Abstract
Colletotrichum graminicola, the causative agent of maize anthracnose leaf blight and stalk rot, severely jeopardizes the healthy development of the maize industry. Auxiliary activity enzymes (AAs), a vital subclass of carbohydrate-active enzymes, act as beneficial accessory proteins for fungi in degrading lignocellulose. This study identified 127 AA genes from the genome of C. graminicola strain TZ-3 and further analyzed the subcellular localization, conserved motifs, and domains of the proteins encoded by these genes. The CgAA genes exhibited significant variations in gene structure, and the structural motifs within their encoded proteins also differed. Subcellular localization analysis revealed that most CgAA proteins were localized in the extracellular space. Moreover, the CgAA gene family contained abundant conserved domains, suggesting diverse functionalities and potential roles in various fungal biological processes. Multiple cis-acting regulatory elements related to stress responses and plant hormones were detected in the promoter regions of these genes. This study analyzed the expression patterns of CgAA genes during pathogen–host interactions and found that most CgAA genes were differentially expressed in the interaction between C. graminicola and maize. Coupled with GO functional analysis, it was discovered that CgAAs are deeply involved in the interaction between C. graminicola and maize, closely associated with the pathogenic mechanisms of the pathogen, and may play crucial roles in the initiation and expansion of fungal infections. These results provide valuable resources for elucidating the functions of AA genes and lay the groundwork for sustainable agricultural development through the utilization of AA genes in disease control and the breeding of stress-resistant, high-yield crop varieties.
1. Introduction
The first line of defense for plants against pathogens is the plant cell wall, primarily composed of polysaccharides such as cellulose and pectin. During the infection process, plant pathogenic fungi secrete cell wall-degrading enzymes to disrupt the plant cell wall, thereby facilitating successful invasion of the host [1,2]. Fungi typically utilize various cellulose-degrading enzymes that synergistically degrade cellulose. The carbohydrate-active enzyme (CAZy) database classifies these enzymes based on sequence similarity into six main classes: glycoside hydrolases (GHs), glycosyltransferases (GTs), polysaccharide lyases (PLs), carbohydrate esterases (CEs), auxiliary activity enzymes (AAs), and carbohydrate-binding modules (CBMs) [2]. Some studies have indicated that members of GHs, PLs, and CEs participate in the degradation of plant cell walls and the pathogenic processes of pathogens [3,4,5,6]. While GHs, PLs, and CEs are well studied, the role of AAs, a key class of redox enzymes, remains poorly understood in fungal pathogenicity.
AAs, a core subclass of CAZymes, focus on redox enzymes that aid in the degradation of recalcitrant polysaccharides via oxidative mechanisms. These enzymes frequently depend on coenzymes to catalyze the generation of reactive oxygen species, synergistically enhancing the accessibility of substrates along with other CAZymes. AAs are indispensable during the process of pathogen infection, as they can disrupt plant cell walls to facilitate successful invasion by pathogens. The AA class is currently divided into nine lignin-degrading enzyme families (AA1–8, AA12) and eight lytic polysaccharide monooxygenase families (AA9–11, AA13–17) [2,7,8]. Members of different AA families have been shown to possess multiple substrate activities, including the oxidative cleavage of crystalline cellulose, hemicellulose, chitin, xylan, xyloglucan, and starch [7,8]. Many AAs in pathogenic fungi possess multiple substrate activities, which are also components of plant cell walls [9,10]. Numerous fungal organisms encode various AA proteins, known for their diversity and abundance in fungal genomes. During the infection of host plants, AAs can not only oxidize and cleave recalcitrant polysaccharides in cell walls to disrupt them but also act as effector molecules involved in the pathogenic process and immune response of plants [11,12,13,14]. Additionally, they can serve as coordinating factors to promote mutualistic symbiosis between different species [15,16].
Colletotrichum graminicola, the causative agent of maize anthracnose stem rot, is a worldwide maize disease that can lead to leaf blight, stalk rot, and plant lodging, resulting in yield losses of 30–50% in severe cases [17,18]. Affected by global climate change, this disease has emerged in major maize-producing areas in Northeast and North China in recent years, posing a serious threat to maize production security in China [19]. C. graminicola belongs to hemibiotrophic pathogenic fungi, with its infection process initially being biotrophic and gradually transitioning to necrotrophic. During the biotrophic stage, spherical invasion hyphae enveloped by the host plasma membrane develop within living epidermal cells, with hyphae rapidly growing and spreading to kill host cells, followed by the entry of fungi into the necrotrophic stage [20]. Currently, the prevention and control of maize anthracnose primarily rely on benzimidazole chemical agents, which can lead to issues such as pathogen resistance, pesticide residues, and environmental pollution [21]. The increasing occurrence of pathogen resistance undermines the effectiveness of chemical control measures. Given the extensive global cultivation of maize, sustainable solutions are imperative to safeguard food security.
Recently, with the development of molecular biology and genomic technologies, researchers have prompted researchers to delve into the genomic architectures and pathogenicity mechanisms of crop pathogenic fungi [22,23,24]. The completion of the whole-genome sequencing of C. graminicola has provided conditions for analyzing genes related to pathogenesis [25]. Currently, research on AA gene families in pathogenic fungi is still limited. This study identified and analyzed all AA-family genes encoding proteins and their biochemical characteristics by searching the genome of C. graminicola TZ-3. Phylogenetic analysis was used to investigate the relationships among AA genes, the Gene Structure Display Server (GSDS) for predicting the structural features of AA genes, and Multiple Em for Motif Elicitation (MEME) for predicting conserved motifs in AA proteins. We analyzed the response of the CgAA gene family during C. graminicola infection, and the results indicated that CgAA genes may play crucial roles in the pathogenesis of C. graminicola. This study expands the available genetic resources of AA genes and broadens their potential applications in sustainable agriculture.
