
Modulating the Plant Microbiome: Effects of Seed Inoculation with Endophytic Bacteria on Microbial Diversity and Growth Enhancement in Pea Plants



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Isolation and Selection of Endophytic Bacteria
2.2. Seed Inoculation with Bacterial Strains
2.3. Soil Chemical Analysis
2.4. DNA Extraction from Pea Plants
2.5. Microbial Community Analysis
2.5.1. High-Throughput Sequencing
2.5.2. Alpha Diversity Analysis
2.5.3. Beta Diversity Analysis
2.5.4. Taxonomic Annotation
2.6. Microbial Functional Profiling
2.7. Microbial Community Network Analysis
2.8. Statistical Analysis
2.9. Data Availability
3. Results
3.1. Soil Chemical Properties
3.2. Impact of Early Inoculation on Microbial Community and Composition
3.2.1. Sequencing Quality and OTU Diversity
- Shared and Unique Microbial Communities Across Treatments
3.3. Impact of Bacteria Inoculation (AR11 and AR32) on Bacterial Diversity and Richness in Pea Plants
3.3.1. Alpha Diversity Results
3.3.2. Impact of Inoculation on Microbial Richness and Community Structure
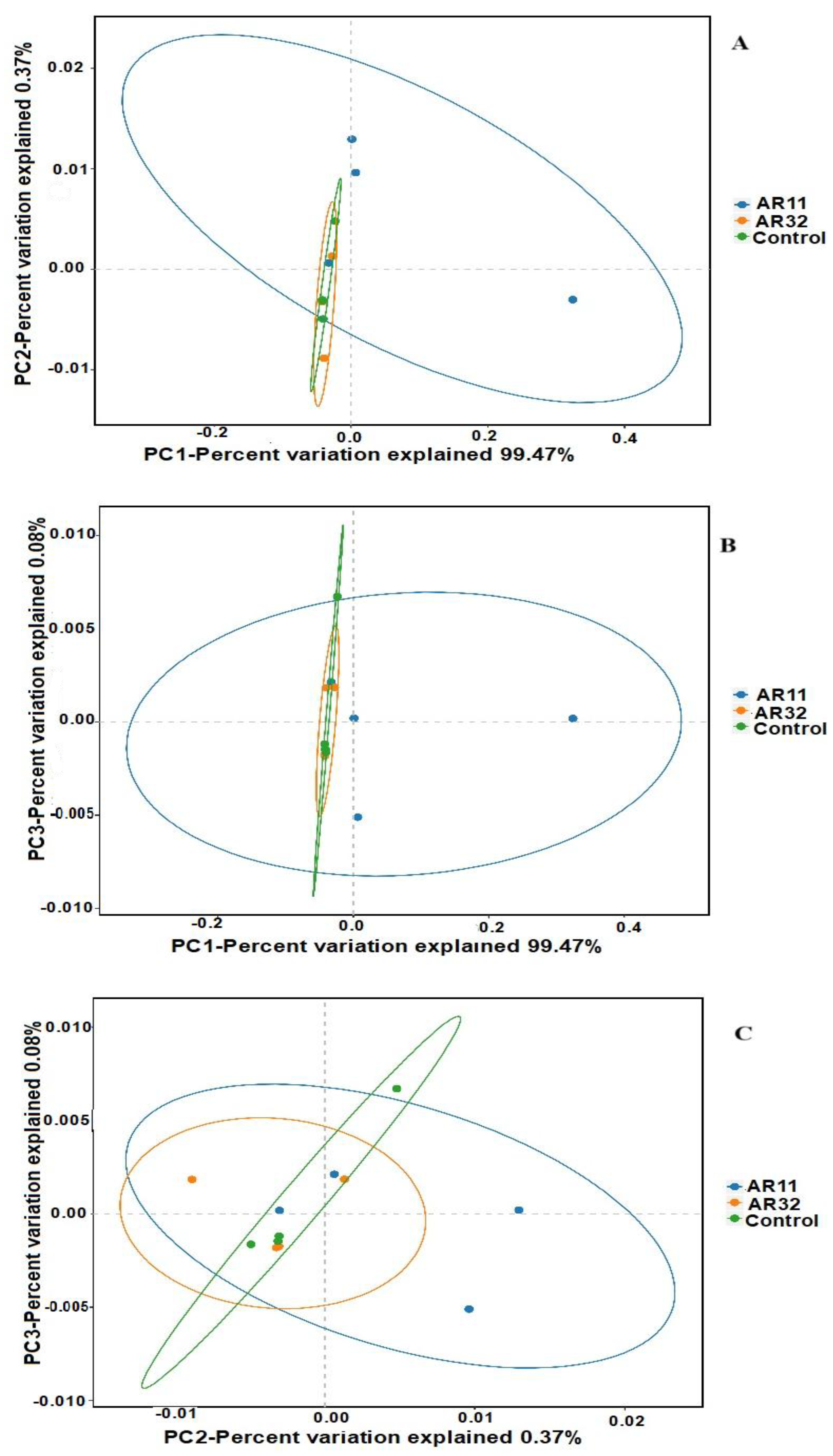
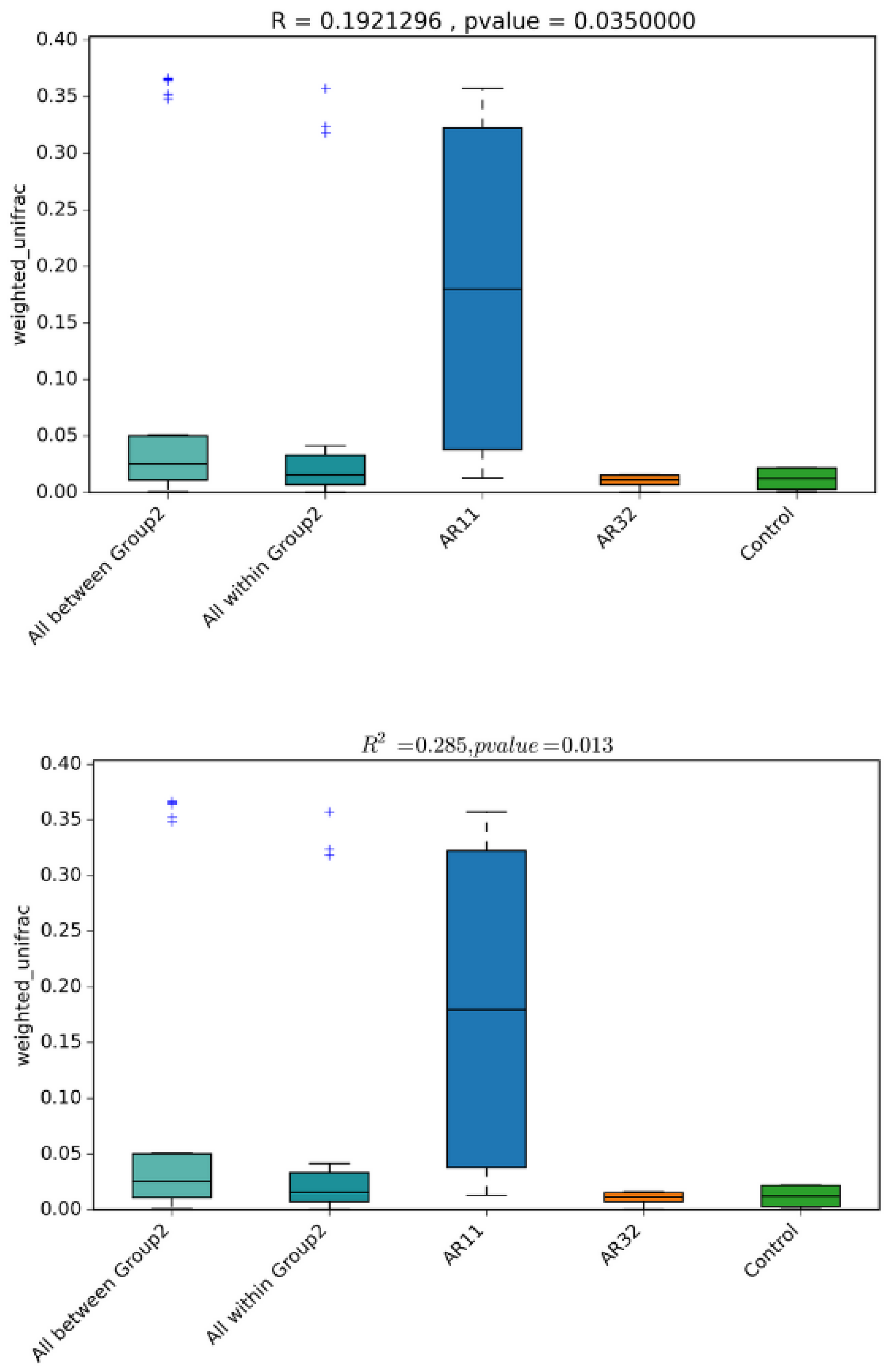
3.3.3. Beta Diversity Results
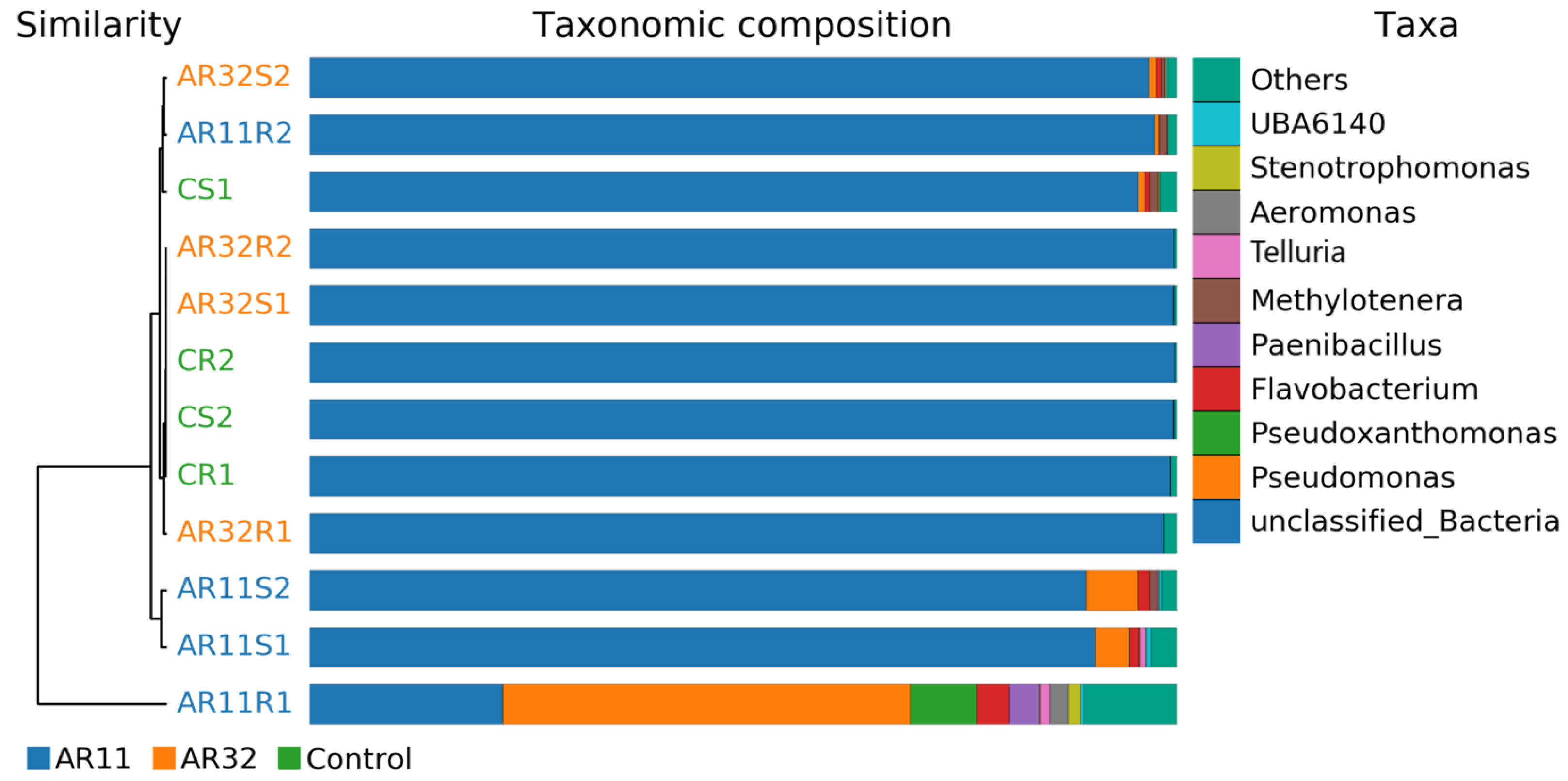
3.4. Taxonimic Anotation
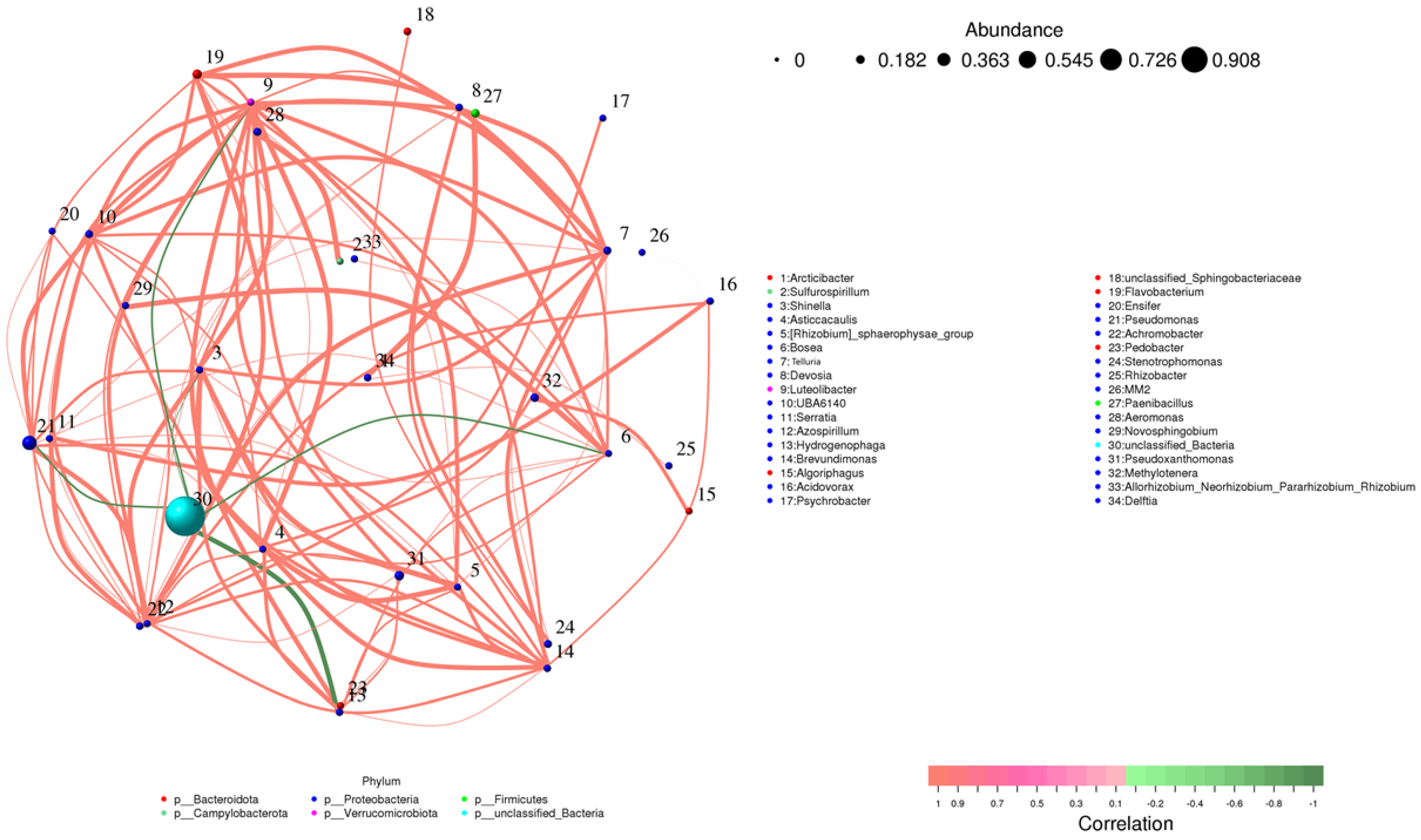
3.5. Correlation Network of Microbial Communities
3.6. Bacterial Community Composition at the Phylum Level Across Treatments
