Courties, C.; Kormas, K.A. Changes in Microbial (Bacteria and Archaea) Plankton Community Structure after Artificial Dispersal in Grazer-Free Microcosms. Microorganisms 2017, 5, 31. https://doi.org/10.3390/microorganisms5020031
Karayanni H, Meziti A, Spatharis S, Genitsaris S, Courties C, Kormas KA. Changes in Microbial (Bacteria and Archaea) Plankton Community Structure after Artificial Dispersal in Grazer-Free Microcosms. Microorganisms. 2017; 5(2):31. https://doi.org/10.3390/microorganisms5020031
Chicago/Turabian StyleKarayanni, Hera, Alexandra Meziti, Sofie Spatharis, Savvas Genitsaris, Claude Courties, and Konstantinos A. Kormas. 2017. "Changes in Microbial (Bacteria and Archaea) Plankton Community Structure after Artificial Dispersal in Grazer-Free Microcosms" Microorganisms 5, no. 2: 31. https://doi.org/10.3390/microorganisms5020031
APA StyleKarayanni, H., Meziti, A., Spatharis, S., Genitsaris, S., Courties, C., & Kormas, K. A. (2017). Changes in Microbial (Bacteria and Archaea) Plankton Community Structure after Artificial Dispersal in Grazer-Free Microcosms. Microorganisms, 5(2), 31. https://doi.org/10.3390/microorganisms5020031