2. Materials and Methods
2.1. Identification and Phylogenetic Analysis of CgAA-Family Genes
To identify AA-family genes in C. graminicola TZ-3, genome data were obtained from the genome sequencing databases based on previously published information [25]. In the CAZy database, AA genes in the C. graminicola genome are identified using an e-value cutoff of 1 × 10−5. The physicochemical properties of CgAA proteins, including molecular weight, isoelectric point, and GRAVY, were predicted using the online ExPASy website (https://web.expasy.org/protparam/) (accessed on 10 May 2025) [26]. Protein subcellular localization was determined using the PSORT web tool (https://wolfpsort.hgc.jp/) (accessed on 10 May 2025).
2.2. Multiple Sequence Alignment and Phylogenetic Analysis
Multiple sequence alignment of AA-family protein sequences from C. graminicola TZ-3 was performed using Clustal W with default parameters. Based on the multiple sequence alignment results, a phylogenetic tree was constructed using the maximum likelihood (ML) method in MEGA 7.0. To obtain relatively stable and accurate results, the bootstrap value was set to 1000. The 127 CgAA proteins were grouped according to their clustering in the phylogenetic tree. The evolutionary tree was visually optimized using the Evolview website (http://evolgenius.info/evolview-v2/) (accessed on 20 May 2025) [27].
2.3. Gene Structure and Protein Motif Analysis
The GSDS (https://gsds.gao-lab.org/index.php) (accessed on 20 May 2025) was used to predict exons and introns in CgAA genes [28]. Conserved motifs in CgAA protein sequences were predicted using the MEME website (http://meme-suite.org) (accessed on 20 May 2025) [29]. To ensure the reliability of the analysis results, the number of motifs in MEME was set to 10, and other parameters were set to default values.
2.4. CARE and Gene Ontology (GO) Analysis
The 2000 bp upstream sequences of CgAA genes were obtained from the C. graminicola TZ-3 genome. CAREs were identified using the PlantCARE online server (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/) (accessed on 20 May 2025) [30]. Visualization was completed using TBtools II v2.142 software [31]. GO functional enrichment analysis was performed using the website (https://www.omicshare.com/tools) (accessed on 20 May 2025) [32].
2.5. Analysis of Expression Patterns of CgAA-Family Genes
To study the expression of CgAA genes at different infection time points, RNA-seq data (accession number: GSE34632) from C. graminicola were collected from previous reports [20]. The selected time points delineate three key infection stages: 24 h post-infection (hpi) marks the initial attachment phase, 36 hpi corresponds to intracellular biotrophic hyphal development, and 60 hpi signifies the emergence of necrotrophic hyphal specialization [20]. A heatmap was generated based on FPKM values using the online website (https://www.omicshare.com/tools) (accessed on 25 May 2025) [32].
2.6. RT-qPCR
Transcriptome data validation was performed via RT-qPCR. Primer design utilized Primer Premier 5.0 software. Total RNA isolation from C. graminicola-infected maize leaves employed the Tiangen DP441 assay kit (Tiangen, Beijing, China). First-strand cDNA synthesis was conducted using the HiScript III First Strand cDNA Synthesis Kit (Vazyme, Nanjing, China). RT-qPCR amplification utilized the Taq Pro Universal SYBR qPCR Master Mix Kit (Vazyme, China) on an ABI 7500 Real-Time System (Applied Biosystems, Waltham, MA, USA). Gene expression levels, normalized to the UBQ reference gene, were quantified using the 2−∆∆CT method [33]. The dataset was statistically analyzed using IBM SPSS Statistics software (v29.0.2.0) and Student’s t-test method. Data visualization was created using GraphPad Prism (v9.3).
3. Results
3.1. Identification and Physicochemical Property Analysis of CgAA Genes
A total of 127 AA-family genes (named CgAA1–CgAA127) were detected in the genome of C. graminicola TZ-3. Table 1 lists their detailed biochemical characteristics. The proteins encoded by these genes vary in size, with CgAA76 being the smallest (21.85 kDa) and CgAA106 being the largest (151.92 kDa). These proteins include 90 acidic proteins (PI value < 6.5), 25 basic proteins (PI value > 7.5), and 12 neutral proteins (6.5 < PI value < 7.5). All CgAA proteins belong to 12 different superfamilies, among which 34 CgAA proteins are classified into the AA7 superfamily, 30 into the AA3 superfamily, and 28 into the AA9 superfamily. The grand average of hydropathicity (GRAVY) range of CgAAs is from −0.625 to 0.201, with only seven CgAA proteins having a GRAVY greater than 0, indicating that most CgAA proteins are hydrophilic. Additionally, we predicted the subcellular localization of CgAA proteins and found that 88 CgAA proteins are localized in the extracellular space, 23 in the cytoplasm, 11 in mitochondria, 4 in the plasma membrane, and 1 in the nucleus (Table 1), suggesting that most CgAA proteins exert their biological functions extracellularly.

Table 1.
Nomenclature and characteristics of the putative auxiliary activity enzymes (AAs) in Colletotrichum graminicola.