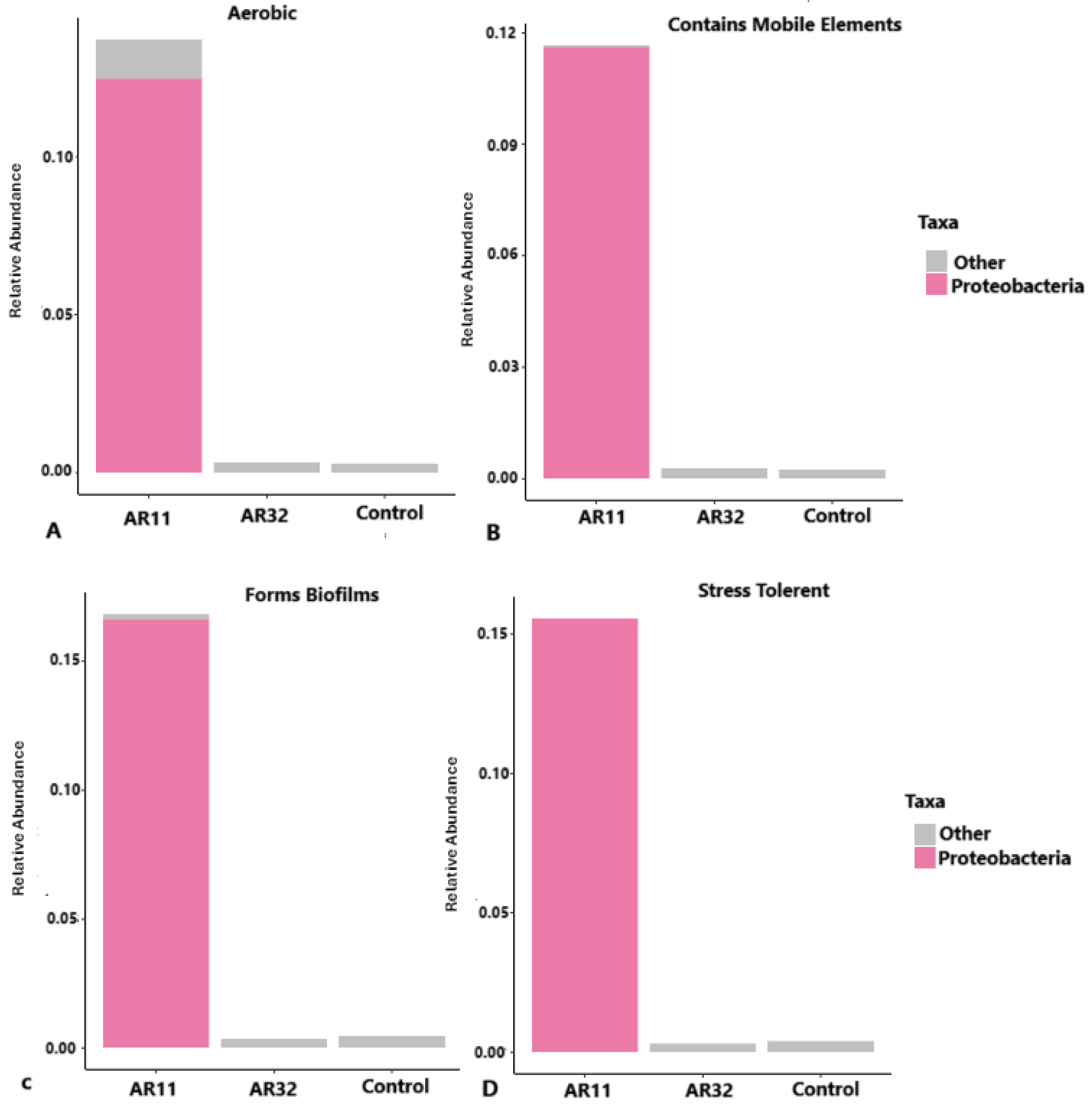
3.7. Functional Profiles of Bacterial Communities Across Treatments
4. Discussion
4.1. Enhancing Microbial Diversity with Bacillus Strains
4.2. Shifts in Beta Diversity and Microbial Community Composition
4.3. Functional Contributions of the Microbial Community
4.4. Practical Applications and Future Research
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, X.; He, S.-W.; He, Q.; Ju, Z.-C.; Ma, Y.-N.; Wang, Z.; Han, J.-C.; Zhang, X.-X. Early Inoculation of an Endophyte Alters the Assembly of Bacterial Communities across Rice Plant Growth Stages. Microbiol. Spectr. 2023, 11, e0497822. [Google Scholar] [CrossRef] [PubMed]
- Chialva, M.; Lanfranco, L.; Bonfante, P. The Plant Microbiota: Composition, Functions, and Engineering. Curr. Opin. Biotechnol. 2022, 73, 135–142. [Google Scholar] [CrossRef]
- Dwibedi, V.; Rath, S.K.; Joshi, M.; Kaur, R.; Kaur, G.; Singh, D.; Kaur, G.; Kaur, S. Microbial Endophytes: Application towards Sustainable Agriculture and Food Security. Appl. Microbiol. Biotechnol. 2022, 106, 5359–5384. [Google Scholar] [CrossRef]
- Santoyo, G.; Moreno-Hagelsieb, G.; del Carmen Orozco-Mosqueda, M.; Glick, B.R. Plant Growth-Promoting Bacterial Endophytes. Microbiol. Res. 2016, 183, 92–99. [Google Scholar] [CrossRef]
- Ali, M.; Ali, Q.; Sohail, M.A.; Ashraf, M.F.; Saleem, M.H.; Hussain, S.; Zhou, L. Diversity and Taxonomic Distribution of Endophytic Bacterial Community in the Rice Plant and Its Prospective. Int. J. Mol. Sci. 2021, 22, 10165. [Google Scholar] [CrossRef]