3.2. Phylogenetic Analysis
To understand the phylogenetic relationships among CgAA-family proteins, we constructed a phylogenetic tree using the maximum likelihood (ML) method with 127 CgAA protein sequences (Table S1). The phylogenetic tree clusters into six groups, namely I, II, III, IV, V, and VI, with 27, 23, 26, 27, 12, and 12 members, respectively (Figure 1). Groups I and II are phylogenetically close, indicating a relatively tight homologous relationship between these two groups. The distance between Groups I and VI is relatively far, suggesting a distant genetic relationship between them.
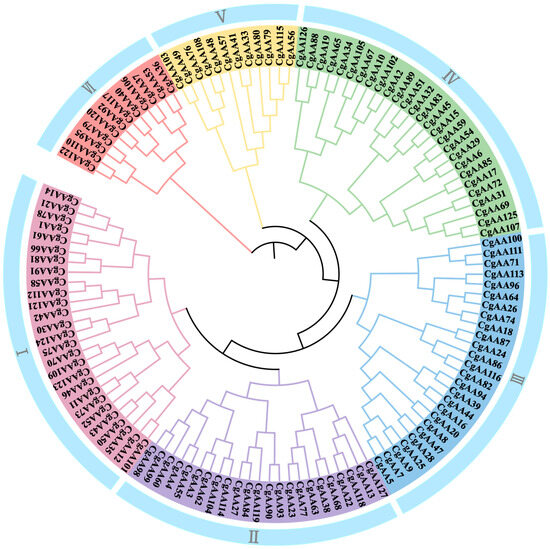
Figure 1.
Phylogenetic analysis of AA proteins from Colletotrichum graminicola. Note: “Cg” represents C. graminicola. The tree reveals that all CgAA proteins can be segregated into six distinct groups (I–VI).
3.3. Sequence and Structural Analysis
To further elucidate the characteristics of the AA-family genes in C. graminicola TZ-3, we analyzed the gene structures of CgAAs and the conserved domains of their encoded protein sequences (Figure 2, Table S1). As shown in Figure 2A, the 127 CgAAs contain zero to nine introns, with the number of introns varying among genes. Among them, CgAA109 has the most introns (nine), while 19 CgAAs have no introns. This indicates that CgAA genes may have experienced intron loss or gain events during evolution. The conserved domain analysis of CgAA protein sequences revealed that CgAAs harbor rich domains, with the BetA superfamily domain being the most abundant (34), followed by the GlcD superfamily (28) and LPMO_auxiliary superfamily (21) (Figure 2B).
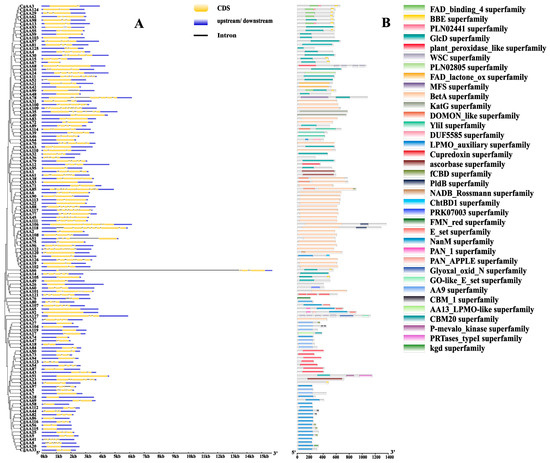
Figure 2.
Analysis of gene structures and protein domains of CgAAs. Note: (A) Exon–intron organization of CgAA genes. Untranslated regions (UTRs) are depicted by blue boxes, exons by yellow boxes, and introns by black lines. (B) Domain architecture of CgAA proteins. Distinct color rectangles designate the respective superfamily assignments.
To deepen our understanding of the functions of CgAA genes, we used the MEME website to predict conserved motifs in CgAA sequences, identifying a total of 10 motifs named motifs 1–10 (Figure 3 and Figure S1). Among them, motif 9 is the most common, present in 33 CgAA proteins. Motifs 1 and 4 are present in 32 CgAA proteins, motif 6 in 31, motif 5 in 28, motifs 2 and 3 in 27, motif 7 in 25, motif 8 in 23, and motif 10 in 12. Twenty-one CgAA proteins contain motifs 1, 2, 4, and 8 simultaneously, while 12 CgAA proteins contain motifs 5, 7, 9, and 10 simultaneously. These motifs play crucial roles in the biological functions of these proteins, and proteins with the same conserved motifs may share similar functions.

Figure 3.
Conserved motif architecture of CgAAs predicted by MEME analysis. Note: Distinctively colored boxes represent diverse conserved motifs varying in length and sequence composition.
3.4. Cis-Acting Regulatory Element (CARE) Analysis
Analyzing CAREs is crucial for understanding gene regulation [34]. We used the PlantCARE online server to examine the 2000 bp upstream sequences of CgAA genes (Table S1) [30]. The results revealed that the promoter regions of CgAA genes harbor many cis-regulatory elements related to development, pathogenicity, and stress responses (Figure 4, Table S2). Further analysis showed that the promoter regions of CgAA genes contain varying numbers of cis-acting elements, including low-temperature response elements (LTR), drought response elements (MBS), and defense response elements (TC-rich repeats). Hormone-related response elements include MeJA response elements (TGACG-motif and CGTCA-motif), salicylic acid response elements (TCA-element), abscisic acid response elements (ABRE), gibberellin response elements (P-box, GARE-motif, and TATC-box), and auxin response elements (TGA-element and AuxRR-core). Additionally, CgAA genes possess CAREs associated with light response (G-Box, Sp1, TCT-motif, GATA-motif, and GT1-motif), zein metabolic regulation (O2-site), anoxic specific inducibility (GC-motif), meristem expression (CAT-box), enhancer-binding protein (CCAAT-box), and seed-specific regulation (RY-element). Among them, MeJA response elements (1064) are the most abundant, followed by light response elements (934) and abscisic acid response elements (486) (Figure S2, Table S2). These findings suggest that CgAAs may play important roles in fungal growth, pathogenicity, and responses to various stresses, providing valuable information for understanding the complex regulatory networks constructed by CgAA genes in different developmental stages, during pathogenic processes, and under multifactorial stress conditions.
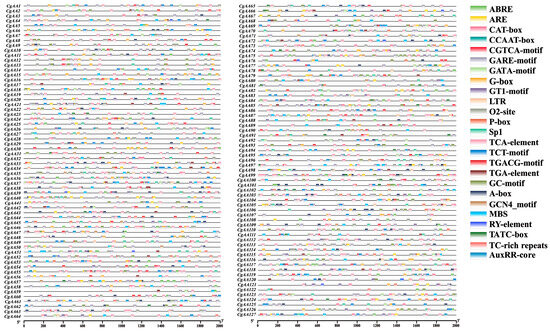
Figure 4.
Characterization of cis-acting regulatory elements (CAREs) within the 2 kb promoter regions of CgAA genes. Color-coded blocks indicate the distribution and positional arrangement of functionally distinct CAREs.
3.5. Gene Ontology (GO) Enrichment Analysis
GO enrichment analysis was performed by comparing protein sequences with known functions of species proteins to analyze the functions of these genes. We conducted GO enrichment analysis on 127 CgAA-family genes. The results showed that CgAA genes are mainly enriched in multiple GO terms, such as oxidation–reduction processes (GO:0055114), single-organism metabolic processes (GO:0044710), cellular oxidation detoxification (GO:0098869), and cellular detoxification (GO:1990748) (Figure 5, Table S3). These results indicate that CgAA genes primarily function in oxidation–reduction processes and cellular detoxification.
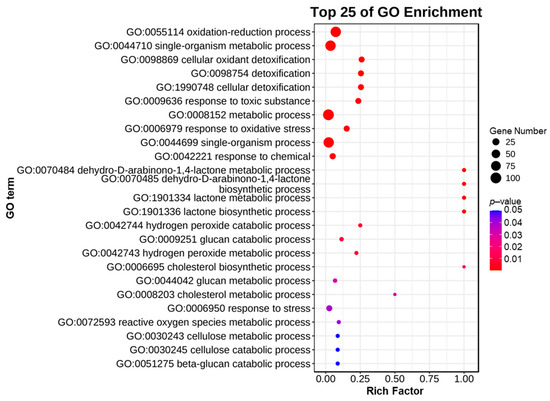
Figure 5.
Gene ontology enrichment analysis of CgAA genes.
3.6. Response of CgAA-Family Genes During C. graminicola Infection
To analyze CgAA-family genes associated with C. graminicola infection, we utilized transcriptome data to examine the expression of 127 CgAA genes during C. graminicola infection of maize. A heatmap was generated based on the FPKM values of 127 AA-family genes, showing the expression levels of CgAAs at different time points (24 h, 36 h, 60 h) after C. graminicola inoculation (Figure 6). Based on the expression profiles of 127 CgAA-family genes, they can be clustered into seven categories. These CgAA-family genes exhibit significant changes in expression levels during pathogen infection, suggesting their important roles in the process of C. graminicola infection. For example, group II contains 39 CgAA-family genes, and group IV contains 19 CgAA-family genes, both of which have higher transcription levels at 60 h post-inoculation with C. graminicola. Group VI contains 45 CgAA-family genes, which have higher transcription levels at 24 h post-inoculation. Additionally, we validated the transcriptome data by performing RT-qPCR on eight selected CgAA genes (Table S4), confirming the accuracy of the transcriptome data (Figure 7). It should be noted that we selected these representative genes to further validate the significant changes in CgAA genes during the stages of pathogen infection and lesion expansion. Among them, CgAA21, CgAA109, and CgAA118 are significantly upregulated at 24 h after infection, while CgAA46, CgAA85, CgAA89, CgAA115 and CgAA126 are significantly upregulated at 60 h after infection.
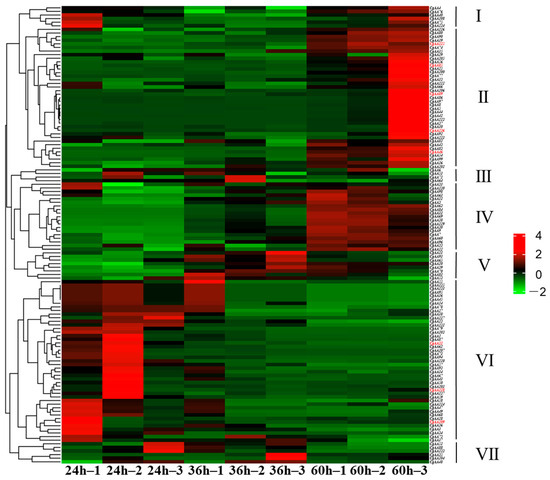
Figure 6.
Transcript abundance profiles of CgAAs derived from RNA sequencing. Color gradients reflect FPKM values, with red and green hues indicating high and low expression levels, respectively. Genes validated through RT-qPCR are distinguished by red font.