- Hadian, S.; Smith, D.L.; Kopriva, S.; Norkevičienė, E.; Supronienė, S. Exploring Endophytic Bacteria from Artemisia Spp. and Beneficial Traits on Pea Plants. Plants 2024, 13, 1684. [Google Scholar] [CrossRef] [PubMed]
- Hadian, S.; Supronienė, S. The Potential of Artemisia Spp. Plant Extracts and Endophytic Bacteria to Increase Plant Productivity: A Review. Zemdirb.-Agric. 2023, 110, 87–94. [Google Scholar] [CrossRef]
- Szparaga, A.; Kocira, S.; Kapusta, I.; Zaguła, G. Prototyping Extracts from Artemisia absinthium L. for Their Biostimulating Properties Yield-Enhancing, and Farmer Income-Increasing Properties. Ind. Crops Prod. 2021, 160, 113125. [Google Scholar] [CrossRef]
- Angulo, V.; Beriot, N.; Garcia-Hernandez, E.; Li, E.; Masteling, R.; Lau, J.A. Plant–Microbe Eco-evolutionary Dynamics in a Changing World. New Phytol. 2022, 234, 1919–1928. [Google Scholar] [CrossRef]
- Berg, G.; Smalla, K. Plant Species and Soil Type Cooperatively Shape the Structure and Function of Microbial Communities in the Rhizosphere. FEMS Microbiol. Ecol. 2009, 68, 1–13. [Google Scholar] [CrossRef]
- Kandel, S.; Joubert, P.; Doty, S. Bacterial Endophyte Colonization and Distribution within Plants. Microorganisms 2017, 5, 77. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast Length Adjustment of Short Reads to Improve Genome Assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME Improves Sensitivity and Speed of Chimera Detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, P.; Bose, S.K.; Park, K.-I.; Dufossé, L.; Fouillaud, M. Plant-Microbe Interactions under the Extreme Habitats and Their Potential Applications. Microorganisms 2024, 12, 448. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, Y.; Zhao, C.; Li, H.; Shen, X.; Zhou, M.; Daigger, G.T.; Zhang, P.; Song, G. Effects of Nitrogen and Carbon Source Addition on Biomass and Protein Production by Rhodopseudomonas via the RSM-CCD Approach. Desalination Water Treat. 2024, 319, 100438. [Google Scholar] [CrossRef]
- Fierer, N.; Jackson, R.B. The Diversity and Biogeography of Soil Bacterial Communities. Proc. Natl. Acad. Sci. USA 2006, 103, 626–631. [Google Scholar] [CrossRef]
- Khan, F.; Siddique, A.B.; Shabala, S.; Zhou, M.; Zhao, C. Phosphorus Plays Key Roles in Regulating Plants’ Physiological Responses to Abiotic Stresses. Plants 2023, 12, 2861. [Google Scholar] [CrossRef]
- Yadav, A.; Yadav, K. Regulation of Plant-Microbe Interactions in the Rhizosphere for Plant Growth and Metabolism: Role of Soil Phosphorus. In Phosphorus in Soils and Plants; IntechOpen: London, UK, 2024. [Google Scholar] [CrossRef]
- Azarbad, H.; Junker, R.R. Biological and Experimental Factors That Define the Effectiveness of Microbial Inoculation on Plant Traits: A Meta-Analysis. ISME Commun. 2024, 4, ycae122. [Google Scholar] [CrossRef]