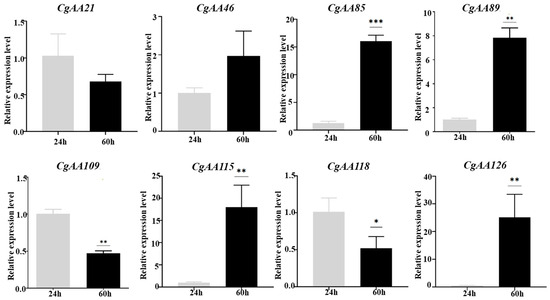
Figure 7.
Validation of CgAA transcript profiles via RT-qPCR. Histograms depict mean expression levels (±standard error) from three technical replicates. Statistical significance was determined by Student’s t-test (n = 3; * p < 0.05; ** p < 0.01; *** p < 0.001).
4. Discussion
AAs, as an important class of the CAZy database, primarily collaborate with GHs to degrade complex polysaccharides such as lignocellulose through oxidation–reduction reactions, exhibiting significant potential in biodegradation, bioenergy, and disease control [1,2,8]. Belonging to the redox enzyme category, AA-family enzymes disrupt the aromatic polymer structures in lignocellulose through oxidation reactions, providing sites of action for GHs and thus synergistically promoting polysaccharide degradation [35]. AAs secreted by pathogenic fungi participate in the degradation of plant cell walls, and their functions during infection require further investigation [1,2,8]. To explore the diversity of AAs in nature and improve enzyme preparations, it is necessary to further study AA families present in various pathogenic fungi, thereby providing a basis for identifying new targets for disease control.
In this study, we identified 127 AA-family genes in C. graminicola TZ-3, which encode proteins of diverse sizes, and most CgAA proteins categorized into the lignin-degrading enzyme family. Phylogenetic analysis classified CgAA proteins into different groups, with genetically closely related CgAA proteins located close together on the phylogenetic tree. Subcellular localization analysis revealed that CgAA proteins are widely distributed in five subcellular structures: plasma membrane, cytoplasm, nucleus, mitochondria, and extracellular space. This distribution suggests their potential diverse functions. Notably, most CgAA proteins are localized in the extracellular space, indicating they may be secreted to the outer side of the cell membrane to exert specific functions extracellularly. These extracellular secreted proteins may play a central role in fungal pathogenesis by degrading cell wall components such as cellulose, hemicellulose, and pectin, causing the cell wall structure to disintegrate and creating conditions for pathogen invasion and colonization [36].
Introns play crucial regulatory roles in gene function during evolution, and genes with fewer introns may respond faster to stress factors such as environmental pressures [37,38]. The distribution of introns in microbial genes often exhibits a discrete pattern [39]. In this study, the number of introns varied greatly among the 127 CgAAs, with CgAA109 having 9 introns and 19 CgAAs having no introns, suggesting that CgAA genes might have undergone intron gain or loss events during evolution. Some CgAA proteins share the same conserved motifs, indicating that these proteins may have similar functions. Additionally, CgAAs possess abundant superfamily domains, combined with the diverse subcellular localization of CgAA proteins, suggesting that CgAAs may have diverse functions and can intervene in fungal biological processes in multiple ways. These rich structural domains also ensure that they can degrade crystalline polysaccharides in lignocellulose through oxidative degradation, providing a carbon source for fungi and disrupting plant cell wall structure [1,9,10].
The presence of CAREs in promoter regions can significantly influence gene function and regulation [40,41]. Studies have shown that many CAREs play crucial roles in multiple gene families [42,43]. CAREs related to development, pathogenicity, and stress responses were found in the promoter regions of CgAAs. Stress response elements include low-temperature response elements, drought response elements, and defense response elements. Hormone response elements include MeJA response elements, salicylic acid response elements, abscisic acid response elements, gibberellin response elements, and auxin response elements. Furthermore, CgAA genes possess light response elements, zein metabolic regulation response elements, meristem expression response elements, etc. CAREs are key DNA sequences that regulate the expression of AA-family genes, and their optimization can improve enzyme production efficiency [40]. These abundant CAREs in the study provide valuable information for understanding the gene functions of CgAAs. In-depth exploration of CAREs is essential for uncovering functional genes related to fungal developmental processes, adaptation mechanisms, and pathogenic processes.
The GO analysis of CgAAs revealed that they primarily function in oxidation–reduction processes and cellular detoxification. Gene functions are often associated with their expression characteristics [44,45,46]. For example, researchers identified genes related to sexual development and virulence by examining infection-specific transcriptional patterns in the maize pathogen Cochliobolus heterotrophus [47]. CgAA-family genes exhibit significant differences in expression during C. graminicola infection of maize, with some CgAA genes having higher transcription levels at 24 h post-infection, while others have higher transcription levels at 60 h. They may play roles at different stages of C. graminicola infection. These CgAA genes participate in host–microbe interactions and are closely related to the pathogenic mechanisms of pathogens, potentially playing important roles in the establishment and expansion of fungal infections. To elucidate the precise roles of these genes in fungal pathogenesis, further functional characterization of these genes is required. These genes can serve as candidate targets for the development of RNAi-based biopesticides and provide scientific evidence for the control of maize diseases caused by C. graminicola.
In recent years, the interaction between C. graminicola and maize has been a research focus, with multiple studies providing insights into the pathogenic mechanisms of C. graminicola [23,48]. Our study provides comprehensive information on CgAAs, which will help ultimately reveal the interaction network between C. graminicola and maize. Through genome-wide analysis, we have initially characterized these CgAAs. However, these results are primarily based on bioinformatics analyses, and extensive research endeavors are imperative to unravel the functions and molecular mechanisms associated with CgAA genes. In addition, AA genes play a pivotal role in sustainable agriculture due to their ability to confer resistance against both biotic and abiotic stresses. Through genetic engineering approaches, scientists can enhance or incorporate specific AA genes to improve crop stress tolerance, thereby reducing reliance on chemical pesticides. Progress in RNA interference biopesticides and genetic engineering methodologies has established the technical foundation for AA gene utilization in sustainable agricultural systems. This study provides conditions for future researchers to further explore the new uses of AA-family enzymes in disease prevention and control. It should be noted that some AA proteins may interfere with the host’s immune response, and their safety as biopesticides needs to be evaluated in the future.
5. Conclusions
This study conducted a comprehensive and systematic bioinformatics analysis of the CgAA gene family, elucidating their physicochemical properties and potential biological functions. We successfully identified 127 AA genes in the genome of C. graminicola TZ-3 and clarified the expression patterns of CgAA genes during C. graminicola infection of maize using transcriptome data and quantitative PCR. CgAA genes were closely related to the pathogenic mechanisms of pathogens and might play important roles in the initiation and expansion of fungal infections. These analysis of CgAA genes also revealed potential targets for fungicide development and maize breeding for enhanced resistance.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/microorganisms13092080/s1, Table S1. CgAA genomic, CDS, protein, and promoter sequences. Table S2. Cis-acting regulatory elements are present in the CgAA gene promoter regions. Table S3. Gene ontology enrichment analysis of CgAA genes. Table S4. The primers used in this study. Figure S1. Sequence logo conserved motif of CgAA proteins. Figure S2. The numbers of predicted CAREs in the CgAA promoter regions.
Author Contributions
Y.W. and Y.G. designed and directed the research. Y.W., J.C., D.Z., Y.C., J.L., H.L., M.L. and Y.Z. performed the experiments and wrote the original draft. Y.W., Y.C. and Y.G. revised and polished the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (No. 32102159), Science and Technology Key Project of Henan Province (No. 252102111100), Natural Science Foundation of Henan Province (No. 242300420158), and High-Level Talents of the Henan Agricultural University (No. 30501380).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The contributions presented in the study are included in the article/Supplementary Materials, and further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Kubicek, C.P.; Starr, T.L.; Glass, N.L. Plant cell wall–degrading enzymes and their secretion in plant-pathogenic fungi. Annu. Rev. Phytopathol. 2014, 52, 427–451. [Google Scholar] [CrossRef]
- Lombard, V.; Ramulu, H.G.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2013, 42, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Kang, Z.; Zingen-Sell, I.; Buchenauer, H. Infection of wheat spikes by Fusarium avenaceum and alterations of cell wall components in the infected tissue. Eur. J. Plant Pathol. 2005, 111, 19–28. [Google Scholar] [CrossRef]
- Van Vu, B.; Itoh, K.; Nguyen, Q.B.; Tosa, Y.; Nakayashiki, H. Cellulases belonging to glycoside hydrolase families 6 and 7 contribute to the virulence of Magnaporthe oryzae. Mol. Plant Microbe Interact. 2012, 25, 1135–1141. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.X.; Li, B.H.; Zhou, S.Y. A novel glycoside hydrolase 74 xyloglucanase cvgh74a is a virulence factor in Coniella vitis. J. Integr. Agric. 2020, 19, 2725–2735. [Google Scholar] [CrossRef]
- Tan, X.; Hu, Y.; Jia, Y.; Hou, X.; Xu, Q.; Han, C.; Wang, Q. A conserved glycoside hydrolase family 7 cellobiohydrolase PsGH7a of Phytophthora sojae is required for full virulence on soybean. Front. Microbiol. 2020, 11, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Bissaro, B.; Streit, B.; Isaksen, I.; Eijsink, V.G.H.; Beckham, G.T.; DuBois, J.L.; Røhr, A.K. Molecular mechanism of the chitinolytic peroxygenase reaction. Proc. Natl. Acad. Sci. USA 2020, 117, 1504–1513. [Google Scholar] [CrossRef]