- Liu, X.; Le Roux, X.; Salles, J.F. The Legacy of Microbial Inoculants in Agroecosystems and Potential for Tackling Climate Change Challenges. iScience 2022, 25, 103821. [Google Scholar] [CrossRef]
- Bergna, A.; Cernava, T.; Rändler, M.; Grosch, R.; Zachow, C.; Berg, G. Tomato Seeds Preferably Transmit Plant Beneficial Endophytes. Phytobiomes J. 2018, 2, 183–193. [Google Scholar] [CrossRef]
- Nizamani, M.M.; Hughes, A.C.; Qureshi, S.; Zhang, Q.; Tarafder, E.; Das, D.; Acharya, K.; Wang, Y.; Zhang, Z.-G. Microbial Biodiversity and Plant Functional Trait Interactions in Multifunctional Ecosystems. Appl. Soil Ecol. 2024, 201, 105515. [Google Scholar] [CrossRef]
- Trivedi, P.; Leach, J.E.; Tringe, S.G.; Sa, T.; Singh, B.K. Plant–Microbiome Interactions: From Community Assembly to Plant Health. Nat. Rev. Microbiol. 2020, 18, 607–621. [Google Scholar] [CrossRef]
- Wei, X.; Xie, B.; Wan, C.; Song, R.; Zhong, W.; Xin, S.; Song, K. Enhancing Soil Health and Plant Growth through Microbial Fertilizers: Mechanisms, Benefits, and Sustainable Agricultural Practices. Agronomy 2024, 14, 609. [Google Scholar] [CrossRef]
- Ahmed, W.; Yang, J.; Tan, Y.; Munir, S.; Liu, Q.; Zhang, J.; Ji, G.; Zhao, Z. Ralstonia Solanacearum, a Deadly Pathogen: Revisiting the Bacterial Wilt Biocontrol Practices in Tobacco and Other Solanaceae. Rhizosphere 2022, 21, 100479. [Google Scholar] [CrossRef]