- Chen, J.; Guo, X.; Zhu, M.; Chen, C.; Li, D. Polysaccharide monooxygenase-catalyzed oxidation of cellulose to glucuronic acid-containing cello-oligosaccharides. Biotechnol. Biofuels 2019, 12, 42–58. [Google Scholar] [CrossRef]
- Polonio, Á.; Fernández-Ortuño, D.; de Vicente, A.; Pérez-García, A. A haustorial-expressed lytic polysaccharide monooxygenase from the cucurbit powdery mildew pathogen Podosphaera xanthii contributes to the suppression of chitin-triggered immunity. Mol. Plant Pathol. 2021, 22, 580–601. [Google Scholar] [CrossRef]
- Sabbadin, F.; Urresti, S.; Henrissat, B.; Avrova, A.O.; Welsh, L.R.J.; Lindley, P.J.; Csukai, M.; Squires, J.N.; Walton, P.H.; Davies, G.J.; et al. Secreted pectin monooxygenases drive plant infection by pathogenic oomycetes. Science 2021, 373, 774–779. [Google Scholar] [CrossRef]
- Li, Y.; Liu, X.; Liu, M.; Wang, Y.; Zou, Y.; You, Y.; Yang, L.; Hu, J.; Zhang, H.; Zheng, X.; et al. Magnaporthe oryzae auxiliary activity protein MoAa91 functions as chitin–binding protein to induce appressorium formation on artificial inductive surfaces and suppress plant immunity. mBio 2020, 11, e03304-19. [Google Scholar] [CrossRef] [PubMed]
- Zarattini, M.; Corso, M.; Kadowaki, M.A.; Monclaro, A.; Magri, S.; Milanese, I.; Jolivet, S.; de Godoy, M.O.; Hermans, C.; Fagard, M.; et al. LPMO-oxidized cellulose oligosaccharides evoke immunity in Arabidopsis conferring resistance towards necrotrophic fungus B. cinerea. Commun. Biol. 2021, 4, 727–739. [Google Scholar] [CrossRef]
- Vandhana, T.M.; Reyre, J.L.; Sushmaa, D.; Berrin, J.G.; Bissaro, B.; Madhuprakash, J. On the expansion of biological functions of lytic polysaccharide monooxygenases. New Phytol. 2022, 233, 2380–2396. [Google Scholar] [CrossRef] [PubMed]
- Yue, H.; Jiang, J.; Taylor, A.J.; Leite, A.L.; Dodds, E.D.; Du, L. Outer membrane vesicle-mediated codelivery of the antifungal HSAF metabolites and lytic polysaccharide monooxygenase in the predatory lysobacter enzymogenes. ACS Chem. Biol. 2021, 16, 1079–1089. [Google Scholar] [CrossRef]
- Zhang, F.; Anasontzis, G.E.; Labourel, A.; Champion, C.; Haon, M.; Kemppainen, M.; Commun, C.; Deveau, A.; Pardo, A.; Veneault-Fourrey, C.; et al. The ectomycorrhizal basidiomycete Laccaria bicolor releases a secreted β-1,4 endoglucanase that plays a key role in symbiosis development. New Phytol. 2018, 220, 1309–1321. [Google Scholar] [CrossRef]
- Green, K.A.; Becker, Y.; Tanaka, A.; Takemoto, D.; Fitzsimons, H.L.; Seiler, S.; Lalucque, H.; Silar, P.; Scott, B. SymB and SymC, two membrane associated proteins, are required for Epichloë festucae hyphal cell-cell fusion and maintenance of a mutualistic interaction with Lolium perenne. Mol. Microbiol. 2017, 103, 657–677. [Google Scholar] [CrossRef]
- Balmer, D.; de Papajewski, D.V.; Planchamp, C.; Glauser, G.; Mauch-Mani, B. Induced resistance in maize is based on organ-specific defence responses. Plant J. 2013, 74, 213–225. [Google Scholar] [CrossRef]
- Miranda, V.J.; Porto, W.F.; Fernandes, G.D.R.; Pogue, R.; Nolasco, D.O.; Araujo, A.C.G.; Cota, L.V.; Freitas, C.G.; Dias, S.C.; Franco, O.L.; et al. Comparative transcriptomic analysis indicates genes associated with local and systemic resistance to Colletotrichum graminicola in maize. Sci. Rep. 2017, 7, 2483–2496. [Google Scholar] [CrossRef] [PubMed]
- Duan, C.X.; Guo, C.; Yang, Z.H.; Sun, S.L.; Zhu, Z.D.; Wang, X.M. First report of Anthracnose leaf blight of maize caused by Colletotrichum graminicola in China. Plant Dis. 2019, 103, 1770. [Google Scholar] [CrossRef]
- O’Connell, R.J.; Thon, M.R.; Hacquard, S.; Amyotte, S.G.; Kleemann, J.; Torres, M.F.; Damm, U.; Buiate, E.A.; Epstein, L.; Alkan, N.; et al. Lifestyle transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses. Nat. Genet. 2012, 44, 1060–1065. [Google Scholar] [CrossRef]
- Jiao, C.; Chen, L.; Sun, C.; Jiang, Y.; Zhai, L.; Liu, H.; Shen, Z. Evaluating national ecological risk of agricultural pesticides from 2004 to 2017 in China. Environ. Pollut. 2020, 259, 113778–113786. [Google Scholar] [CrossRef]
- Gong, A.; Jing, Z.; Zhang, K.; Tan, Q.; Wang, G.; Liu, W. Bioinformatic analysis and functional characterization of the CFEM proteins in maize anthracnose fungus Colletotrichum graminicola. J. Integr. Agric. 2020, 19, 541–550. [Google Scholar] [CrossRef]
- Mei, J.; Li, Z.; Zhou, S.; Chen, X.; Wilson, R.; Liu, W. Effector secretion and stability in the maize anthracnose pathogen Colletotrichum graminicola requires N-linked protein glycosylation and the ER chaperone pathway. New Phytol. 2023, 240, 1449–1466. [Google Scholar] [CrossRef] [PubMed]
- Eisermann, I.; Weihmann, F.; Krijger, J.J.; Kröling, C.; Hause, G.; Menzel, M.; Pienkny, S.; Kiesow, A.; Deising, H.B.; Wirsel, S.G.R. Two genes in a pathogenicity gene cluster encoding secreted proteins are required for appressorial penetration and infection of the maize anthracnose fungus Colletotrichum graminicola. Environ. Microbiol. 2019, 21, 4773–4791. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Xia, X.; Mei, J.; Gong, Z.; Zhang, J.; Xiao, Y.; Duan, C.; Liu, W. Genome sequence resource of a Colletotrichum graminicola field strain from China. Mol. Plant Microbe Interact. 2023, 36, 447–451. [Google Scholar] [CrossRef]
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 1999, 112, 531–552. [Google Scholar]
- Subramanian, B.; Gao, S.; Lercher, M.J.; Hu, S.; Chen, W.H. Evolview v3: A webserver for visualization, annotation, and management of phylogenetic trees. Nucleic Acids Res. 2019, 47, W270–W275. [Google Scholar] [CrossRef]
- Hu, B.; Jin, J.; Guo, A.Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y.; et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef]
- Mu, H.; Chen, J.; Huang, W.; Huang, G.; Deng, M.; Hong, S.; Ai, P.; Gao, C.; Zhou, H. OmicShare tools: A zero-code interactive online platform for biological data analysis and visualization. Imeta 2024, 3, e228. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, J.; Dong, D.; Lou, C.; Zhang, Y.; Wang, Y.; Yu, B.; Wang, F.; Kang, G. Comparative analysis of TaPHT1;9 function using CRISPR-edited mutants, ectopic transgenic plants and their wild types under soil conditions. Plant Soil 2025, 509, 249–260. [Google Scholar] [CrossRef]
- Higo, K.; Ugawa, Y.; Iwamoto, M.; Korenaga, T. Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Res. 1999, 27, 297–300. [Google Scholar] [CrossRef]
- Chirania, P.; Holwerda, E.K.; Giannone, R.J.; Liang, X.; Poudel, S.; Ellis, J.C.; Bomble, Y.J.; Hettich, R.L.; Lynd, L.R. Metaproteomics reveals enzymatic strategies deployed by anaerobic microbiomes to maintain lignocellulose deconstruction at high solids. Nat. Commun. 2022, 13, 3870–3882. [Google Scholar] [CrossRef]
- Wei, W.; Xu, L.; Peng, H.; Zhu, W.; Tanaka, K.; Cheng, J.; Sanguinet, K.A.; Vandemark, G.; Chen, W. A fungal extracellular effector inactivates plant polygalacturonase-inhibiting protein. Nat. Commun. 2022, 13, 2213–2227. [Google Scholar] [CrossRef]
- Roy, S.W.; Penny, D. Patterns of intron loss and gain in plants: Intron loss–dominated evolution and genome-wide comparison of O. sativa and A. thaliana. Mol. Biol. Evol. 2007, 24, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.W.; Gilbert, W. The evolution of spliceosomal introns: Patterns, puzzles and progress. Nat. Rev. Genet. 2006, 7, 211–221. [Google Scholar] [CrossRef]
- Xu, G.; Guo, C.; Shan, H.; Kong, H. Divergence of duplicate genes in exon–Intron structure. Proc. Natl. Acad. Sci. USA 2012, 109, 1187–1192. [Google Scholar] [CrossRef]
- Hernandez-Garcia, C.M.; Finer, J.J. Identification and validation of promoters and cis-acting regulatory elements. Plant Sci. 2014, 217–218, 109–119. [Google Scholar] [CrossRef]
- Xuan, C.; Feng, M.; Li, X.; Hou, Y.; Wei, C.; Zhang, X. Genome-wide identification and expression analysis of chitinase genes in watermelon under abiotic stimuli and Fusarium oxysporum infection. Int. J. Mol. Sci. 2024, 25, 638–658. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, Q.; Chen, X.; Li, H.; Chang, J.; Zhang, Y.; Wang, Y.; Shi, Y. Genome-wide Identification and analysis of carbohydrate-binding modules in Colletotrichum graminicola. Int. J. Mol. Sci. 2025, 26, 919–933. [Google Scholar] [CrossRef]
- Li, L.; Tang, J.; Wu, A.; Fan, C.; Li, H. Genome-wide identification and functional analysis of the GUX gene family in Eucalyptus grandis. Int. J. Mol. Sci. 2024, 25, 8199–8214. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, H.; Yu, D. Arabidopsis WRKY transcription factors WRKY12 and WRKY13 oppositely regulate flowering under Short-Day conditions. Mol. Plant 2016, 9, 1492–1503. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Liu, Z.; Wang, L.; Kim, S.G.; Seo, P.J.; Qiao, M.; Wang, N.; Li, S.; Cao, X.; Park, C.M.; et al. WRKY71 accelerates flowering via the direct activation of FLOWERING LOCUS T and LEAFY in Arabidopsis thaliana. Plant J. 2016, 85, 96–106. [Google Scholar] [CrossRef]
- Zhang, C.Q.; Xu, Y.; Lu, Y.; Yu, H.X.; Gu, M.H.; Liu, Q.Q. The WRKY transcription factor OsWRKY78 regulates stem elongation and seed development in rice. Planta 2011, 234, 541–554. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Zhang, J.; Fan, J.; Jia, W.; Lv, Y.; Pan, H.; Zhang, X. Infection-specific transcriptional patterns of the maize pathogen Cochliobolus heterostrophus unravel genes involved in asexual development and virulence. Mol. Plant Pathol. 2024, 25, e13413. [Google Scholar] [CrossRef]
- Sanz-Martín, J.M.; Pacheco-Arjona, J.R.; Bello-Rico, V.; Vargas, W.A.; Monod, M.; Díaz-Mínguez, J.M.; Thon, M.R.; Sukno, S.A. A highly conserved metalloprotease effector enhances virulence in the maize anthracnose fungus Colletotrichum graminicola. Mol. Plant Pathol. 2016, 17, 1048–1062. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).