- Pandey, S.; Gupta, S. Diversity Analysis of ACC Deaminase Producing Bacteria Associated with Rhizosphere of Coconut Tree (Cocos nucifera L.) Grown in Lakshadweep Islands of India and Their Ability to Promote Plant Growth under Saline Conditions. J. Biotechnol. 2020, 324, 183–197. [Google Scholar] [CrossRef]
- de Souza, R.; Ambrosini, A.; Passaglia, L.M.P. Plant Growth-Promoting Bacteria as Inoculants in Agricultural Soils. Genet Mol. Biol. 2015, 38, 401–419. [Google Scholar] [CrossRef]
- Azarbad, H. Conventional vs. Organic Agriculture–Which One Promotes Better Yields and Microbial Resilience in Rapidly Changing Climates? Front. Microbiol. 2022, 13, 903500. [Google Scholar] [CrossRef]
- Dong, W.; Liu, H.; Ning, Z.; Bian, Z.; Zeng, L.; Xie, D. Inoculation with Bacillus Cereus DW019 Modulates Growth, Yield and Rhizospheric Microbial Community of Cherry Tomato. Agronomy 2023, 13, 1458. [Google Scholar] [CrossRef]
- Kumar, M.; Kour, D.; Yadav, A.N.; Saxena, R.; Rai, P.K.; Jyoti, A.; Tomar, R.S. Biodiversity of Methylotrophic Microbial Communities and Their Potential Role in Mitigation of Abiotic Stresses in Plants. Biologia 2019, 74, 287–308. [Google Scholar] [CrossRef]
- Daud, N.S.; Mohd Din, A.R.J.; Rosli, M.A.; Azam, Z.M.; Othman, N.Z.; Sarmidi, M.R. Paenibacillus Polymyxa Bioactive Compounds for Agricultural and Biotechnological Applications. Biocatal. Agric. Biotechnol. 2019, 18, 101092. [Google Scholar] [CrossRef]
- Choi, A.; Cha, I.-T.; Lee, K.-E.; Son, Y.K.; Yu, J.; Seol, D. The Role of Flavobacterium Enshiense R6S-5-6 in the Wetland Ecosystem Revealed by Whole-Genome Analysis. Curr. Microbiol. 2023, 80, 83. [Google Scholar] [CrossRef]
- Lee Díaz, A.S.; Macheda, D.; Saha, H.; Ploll, U.; Orine, D.; Biere, A. Tackling the Context-Dependency of Microbial-Induced Resistance. Agronomy 2021, 11, 1293. [Google Scholar] [CrossRef]
- Lee Díaz, A.S.; Minchev, Z.; Raaijmakers, J.M.; Pozo, M.J.; Garbeva, P. Impact of Bacterial and Fungal Inoculants on the Resident Rhizosphere Microbiome and the Volatilome of Tomato Plants under Leaf Herbivory Stress. FEMS Microbiol. Ecol. 2024, 100, fiad160. [Google Scholar] [CrossRef] [PubMed]
- Zengler, K.; Hofmockel, K.; Baliga, N.S.; Behie, S.W.; Bernstein, H.C.; Brown, J.B.; Dinneny, J.R.; Floge, S.A.; Forry, S.P.; Hess, M.; et al. EcoFABs: Advancing Microbiome Science through Standardized Fabricated Ecosystems. Nat. Methods 2019, 16, 567–571. [Google Scholar] [CrossRef]
- Kong, Z.; Hart, M.; Liu, H. Paving the Way From the Lab to the Field: Using Synthetic Microbial Consortia to Produce High-Quality Crops. Front. Plant Sci. 2018, 9, 1467. [Google Scholar] [CrossRef]
- Li, C.; Li, Y.; Ma, J.; Wang, Y.; Wang, Z.; Liu, Y. Microbial Community Variation and Its Relationship with Soil Carbon Accumulation during Long-Term Oasis Formation. Appl. Soil Ecol. 2021, 168, 104126. [Google Scholar] [CrossRef]
- Brown, S.P.; Grillo, M.A.; Podowski, J.C.; Heath, K.D. Soil Origin and Plant Genotype Structure Distinct Microbiome Compartments in the Model Legume Medicago Truncatula. Microbiome 2020, 8, 139. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hadian, S.; Smith, D.L.; Supronienė, S. Modulating the Plant Microbiome: Effects of Seed Inoculation with Endophytic Bacteria on Microbial Diversity and Growth Enhancement in Pea Plants. Microorganisms 2025, 13, 570. https://doi.org/10.3390/microorganisms13030570
Hadian S, Smith DL, Supronienė S. Modulating the Plant Microbiome: Effects of Seed Inoculation with Endophytic Bacteria on Microbial Diversity and Growth Enhancement in Pea Plants. Microorganisms. 2025; 13(3):570. https://doi.org/10.3390/microorganisms13030570
Chicago/Turabian StyleHadian, Shervin, Donald L. Smith, and Skaidrė Supronienė. 2025. "Modulating the Plant Microbiome: Effects of Seed Inoculation with Endophytic Bacteria on Microbial Diversity and Growth Enhancement in Pea Plants" Microorganisms 13, no. 3: 570. https://doi.org/10.3390/microorganisms13030570
APA StyleHadian, S., Smith, D. L., & Supronienė, S. (2025). Modulating the Plant Microbiome: Effects of Seed Inoculation with Endophytic Bacteria on Microbial Diversity and Growth Enhancement in Pea Plants. Microorganisms, 13(3), 570. https://doi.org/10.3390/microorganisms13030570